
Abstract
Polyketides constitute a large family of natural products that display various biological activities. Polyketides exhibit a high degree of structural diversity, although they are synthesized from simple acyl building blocks. Recent biochemical and structural studies provide a better understanding of the biosynthetic logic of polyketide diversity. This review highlights the biosynthetic mechanisms of structurally unique polyketides, β-amino acid-containing macrolactams, enterocin, and phenolic lipids. Functional and structural studies of macrolactam biosynthetic enzymes have revealed the unique biosynthetic machinery used for selective incorporation of a rare β-amino acid starter unit into the polyketide skeleton. Biochemical and structural studies of cyclization enzymes involved in the biosynthesis of enterocin and phenolic lipids provide mechanistic insights into how these enzymes diversify the carbon skeletons of their products.
Various strategies for production of structurally diverse polyketides.

Polyketides are distributed widely in bacteria, fungi, and plants, and form a large family of natural products with various biological activities. Polyketides are synthesized by polyketide synthases (PKSs).Citation1) PKSs possess ketosynthase (KS) activity, which catalyzes the condensation of extender units onto an acyl starter substrate or a growing polyketide chain (Fig. (A)). The substrates and reaction intermediates of PKSs are maintained as thioester conjugates to an acyl carrier protein (ACP) or a small molecule, coenzyme A (CoA). An acyltransferase (AT) recognizes a specific acyl starter or extender unit and catalyzes its transfer reaction onto the phosphopantetheine arm of ACP. PKS performs particular reduction and dehydration reactions on each resulting β-keto carbon and catalyzes the intramolecular cyclization of the resulting polyketide chain to generate a monocyclic or polycyclic product.
Fig. 1. The structures of polyketides. The starter units and loading modules are shown in red.
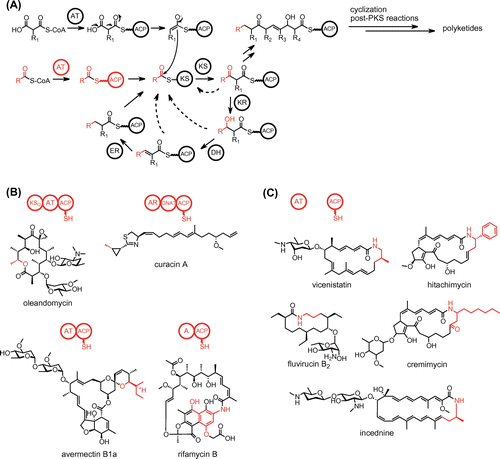
PKSs fall into three groups. Type I PKSs are multifunctional enzymes that are organized into modules, each of which harbors a set of distinct catalytic domains. Modular type I PKSs comprise several modules that are each responsible for a single round of polyketide chain elongation, whereas iterative type I PKSs comprise a single module that acts iteratively for polyketide chain elongation. Type II PKSs consist of a complex of subunits such as KS and ACP. Type III PKSs have the simplest architecture, a homodimer of KS.
PKS systems employ several distinct strategies for the production of structurally diverse polyketides, including the use of various starter and extender units for assembly of the polyketide chain, the number of condensation reactions, the diverse mechanisms of cyclization of the fully elongated polyketide chain, and a wide range of post-PKS tailoring reactions. Understanding how PKS systems diversify their polyketide products is important for engineering metabolic pathways to produce artificial natural products. This review describes the biosynthetic systems of structurally unique polyketides that the author has analyzed: β-amino acid-containing macrolactams synthesized by modular type I PKSs; enterocin synthesized by a type II PKS; and phenolic lipids synthesized by type III PKSs.
