
Abstract
Four new oleanane-type saponins, macrostachyaosides A, B, C, and D (1–4) were isolated from the roots of Acacia macrostachya. Their structures were elucidated on the basis of extensive 1D- and 2D-NMR data and HR-ESI-MS analyses. At concentrations of 100 μM of each compounds, none of the tested compounds caused a significant growth reduction against HL60 cells.
Four new oleanane-type saponins were isolated from Acacia macrostachya. Their structures were elucidated using 1D and 2D NMR and HRESIMS.

Acacia macrostachya (Mimosaceae) is a scrambling shrub, or tree to 20 ft. high, with rusty-pubescent branchlets, armed with recurved prickles distributed in Africa – drier areas from Senegal to Sudan and Angola.Citation1) The plant is used in the traditional treatment of snakebites and pathologies accompanied with oxidative stress such as malaria, inflammation, and painful spasms.Citation2) The young leaves are boiled for treatment of gastro-intestinal disorders, as an anti-helminthic, and as an antidote to snakebite. Acacia macrostachya induced a very significant antiproliferative effect against cancer cells (KB, Vero and MCR-5),Citation3) and developed a strong antioxidant activity.Citation2,3) Although A. machrostachya has proved to be very rich in saponins, flavonoids, tannins, and alkaloids,Citation2) there is no report on the phytochemical study of this plant to date.
As part of an ongoing search for bioactive saponins from Cameroonian medicinal plants,Citation4) four new triterpene saponins, macrostachyaosides A, B, C, and D (1–4) were isolated from the roots of Acacia macrostachya. This paper describes the isolation and structure elucidation of these new compounds.
The air-dried and powdered roots of Acacia macrostachya were extracted with n-hexane, then with MeOH at room temperature. MeOH crude extract was dissolved in H2O, and successively partitioned with EtOAc, and n-BuOH saturated with H2O. The n-BuOH extract was pretreated with Diaion HP-20 column followed by Sephadex LH-20 column before ODS chromatography. Enriched saponin fractions were applied to flash chromatography over a prepack silica gel cartridge and purification over repeated open ODS column chromatography afforded four new triterpene saponins, macrostachyaosides A, B, C, and D (1–4) (Fig. ).
Figure 1. Chemical structures of compounds 1–4.
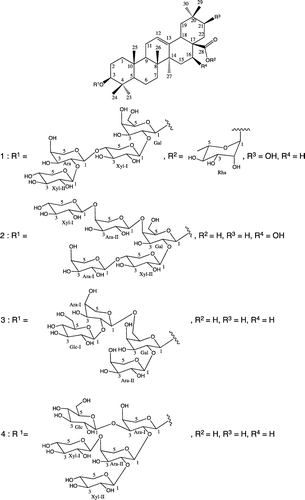
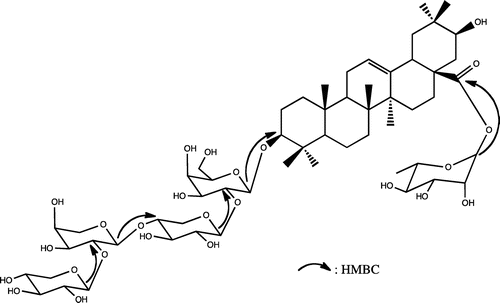
Macrostachyaoside A (1) was obtained as colorless powder. The IR spectrum showed absorption bands for hydroxyl and olefin moiety at νmax 3583 and 1635 cm−1, respectively. Its HR-ESI-MS (positive-ion mode) exhibited an accurate pseudo-molecular ion peak at m/z 1199.58199 [M + Na]+, consistent with the molecular formula C57H92O25Na. The 1H-NMR spectrum of 1 displayed signals of five anomeric protons at δH 4.25 (1H, d, J = 5.9 Hz), 4.31 (1H, d, J = 6.7 Hz), 4.42 (1H, d, J = 6.3 Hz), 4.57 (1H, d, J = 5.9 Hz), and 5.89 (1H, d, J = 1.3 Hz), which correlated, in the HMQC, with