I. Biosynthesis of vicenistatin and other β-amino acid-containing macrolactam polyketides
Bacterial modular type I PKSs use various starter units and adopt several strategies for starter unit loading.Citation1–3) A loading module of modular PKS is responsible for the selection of a starter unit. A KSQ-type loading module consists of KSQ, AT, and ACP domains (Fig. (B)). In this system, the AT domain selects malonyl-CoA or methylmalonyl-CoA as a starter substrate and transfers it onto the ACP domain. The KSQ domain subsequently catalyzes the decarboxylation of the malonyl or methylmalonyl group to construct an acetate or propionate unit, respectively, which is transferred to the downstream extension module for polyketide chain elongation. The acetate starter unit can also be introduced by a GCN5-related N-acetyltransferase (GNAT) domain. The GNAT domain catalyzes the decarboxylation of malonyl-CoA and transfers the resulting acetate group to the ACP domain of the loading module.Citation4) Acyl starter units other than acetate and propionate units can be introduced by an AT-ACP or adenylation (A)-ACP didomain-type loading module (Fig. (B)).Citation1,3) The didomain-type loading AT domain generally selects an acyl-CoA substrate and directly loads the acyl unit onto the ACP domain (Fig. (A)). The loading A domain selects an acyl substrate as a free carboxylic acid and loads the acyl unit onto the ACP domain through activation using ATP. Both AT-ACP and A-ACP didomain-type loading modules can select various starter units, providing structural diversity to the polyketide products.
β-Amino acid-containing macrolactam antibiotics are an important class of macrocyclic polyketides.Citation5,6) They contain various β-amino acid starter units in their polyketide skeletons (Fig. (C)). Vicenistatin isolated from Streptomyces halstedii HC34 contains a 3-aminoisobutyrate unit, fluvirucin B2 isolated from Actinomadura fulva subsp. indica ATCC 53714 contains a β-alanine unit, hitachimycin isolated from Streptomyces scabrisporus JCM 11712 contains a β-phenylalanine unit, cremimycin isolated from Streptomyces sp. MJ635-86F5 contains a 3-aminononanoate unit, and incednine isolated from Streptomyces sp. ML694-90F3 contains a 3-aminobutyrate unit. Most of these β-amino acids are synthesized from proteinogenic α-amino acids. For example, the 3-aminoisobutyrate unit is generated from l-glutamate in vicenistatin biosynthesis.Citation7–9) (S)-β-Phenylalanine is converted from l-phenylalanine by the phenylalanine 2,3-aminomutase HitA in hitachimycin biosynthesis.Citation10) Interestingly, the 3-aminononanoate unit is constructed through the polyketide pathway in cremimycin biosynthesis.Citation11) The dual functional thioesterase (TE) CmiS1 catalyzes the Michael addition of glycine to non-2-enoyl-ACP and the subsequent hydrolysis to release N-carboxymethyl-3-aminononanoate, which is converted to 3-aminononanoate by the flavin adenine dinucleotide (FAD)-dependent oxidase CmiS2. The crystal structure of the CmiS1 homolog SAV606 from Streptomyces avermitilis provides mechanistic insight into how CmiS1 catalyzes the Michael addition of glycine.Citation12)
These macrolactam polyketides are synthesized by a modular PKS that contains only the ACP domain in the loading module.Citation6,7) This minimal loading module employs a trans-acting loading AT for the starter unit. The guanidine-containing polyketides such as azalomycin are also known to be synthesized by a minimal loading module-type modular PKS.Citation13) The incorporation mechanism of the β-amino acid unit into a modular PKS was firstly elucidated in the vicenistatin biosynthetic pathway (Fig. ).Citation9) In the vicenistatin biosynthetic pathway, the cobalamin-dependent glutamate mutase consisting of VinH and VinI catalyzes the conversion of l-glutamate to (2S,3S)-3-methylaspartate (3-MeAsp).Citation8) The adenylation enzyme VinN recognizes 3-MeAsp as a β-amino acid and transfers it onto the stand-alone ACP VinL. After decarboxylation by the pyridoxal 5′-phosphate-dependent enzyme VinO, the resulting 3-aminoisobutyrate unit is aminoacylated with l-alanine by another adenylation enzyme VinM to produce a dipeptidyl-VinL. The trans-acting loading AT VinK then transfers the dipeptidyl group from VinL to the ACP domain of the loading module of PKS VinP1 (VinP1LdACP). The terminal alanyl group remains attached during the polyketide elongation reaction and is removed from the elongated polyketide chain by the amidohydrolase VinJ to allow macrolactam formation by the TE domain of PKS VinP4.