five anomeric carbon signals at δC 102.4, 104.0, 103.8, 104.5, and 93.6, respectively. The 13C-NMR spectrum showed characteristic signals of an olean-12-ene skeleton with seven methyl singlet signals at δC 14.6, 15.2, 16.7, 23.9, 24.4, 26.7, and 26.9 and two ethylenic carbons at δC 123.1 and 143.3. The 1H–1H spin-couplings of the hydroxymethine proton at δH 3.41 (1H, t, J = 9.6 Hz) to methylene protons at δH 1.67 (1H, dd, J = 3.0, 14.4 Hz) and δH 1.87 (1H, d, J = 3.0 Hz), together with the HMBC correlations with C-17 (δC 47.0) and C-19 (δC 40.2), indicated the presence of a hydroxyl group at C-21. Moreover, the upfield shift of the C-19 (δC 40.2) was consistent with the presence of the C-21α-axial hydroxyl group. The three-proton doublet signal at δH 1.22 (3H, d, J = 6.0 Hz) indicated the presence of a deoxyhexopyranosyl unit. Acid hydrolysis of 1 with 2N HCl gave 21α-hydroxyoleanolic acid (machaerinic acid),Citation5,6) together with D-galactose, D-xylose, L-arabinose, and L-rhamnose, which were identified by comparison in HPLC of their retention time and optical rotation with standard sugars.Citation4) The downfield shift of aglycone (Agly) C-3 and the upfield shift of Agly C-28 indicated that 1 was a bidesmosidic saponin of machaerinic acid. Based on the 1H-, 13C-NMR spectral data, and literature,Citation7) the anomeric configurations of the sugar moieties were determined as β for galactose and xylose, and α-configuration for arabinose and rhamnose. The 2D-NMR data allowed the identification of a tetrasaccharide chain of one D-galactose, two D-xylose, and one L-arabinose at C-3 and a unit of one L-rhamnose at C-28. The ring protons of the five monosaccharides were assigned starting from the anomeric protons by means of COSY, TOCSY, HMQC, and HMBC experiments. The sequencing of the glycosidic chains was achieved by analysis of the HMBC. In the HMBC spectrum (Fig. S7), long-range correlation observed between H-1 (δH 4.42) of galactose and Agly C-3, suggested that Gal was attached to C-3 of the aglycone. The HMBC spectrum showed interglycosidic correlations for the tetrasaccharidic chain between H-1 (δH 4.57) of the xylose (Xyl-I) and C-2 (δC 81.0) of the galactose, H-1 (δH 4.25) of the arabinose and C-4 (δC 76.5) of the xylose (Xyl-I), H-1 (δH 4.31) of the xylose (Xyl-II) and C-2 (δC 79.0) of the arabinose (Fig. ). These correlations suggested that 1 has a linear tetrasaccharide chain at Agly C-3 identified as β-D-xylopyranosyl-(1 → 2)-α-L-arabinopyranosyl-(1 → 4)-β-D-xylopyranosyl-(1 → 2)-β-D-galactopyranoside. Further HMBC correlations between H-1 (δH 5.89) of rhamnose and C-28 (δC 175.4), suggested that Rha was directly attached to C-28 of the aglycone (Fig. ). Based on the above evidence, the structure of 1 was established as 3-O-β-D-xylopyranosyl-(1 → 2)-α-L-arabinopyranosyl-(1 → 4)-β-D-xylopyranosyl-(1 → 2)-β-D-galactopyranosylmachaerinic acid-28-O-α-L-rhamnopyranosyl ester, named macrostachyaoside A.
Figure 2. Key HMBC correlations of compound 1.