Fig. 2. The biosynthetic pathway of vicenistatin.
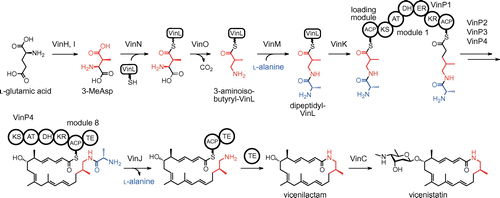
The terminal alanyl group likely functions as a biosynthetic protecting group to avoid spontaneous formation of a six-membered lactam during the PKS reaction.Citation6,9) The terminal alanyl group should be attached before the PKS reaction. Biochemical analysis of VinK showed that VinK strongly prefers dipeptidyl-VinL over 3-aminoisobutyryl-VinL, suggesting that VinK recognizes the terminal alanyl group.Citation9) To obtain insight into the recognition mechanism of the terminal alanyl group, the crystal structure of VinK in the ligand-free form was determined.Citation14) Although the AT domains of the PKS extension modules and KSQ-type loading modules contain a conserved Arg residue in their active sites that interacts with the α-carboxyl group of the dicarboxylic acid thioester substrate,Citation15,16) VinK has Leu131 at the corresponding position to provide sufficient space for the dipeptidyl moiety.Citation14) Based on a docking analysis, the terminal alanyl group appears to be recognized by a few polar residues at the bottom of the substrate-binding tunnel of VinK (Fig. (A)). VinJ seems to regulate the timing of the removal of the terminal alanyl group. VinJ has a long hydrophobic substrate-binding tunnel in the crystal structure, suggesting that a mostly or fully elongated N-alanyl-polyketide intermediate is a suitable substrate for VinJ (Fig. (B)).Citation17)
Fig. 3. The crystal structures of macrolactam biosynthetic enzymes.
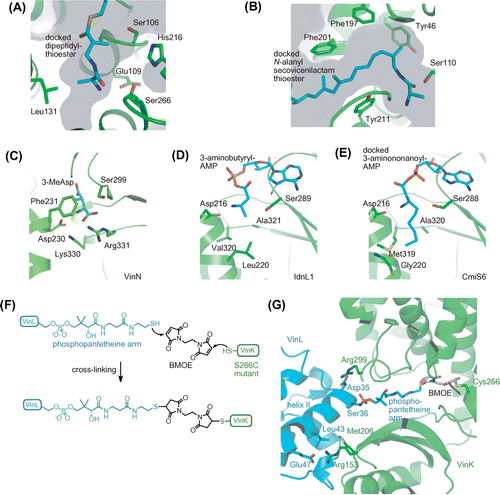
The homologous genes for the five starter-related enzymes, VinN, VinL, VinM, VinK, and VinJ, are fully conserved in biosynthetic gene clusters of other β-amino acid-containing macrolactams, such as fluvirucin B2,Citation18) hitachimycin,Citation10) incednine,Citation19) and cremimycin,Citation11) suggesting that each β-amino acid starter unit is loaded onto a modular PKS by the same mechanism in their biosynthesis. Among these starter-related enzymes, the sequences of VinN-type adenylation enzymes are relatively diverse.Citation6) The substrate specificities of VinN-type enzymes were compared by biochemical and X-ray structural analyses. VinN has a strong preference for 3-MeAsp over other amino acids.Citation9) FlvN, which is involved in fluvirucin B2 biosynthesis, recognizes l-aspartate as a β-amino acid substrate and possesses no activity toward 3-MeAsp.Citation18) The crystal structure of VinN showed that VinN contains two basic residues, Lys330 and Arg331, in the substrate-binding pocket for recognition of the C1 carboxyl group of 3-MeAsp (Fig. (C)).Citation20) These two basic residues are also conserved to FlvN. Phe231 and Ser299 of VinN are located close to the methyl group of 3-MeAsp in the substrate-binding pocket and are replaced by Tyr231 and Thr299 in FlvN, respectively. Substitution of these residues might account for the different substrate specificities between VinN and FlvN. IdnL1 and CmiS6, which are involved in the biosynthesis of incednine and cremimycin, respectively, recognize an aliphatic β-amino acid and show no activity against 3-MeAsp.Citation21) The crystal structures of IdnL1 and CmiS6 show that both enzymes have a hydrophobic substrate-binding pocket. IdnL1 has Leu220 at the bottom of the substrate-binding pocket to recognize short-chain β-amino acids such as 3-aminobutyrate (Fig. (D)), whereas CmiS6 has Gly220 at