Macrostachyaoside B (2) was obtained as colorless powder. Its HR-ESI-MS (positive ion mode) exhibited an accurate pseudo-molecular ion peak at m/z 1185.56629 [M + Na]+, in accordance with the molecular formula C56H90O25Na. The 1H-NMR spectrum showed seven angular methyl signals as singlet at δH, 0.77, 0.80, 0.85, 0.93, 0.94, 1.02, and 1.34 (each 3H), one olefinic proton signal at δH 5.27 (1H, t-like, J = 3.0 Hz), and five anomeric protons at δH 4.25 (1H, d, J = 6.6 Hz), 4.31 (1H, d, J = 7.2 Hz), 4.43 (1H, d, J = 8.4 Hz), 4.47 (1H, d, J = 8.4 Hz), and 4.56 (1H, d, J = 6.6 Hz). The 13C-NMR spectrum showed characteristic signals of an olean-12-ene skeleton with seven methyl singlet signals at δC 14.6, 15.3, 16.3, 23.4, 25.8, 26.7, and 32.0, two ethylenic carbons at δC 122.0 and 143.7, and five anomeric carbons at δC 102.5, 103.7, 103.8, 103.9, and 104.6. Acid hydrolysis with 2 N HCl gave 16α-oleanolic acid (echinocystic acid),Citation8) along with D-galactose, D-xylose, and L-arabinose. In the 13C-NMR spectrum of 2, carbon signals at δC 89.6 and 180.0 for C-3 and C-28, respectively, suggested that 2 is a monodesmosidic saponin with sugar chain linked at C-3 of the aglycone. The relatively large coupling constant values of the galactosyl, xylosyl, and arabinosyl moieties (J = 6.6–8.4 Hz) indicated that the anomeric configurations of the sugar moieties were determined as β for galactose and xylose, and α-configuration for arabinose. HMBC correlations between the anomeric signal at H-1 (δH 4.43) of galactose and C-3 (δC 89.6) of the aglycone revealed a substitution at C-3 of the aglycone (Agly C-3) by a galactopyranosyl unit. Furthermore, in the HMBC spectrum of 2, long-range correlations were observed between H-1 (δH 4.31) of arabinose (Ara-II) and C-4 (δC 78.7) of galactose, and H-1 (δH 4.47) of xylose (Xyl-I) and C-4 (δC 78.6) of arabinose (Ara-II). Further correlations were observed between H-1 (δH 4.25) of arabinose (Ara-I) and C-4 (δC 76.6) of xylose (Xyl-II), and H-1 (δH 4.56) of xylose (Xyl-II) and C-2 (δC 81.1) of galactose. The absence of any glycosylation shifts at Xyl-I and Ara-I indicated that they are terminal sugars and supported the sequence of the sugars units. Hence, macrostachyaoside B (2) was elucidated as echynocystic acid 3-O-β-D-xylopyranosyl-(1 → 4)-α-L-arabinopyranosyl-(1 → 4)-[α-L-arabinopyranosyl-(1 → 4)-β-D-xylopyranosyl-(1 → 2)]-β-D-galactopyranoside.
Macrostachyaoside C (3) was obtained as colorless powder. Its HR-ESI-MS (positive ion mode) exhibited an accurate pseudo-molecular ion peak at m/z 1067.53962 [M + Na]+, in accordance with the molecular formula C52H84O21Na. The 1H-NMR spectrum showed seven methyl singlets at δH 0.78, 0.83, 0.88, 0.91, 0.93, 1.06, and 1.14 (each 3H), one olefinic proton signal at δH 5.21 (1H, t-like, J = 3.6 Hz). The aglycone was identified as oleanolic acid.Citation5,8) Besides, the 1H-NMR of 3 displayed signals of four anomeric protons at δH 4.42 (1H, d, J = 7.2 Hz), 4.47 (1H, d, J = 6.6 Hz), 4.48 (1H, d, J = 6.0 Hz), and 4.65 (1H, d, J = 7.8 Hz), which correlated with the anomeric carbons at δC 104.0, 104.6, 102.3, and 103.1, respectively, in the HMQC spectrum. Acid hydrolysis of 3 afforded L-arabinose, D-glucose, and D-galactose confirmed by specific rotation and retention time in HPLC. The 1H- and 13C-NMR spectra indicated the presence of two α-arabinopyranosyl units (δH 4.47 and 4.48), one β-glucopyranosyl unit (δH 4.65), and β-galactopyranosyl unit (δH 4.42). In the 13C-NMR spectrum, the C-2 and C-6 positions of the galactose (Gal) and C-2 position of arabinose (Ara-I) were shifted to δC 79.3, 68.3, and 79.1, respectively, establishing the site of glycosylation shifts. The sequencing of the sugars chain was achieved by analysis of HMBC and NOESY experiments. In the HMBC spectrum of 3, long-range correlations observed between H-1 (δH 4.42) of the galactose and C-3 (δC 89.7) of the aglycone, and the NOESY correlation between Gal H-1 (δH 4.42) and Agly H-3 (δH 3.25) revealed a linkage between the aglycone and a galactopyranosyl moiety. In the HMBC spectrum, long-range correlations were observed between H-1 (δH 4.48) of the arabinose (Ara-I) and C-6 (δC 68.3) of the galactose (Gal), H-1 (4.65) of glucose (Glc) and C-2 (δC 79.1) of arabinose (Ara-I), and H-1 (δH 4.47) of arabinose (Ara-II) and C-2 (δC 79.3) of galactose (Gal-I). Above observations were confirmed by the NOESY correlations between Ara-I H-1 (δH 4.48) and Gal H-6 (δH 3.69, 4.02), Ara-II H-1 (δH 4.47) and Gal H-2 (δH 3.54). Hence, 3 was determined as oleanolic acid 3-O-β-D-glycopyranosyl-(1 → 2)-α-L-arabinopyranosyl-(1 → 6)-[α-L-arabinopyranosyl-(1 → 2)]-β-D-galactopyranoside, named macrostachyaoside C.
Macrostachyaoside D (4) was obtained as colorless powder. Its HR-ESI-MS (positive ion mode) exhibited an accurate pseudo-molecular ion peak at m/z 1169.57190 [M + Na]+, in accordance with the molecular formula C56H90O24Na. The 1H-NMR spectrum showed seven methyl singlets at δH 0.79, 0.83, 0.88, 0.92, 0.93, 1.06, and 1.14 (each 3H), one olefinic proton signal at δH 5.22 (1H, t-like, J = 3.6 Hz), and five anomeric protons at δH 4.39 (1H, d, J = 7.2 Hz), 4.43 (1H, d, J = 7.8 Hz), 4.48 (1H, d, J = 7.2 Hz), 4.50 (1H, d, J = 7.8 Hz), and 4.69 (1H, d, J = 8.4 Hz). The 1H- and 13C-NMR data (Table ) indicated that 4 was also a C-3 monodesmoside of oleanolic saponin. The signals of the aglycone were superimposable with those of 3 but the sugars moieties were different. Units of one β-glucopyranosyl, two β-xylopyranosyl, and two α-arabinopyranosyl were identified. Acid hydrolysis of 4 yielded D-glucose, D-xylose, and L-arabinose. The sequence of the glycoside chain connected to C-3 was established by analysis of the HMBC correlations. In the HMBC spectrum, the anomeric proton at δH 4.43 (1H, d, J = 7.8 Hz, Ara H-1) showed correlation with Agly C-3 (δC 90.3), hence, Ara was linked to Agly C-3 through ether linkage. The signal of Ara C-3 (δC 78.1) is shifted downfield, due to the glycosylation shift. This is confirmed in the HMBC spectrum by the long-range correlation observed between H-1 (δH 4.50) of the terminal Glc and Ara C-3 (δC 78.1) (Ara-I). The linkage of the three other sugars was confirmed by HMBC correlations of H-1 (δH 4.69) of the xylose (Xyl-I) and C-4 (δC 78.6) of arabinose (Ara-II), H-1 (δH 4.39) of the xylose (Xyl-II) and C-2 (δC 82.5) of the arabinose (Ara-II), H-1 (δH 4.48) of the arabinose (Ara-II) and C-2 (δC 75.0) of the arabinose (Ara-I). Therefore, the structure of 4 was determined as oleanolic acid 3-O-β-D-glucopyranosyl-(1 → 3)-[β-D-xylopyranosyl-(1 → 4)-[β-D-xylopyranosyl-(1 → 2)]-α-L-arabinopyranosyl-(1 → 2)]-α-L-arabinopyranoside, named macrostachyaoside D.
Table 1. 1H- and 13C-NMR data for the aglycone moieties of 1–4 (in MeOH-d4).Table Footnotea
The cytotoxicity activities of 1, 2, 3, and 4 against HL60 cells were evaluated by MTT method. However, none of the tested compounds caused a significant reduction of the cell number. As a result, they did not show cytotoxicity (IC50 > 100 μM).
Experimental
General procedures
Optical rotation was measured with a Horiba SEPA-300 polarimeter (HORIBA, Kyoto, Japan). IR spectrum was recorded with a Jasco J-20A (JASCO Cooperation, Tokyo, Japan) and Shimadzu UV mini-1240 (SHIMADZU, Kyoto, Japan) spectrophotometers. The ESI-MS was measured on a Varian AAT 311A mass spectrometer, and the HRESIMS was taken on a JEOL HX110 mass spectrometer (JEOL, Tokyo, Japan). 1D and 2D NMR spectra were run on JEOL JNM-ECZ600R/S1 600 MHz NMR spectrometer. The 1H (600 MHz) and 13C (150 MHz) chemical shifts were referenced to the residual solvent peak of methanol-d4 at δH 3.30 for proton and δC 49.0 ppm for carbon. Coupling constants (J) are in hertz. The 1H sweep width was set at 11282 Hz for all experiments with a 45° pulse for 1H and a 30° pulse for 13C. The pulse programs of the COSY, HMQC, and HMBC experiments were taken from the Varian Software Library and standard pulse sequences were used for 2D spectra. Semipreparative HPLC was carried out with Shimadzu pump and UV LC-10A detector (set at 210 nm) on Mightysil RP-18 column (250 × 6.0 mm i.d.) at the flow rate of 0.8 and 1.0 mL/min−1. Column chromatography was conducted on silica gel 60 (Kanto Chemical Co, Inc., Japan). TLC was carried out on Merck precoated silica gel plates (silica gel 60 F254, 20 × 20 cm, Merck, Darmstadt, Germany), and spots were detected by spraying with H2SO4 followed by heating.