the corresponding position to accommodate medium-chain β-amino acids such as 3-aminonnanoate (Fig. (E)). A mutational study confirmed that the size of the amino acid residue at position 220 is crucial for the selection of a short-chain and medium-chain aliphatic β-amino acid in IdnL1 and CmiS6, respectively. Thus, VinN-type enzymes possess a different substrate-binding pocket to recognize a specific β-amino acid in respective biosynthetic systems. The recognition of a specific β-amino acid by the VinN-type enzyme appears to be important for the selective incorporation of a β-amino acid into the macrolactam polyketide skeleton. Therefore, the VinN-type enzyme could represent an engineering target for changing the β-amino acid starter moiety in the macrolactam polyketide skeleton.
II. Protein–protein interaction between AT and ACP in vicenistatin biosynthesis
ATs are responsible for the selection and incorporation of acyl starter and extender units in polyketide biosynthesis.Citation22) Therefore, ATs could be attractive targets to alter the specificity of the acyl building block to obtain biologically active unnatural polyketide analogs. However, the replacement of an AT domain by a homologous AT domain possessing different acyl substrate specificity resulted in reduced or abolished production of polyketide analogs in many cases.Citation22,23) AT was reported to recognize its cognate ACP from other ACPs through a protein–protein interaction.Citation24,25) Thus, a proper protein–protein interaction between AT and ACP is important for functional transfer of acyl groups in polyketide biosynthesis. However, the mechanism of ACP recognition is not well understood because structural determination of the AT–ACP complex is hampered by the weak and transient interaction between them.Citation26) Structural determination of the AT–ACP complex is necessary for understanding the ACP recognition mechanism during the acyl transfer reaction.
In vicenistatin biosynthesis, VinK is supposed to recognize VinL and VinP1LdACP from other ACPs for the transfer of the dipeptidyl group. To visualize the specific protein–protein interaction between VinK and two ACPs, we attempted to cocrystallize VinK with VinL and VinP1LdACP. To trap the transient VinK–ACP complexes, a covalent cross-linking method using a bifunctional maleimide reagent was designed.Citation14) The complex structure of P450cam with the redox partner putidaredoxin was determined previously by trapping the transient complex using 1,6-bismaleimidohexane.Citation27) In this case, surface residues of these proteins were mutated to Cys, enabling site-specific cross-linking to occur at their binding interface. In VinK–ACP, a cross-link with the thiol group of the phosphopantetheine arm of ACP at the substrate-binding tunnel of VinK was proposed (Fig. (F)). For this purpose, a Cys mutation at Ser266 was introduced, which is located at the bottom of the substrate-binding tunnel in VinK. The cross-linking reaction between the VinK S266C mutant and VinL in the presence of 1,2-bismaleimidoethane (BMOE) gave a covalent complex, as expected. Finally, the VinK–VinL complex structure was determined successfully, which is the first crystal structure of an AT–ACP complex.Citation14) Similarly, the cross-linking reaction between the VinK S266C mutant and VinP1LdACP in the presence of BMOE gave a covalent complex; however, obtaining a crystal of VinK–VinP1LdACP complex failed.