Plant material
Acacia macrostachya was collected in November 2015 in Figuil, North Region of Cameroon and identified by Dr. Guidawa Fawa, Department of Biological Sciences, University of Ngaoundere, Cameroon. A voucher specimen N° 38107 HNC has been deposited at the National Herbarium Cameroon.
Extraction and isolation
A. macrostachya (liana, 1.5 kg) was extracted with n-hexane (2 × 5 L), then with MeOH (3 × 5 L) at room temperature. After filtration and evaporation procedures, n-hexane (4 g) and MeOH (50 g) extracts were obtained. The MeOH extract was dissolved in H2O (400 mL), and successively partitioned with EtOAc (3 × 200 mL), and n-BuOH (3 × 200 mL) saturated with H2O. The n-BuOH extract (20 g) was pretreated with Diaion HP-20 column followed by Sephadex LH-20 column before ODS chromatography. This reverse phase ODS chromatography was used with the eluent MeOH–H2O (6:4), repeated two times and combined the same zones (fractions) to give six main fractions (Fr. A-1 to A-6). Two enriched saponin fractions were applied separately to Flash chromatography using a prepack silica gel cartridge, 80 g. Fr. A-1 was subjected to flash chromatography with the following eluates, CHCl3–MeOH–H2O (80:20:2 and 70:30:3) yielded 505 fractions (Fr. B-1 to B-505). Frs. B-180 to B-195 were combined and chromatographed on ODS with MeOH–H2O (7:3) yielding 1 (17.3 mg, HPLC chart: Supplementary Figure 34a). Frs. B-196 to B-215 were combined and applied to an open ODS column with solvent system MeOH-H2O (7:3) to give 2 (7.6 mg, HPLC chart: Supplementary Figure 34b). Fr. A-3 was applied to flash chromatography with CHCl3-MeOH-H2O (80:20:2 and 70:30:3) yielding 368 fractions (Fr. C-1 to C-368). Frs. C-126 to C-145 were combined and separated by ODS column with MeOH–H2O (8:2) to yield 3 (6.6 mg, HPLC chart: Supplementary Figure 34c). Frs. C-160 to C-169 were combined and separated by ODS column with MeOH–H2O (8:2) to give 4 (8.5 mg, HPLC chart: Supplementary Figure 34d).
Macrostachyaoside A (1)
White amorphous powder; − 6.7° (c 0.5, MeOH); IR (KBr) νmax 3737, 3583, 3467, 3436, 2302, 1635, 1519, 1373, and 1045 cm−1; 1H- and 13C-NMR data, see Tables and ; HR-ESI-MS (positive-ion mode) m/z 1199.58199 [M + Na]+ calcd for C57H92O25Na 1199.58199).
Table 2. 1H- and 13C- NMR data for the sugar moieties of 1–4 (in MeOH-d4).Table Footnotea
Macrostachyaoside B (2)
White amorphous powder; − 4.7° (c 0.4, MeOH); IR (KBr) νmax 3737, 3586, 3293, 2981, 2321, 1650, 1523, 1376, and 1083 cm−1; 1H- and 13C-NMR data, see Tables and ; HR-ESI-MS (positive-ion mode) m/z 1185.56629 [M + Na]+ calcd for C56H90O25Na 1185.56634)
Macrostachyaoside C (3)
White amorphous powder; + 7.3° (c 0.6, MeOH); IR (KBr) νmax 3745, 3583, 3467, 3444, 2306, 1635, 1519, and 1079 cm−1; 1H- and 13C-NMR data, see Tables and ; HR-ESI-MS (positive-ion mode) m/z 1067.53962 [M + Na]+ calcd for C52H84O21Na 1067.53973).
Macrostachyaoside D (4)
White amorphous powder; + 3.4° (c 0.5, MeOH); IR (KBr) νmax 3598, 3390, 3270, 2375, 1700, 1542, 1376, and 1045 cm−1; 1H- and 13C-NMR data, see Tables and ; HR-ESI-MS (positive-ion mode) m/z 1169.57190 [M + Na]+ calcd for C56H90O24Na 1169.57142.