In the VinK–VinL complex structure, the phosphopantetheine arm of VinL is orientated into the VinK substrate-binding pocket and covalently attached to the mutated Cys266 of VinK through BMOE (Fig. (G)). The binding interface between VinK and VinL comprises approximately 650 Å2. This small contact area is consistent with the transient nature of the AT–ACP interaction. VinK mainly recognizes the helix II region of VinL through salt bridges and hydrophobic interactions (Fig. (G)). Arg153 and Arg299 of VinK form salt bridges with Glu47 and Asp35 of VinL, respectively. Met206 of VinK forms hydrophobic contacts with Thr39, Leu43, Leu59, and Phe64 of VinL. VinK R153A, M206A, and R299A mutants showed significantly reduced affinities for VinL, confirming the importance of these VinK residues for the interaction with VinL. Based on the VinK–VinL complex structure, insight into the recognition mechanism of VinP1LdACP by VinK was also obtained. The VinK–VinL complex structure could be useful as a model for predicting the interaction of other AT–ACP complexes.
Most ATs accept acyl-CoA as a substrate for transfer of the acyl group to the partner ACP. These acyl-CoA-specific ATs generally have an Arg or Lys residue near the entrance of the substrate-binding tunnel for interaction with the phosphate group of the ribose moiety of CoA.Citation15) In contrast, VinK has a hydrophobic Met206 residue, which interacts with VinL residues, at the corresponding position. Other acyl-ACP-specific ATs such as ZmaA,Citation28) ZmaF,Citation28) and ClbGCitation29) also contain a hydrophobic residue at this position. Thus, the presence of a hydrophobic residue at this position might be a conserved feature in acyl-ACP-specific ATs.
III. Biosynthesis of enterocin
Type II PKSs typically produce polycyclic aromatic products, such as the antibiotic tetracycline and the anticancer agent doxorubicin in bacteria.Citation30) Type II PKSs minimally consist of two KS subunits (KSα and KSβ) and a stand-alone ACP. KSα and KSβ form a heterodimer and catalyze the iterative decarboxylative condensation of malonyl-CoA extender units with an acyl starter unit to produce the poly-β-keto intermediate. ACP serves as an anchor for the growing poly-β-keto chain. Type II PKS systems generally have additional PKS subunits including ketoreductase, cyclase, and aromatase to define the cyclization pattern of the poly-β-keto intermediate. The resulting polycyclic aromatic core is further modified by post-PKS tailoring enzymes such as oxygenase, glycosyltransferase, and methyltransferase for diversification of the final polyketide structure.
Enterocin is the most unusual compound among type II PKS-derived products because enterocin contains a nonaromatic tricyclic caged core. An initial isotope labeling study suggested the involvement of a Favorskii-type rearrangement in enterocin biosynthesis.Citation31) The identification, heterologous expression, and biochemical analysis of the enterocin biosynthetic gene cluster from Streptomyces maritimus provide insight into its biosynthetic mechanism.Citation32–34) In the enterocin biosynthetic pathway, the benzoate starter unit derived from l-phenylalanine is loaded onto the stand-alone ACP EncC (Fig. (A)). Similar to other type II PKS systems, the KSα EncA and KSβ EncB elongate the poly-β-keto chain through successive decarboxylative condensations of malonyl-CoA extender units. The ketoreductase EncD catalyzes the reduction of the C7 keto group during elongation,Citation35) leading to the formation of a linear C7-reduced octaketide (C7,O4-dihydrooctaketide) intermediate. Interestingly, the enterocin biosynthetic system lacks typical cyclase- and aromatase-type enzymes that are found in all other known type II PKS systems. In vivo and in vitro experiments showed that the FAD-dependent enzyme EncM is important in the construction of the tricyclic core structure.Citation33,34) In the absence of EncM, the linear octaketide intermediate spontaneously cyclizes through C1−C10 and C4−C9 aldol condensations, resulting in the formation of aromatic polyketides, such as wailupemycin F and wailupemycin G. Conversely, in the presence of EncM, the linear octaketide intermediate is converted to desmethyl-5-deoxyenterocin, which has a non-aromatic tricyclic core. Finally, two post-PKS tailoring enzymes, EncK and EncR, catalyze methylation and hydroxylation, respectively, to produce enterocin.
Fig. 4. The EncM reaction in enterocin biosynthesis.
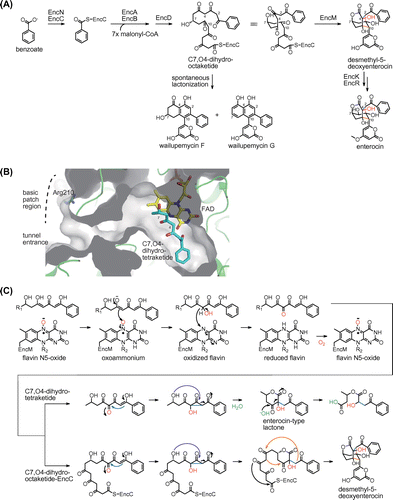
The crystal structure of EncM was determined to understand how EncM constructs the tricyclic core structure.Citation36) The structure of EncM shows that this enzyme has a long L-shaped substrate-binding tunnel that connects from the surface to the FAD active site for binding of the EncC-bound linear octaketide intermediate (Fig. (B)). EncM has a basic patch region near the entrance of the tunnel, which appears to be complementary to the negatively charged EncC. The disruption of the basic patch region with an R210E mutant decreases the desmethyl-5-deoxyenterocin-forming activity of EncM, supporting the importance of a protein−protein interaction between EncM and EncC during the reaction. In the complex structure with the substrate analog C7,O4-dihydrotetraketide, the aromatic group is located at the bottom of the substrate-binding tunnel, the C4 methylene carbon is positioned 3.3 Å from the N5 atom of FAD, and the C7-hydroxy group is located at the corner of the L-shaped tunnel (Fig. (B)). This structural observation suggests that EncM likely separates the C1−C7 dihydrotetraketide region of the octaketide intermediate from the other poly-β-keto (C8−C15) region to prevent undesired cyclization reactions.
Biochemical analysis of EncM with C7,O4-dihydrotetraketide was conducted. In the EncM reaction, the C7,O4-dihydrotetraketide was converted to ring-opened derivatives of the expected enterocin-type lactone (Fig. (C)). The enterocin-type lactone is supposed to be produced through a Favorskii-type rearrangement and lactonization, and may be subject to hydrolytic retro-Claisen ring cleavage. The analysis of the EncM reaction in the presence of 18O2 showed that EncM incorporates the oxygen atom from molecular oxygen at the C4 position. These observations allowed us to propose the EncM reaction mechanism. EncM first catalyzes the hydroxylation of the C4 methylene group and the dehydrogenation of the introduced alcohol group to give C3,C4,C5 triketone (Fig. (C)). The electrophilic C4 ketone triggers the Favorskii-type rearrangement through cyclopropane formation and subsequent opening by intramolecular attack of the C7 hydroxy group, leading to the formation of the lactone ring. The Favorskii-type rearrangement causes the cleavage of the C3−C4 bond and the formation of the C2−C4 bond. Intramolecular aldol condensations (C5−C10 and C2−C9) prevent hydrolytic retro-Claisen ring cleavage and facilitate construction of two additional rings to complete the formation of the tricyclic core in enterocin biosynthesis.
Interestingly, EncM is active even in the absence of NAD(P)H, which is unlike other known flavin-dependent monooxygenases. Further biochemical studies revealed that the EncM reaction is independent of the peroxy species bound to the C4a position of flavin, which is universally used in flavin-dependent oxygenation reactions.Citation37) Instead, EncM uses an unprecedented flavin-N5-oxide oxygenating species for the hydroxylation−dehydrogenation dual oxidation reactions (Fig. (C)).Citation36,38) In the EncM reaction, the N5-oxide is first protonated by the hydroxy proton of the C5 enol group of the substrate. The resulting N5-hydroxylamine tautomerizes to the electrophilic oxoammonium, which oxygenates the C4 position of the substrate enolate to yield the C4-hydroxylated intermediate and oxidized flavin. The introduced C4 hydroxy group is dehydrogenated by the oxidized flavin to produce the C4 ketone and reduced flavin. The C4 ketone triggers the Favorskii-type rearrangement, while the reduced flavin reacts with molecular oxygen to regenerate flavin-N5-oxide species for the next catalytic cycle. After our identification of flavin-N5-oxide, the involvement of flavin-N5-oxide began to be reported in the reactions of flavin-dependent enzymes.Citation39,40)
IV. Biosynthesis of phenolic lipids
Type III PKSs are involved in the biosynthesis of various aromatic polyketides such as polyphenols and phenolic lipids in plants, bacteria, and fungi.Citation41,42) The starter unit for type III PKS is usually provided as a CoA thioester and this unit is directly transferred to the catalytic Cys residue of type III PKS in an AT-independent manner. Type III PKS catalyzes the iterative decarboxylative condensation with several extender units such as malonyl-CoA in the active site cavity. The resulting linear poly-β-keto intermediate is subsequently cyclized in the same cavity. Type III PKSs synthesize a wide variety of products because they differ in terms of their preference of starter and extender substrates, the number of condensation steps, and the mechanism of intramolecular cyclization of poly-β-keto intermediates.