Acid hydrolysis and HPLC analysis
According to the reported method.Citation4) Briefly, each solution of 1–4 (5 mg) in 0.2 M HCl (dioxane-H2O 1:1, 3 mL) was heated at 95 °C for 30 min under argon. After cooling, the mixture was neutralized by passage through an Amberlite-IRA-93ZU (Organo, Tokyo, Japan) column and chromatographed (Diaion HP-20, 40% MeOH followed by Me2CO-EtOH 1:1) to give aglycone fractions (2.5 mg) and a sugar fraction (1.7 mg). The aglycone was identified as machaerinic acid ( + 81.7°; c 0.5, MeOH) in compound 1, echinocystic acid (
+ 32°; c 0.6, MeOH) in compound 2, and oleanolic acid (
+ 76°; c 0.6, MeOH) in compounds 3 and 4. After the sugar fraction was passed through a Sep-Pak-C18 cartridge (Waters, Milford, MA, USA; with 40% MeOH) and Toyopak-IC-SP-M-cartridge (Tosoh; with 40% MeOH), it was analyzed by HPLC (MeCN-H2O 17: 3, flow rate, 0.9 mL min−1; detection, refractive index (RI) and optical rotation (OR): 7.79 (L-rhamnose, negative OR); 9.35 (L-arabinose, positive OR), 9.73 (D-xylose, positive OR). 15.37 (D-glucose, positive OR); 14.65 (D-galactose, positive OR).
Cell culture and cytotoxicity
HL60 cells (RCB0041, RIKEN BioResource Center, Tsukuba, Japan) were grown in RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Sigma-Aldrich Co. LLC. Mexico origin) and penicillin (50 units/ml)-streptomycin (50 μg/ml) (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) in a humidified atmosphere at 37 °C under 5% CO2. The cytotoxicity of the compounds was examined by MTT assay, as described previously.Citation4) Positive control camptothecin was used as positive control for HL60 with IC50 = 23.6 nM.
Author contributions
The study was designed by AT, TKT, TK, YS, and BTN. The cytotoxic assay was performed and analyzed by NU and KK. The ESI-MS spectra were measured by EK and HM. All authors discussed the results. The manuscript was written by AT, TKT, and YS, and revised by all authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the JSPS RONPAKU (PhD Dissertation) [grant number R11516] to Abdou Tchoukoua.
Supplemental material
The supplemental material for this paper is available at https://doi.org/10.1080/09168451.2017.1390393.
Revised_R3_Supp_info-AMA.doc
Download MS Word (5.1 MB)Related Research Data
References
- Hutchinson J, Dalziel JM. Flora of West Tropical Africa ( Revised by Keay RWJ). London: Millbank; 1958. p. 496–501.
- Tondé I, Fofana S, Gnoula C, et al. Antiplasmodial and DPPH radical scavenging effects in extracts from Acacia macrostachya (Mimosaceae) DC. World J Pharm Res. 2016;5:219–233.
- Sawadogo WR, Maciuk A, Banzouzi JT, et al. Mutagenic effect, antioxidant and anticancer activities of six medicinal plants from Burkina Faso. Nat Prod Res. 2012;26:575–579.10.1080/14786419.2010.534737
- Tchoukoua A, Tabopda TK, Uesugi S, et al. Triterpene saponins from the roots of Acacia albida Del. (Mimosaceae). Phytochemistry. 2017;136:31–38.10.1016/j.phytochem.2016.12.019
- Mimaki Y, Yokosuka A, Hamanaka M, et al. Triterpene saponins from the roots of Clematis chinensis. J Nat Prod. 2004;67:1511–1516.10.1021/np040088 k
- Delgado MCC, Da silva MS, Fot RB. 3β-hydroxy-21β-E-cinnamoyloxyolean-12-en-28-oic acid, a triterpenoid from Enterolobium contorstisiliquum. Phytochemistry. 1984;23:2289–2292.10.1016/S0031-9422(00)80537-3
- Agrawal PK. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry. 1992;31:3307–3330.10.1016/0031-9422(92)83678-R
- Nigam SK, Gopal M, Uddin R, et al. Pithedulosides A-G, oleanane glycosides from Pithecellobium dulce. Phytochemistry. 1997;44:1329–1334.10.1016/S0031-9422(96)00725-X