Alkylresorcinols and alkylpyrones are the major lipids of the cyst membrane in the nitrogen-fixing soil bacterium Azotobacter vinelandii.Citation43) These Azotobacter phenolic lipids have an unusually long saturated C21–C23 alkyl side chain (Fig. (A)), which contrasts with other microbial phenolic lipids possessing C15–C17 alkyl side chains.Citation44,45) Gene disruption analysis showed that the ars gene cluster is essential for the biosynthesis of phenolic lipids.Citation46) The ars gene cluster consists of two type I fatty acid synthase (FAS) genes, arsA and arsD, and two type III PKS genes, arsB and arsC.
Fig. 5. The ArsB and ArsC reactions in the biosynthesis of phenolic lipids.
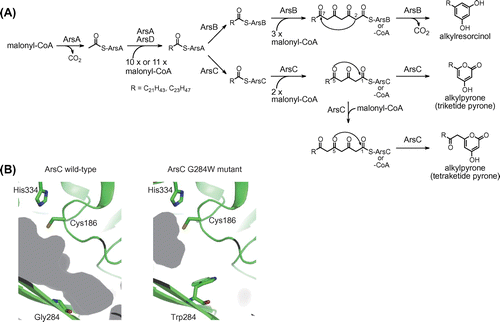
To clarify the biosynthetic mechanism of phenolic lipids, biochemical analysis of a type I FAS consisting of ArsA and ArsD was conducted initially. In the reaction of ArsA and ArsD with malonyl-CoA, the formation of saturated C22 and C24 fatty acids was detected by treatment of the reaction mixture with alkali.Citation47) When low-molecular weight compounds from macromolecules in the reaction mixture were separated by ultrafiltration, fatty acid products were detected only in the macromolecular fraction, suggesting that the fatty acid products remain attached to the ACP domain of ArsA. This is consistent with the domain organization of ArsA and ArsD that lacks TE and malonyl/palmitoyl transferase domains, each of which is responsible for releasing the fatty acid product from the ACP domain. The in vitro phenolic lipid synthesis was then reconstituted using ArsA, ArsB, ArsC, and ArsD proteins. As a result, the reaction of ArsA, ArsB, and ArsD gave alkylresorcinols, whereas the reaction of ArsA, ArsC, and ArsD gave alkylpyrones (Fig. (A)).Citation47) The direct transfer of fatty acid products from ArsA to ArsB and ArsC was also observed by monitoring the localization of radioactivity derived from [14C] malonyl-CoA in the sequential reaction of ArsA, ArsB, ArsC, and ArsD. These results allowed us to propose the biosynthetic mechanism of Azotobacter phenolic lipids. ArsA and ArsD synthesize saturated C22 and C24 fatty acids solely from malonyl-CoA (Fig. (A)). Two type III PKSs, ArsB and ArsC, receive the fatty acid products directly from the ACP domain of ArsA as starter substrates to synthesize alkylresorcinols and alkylpyrones, respectively.
ArsB and ArsC can also use acyl-CoA as a starter substrate and have a promiscuous starter substrate specificity like other type III PKSs.Citation46) ArsB and ArsC accept C10–C22 and C6–C22 fatty acyl-CoAs, respectively. ArsB even accepts 15-phenylpentadecanoyl-N-acetylcysteamine, which has an aromatic ring at the alkyl chain tail.Citation48) However, the direct transfer system suggests that their primary substrate should be acyl-ACP. ArsB and ArsC use C22 and C24 fatty acid starter units, which have longer alkyl chains than the C16 and C18 fatty acids in primary fatty acid metabolism. Therefore, the ars gene cluster encodes a specific type I FAS, ArsA and ArsD, for the synthesis of unusual C22 and C24 fatty acids. Because these long-chain fatty acids are used only by ArsB and ArsC, it is reasonable that ArsB and ArsC accept the long-chain fatty acyl starter unit directly from the ACP domain of ArsA. ArsB and ArsC are hypothesized to recognize the ArsA ACP domain through a protein−protein interaction, although the molecular basis of ACP recognition by a type III PKS remains unclear.
ArsB and ArsC, which share 71% amino acid sequence identity, produce different phenolic lipids, as described above (Fig. (A)). They use the same fatty acyl starter substrate and generate the same tetraketide intermediate by catalyzing three successive condensations with malonyl-CoA. However, the cyclization pattern is different between ArsB and ArsC reactions. ArsB catalyzes the decarboxylative C2–C7 aldol condensation of the tetraketide intermediates to produce alkylresorcinols. In contrast, ArsC catalyzes the C5 oxygen–C1 lactonization of the triketide and tetraketide intermediates to produce alkylpyrones. ArsB has Trp281, which is a conserved aromatic residue among the alkylresorcinol- and alkylresorcylic acid-producing type III PKSs, at the active site cavity, whereas ArsC contains Gly284 at the corresponding position. The amino acid difference at this position may affect cyclization specificity. A mutational study was conducted to examine the importance of this residue in providing specificity. As a result, the ArsB W281G mutant exhibited alkylpyrone-producing activity like the ArsC wild-type and the ArsC G284W mutant exhibited an alkylresorcinol-producing activity like the ArsB wild-type.Citation49) The crystal structures of ArsC wild-type and G284W mutant were then determined. Comparison of these two structures showed that the G284W substitution resulted in a significant reduction of the active site cavity volume (Fig. (B)). The tetraketide intermediate probably folds to a suitable form for cyclization via the aldol condensation in the relatively narrow cavity of ArsB wild-type or the ArsC G284W mutant. Conversely, the large cavity of ArsC wild-type might give flexibility to the tetraketide chain, leading to spontaneous lactonization. Thus, mutational and structural studies showed that only a single amino acid residue at the active site cavity is a crucial determinant of the different cyclization specificities of ArsB and ArsC.
V. Future perspectives
The variety of substrate specificities and reactivities of polyketide biosynthetic enzymes greatly diversify the chemical structures of polyketide natural products. The diverse biological activities of polyketides have engendered researchers to investigate the polyketide biosynthetic machinery, and with such information, researchers have begun to examine the possible manipulation of this machinery to produce unnatural bioactive polyketides. Our studies represent important mechanistic information on macrolactam polyketide biosynthetic enzymes that are responsible for the incorporation of rare β-amino acid starter units. Our studies also add new insights into how cyclization enzymes diversify the carbon skeletons of products in reactions of type II and type III PKSs. In addition to our studies, recent structural and biochemical studies have led to advances in understanding the mechanisms of substrate recognition and the catalytic reaction of polyketide biosynthetic enzymes. However, it remains difficult to predict the function of some polyketide biosynthetic enzymes from their amino acid sequences. The underlying mechanisms for ACP-based interactions that are crucial to the core functions of polyketide biosynthetic enzymes are poorly understood. Therefore, further efforts are necessary to increase understanding of the polyketide biosynthetic machinery at the structural and biochemical levels. Further accumulation of knowledge should be useful for engineering biosynthetic pathways to create novel bioactive unnatural polyketides.
Disclosure statement
No potential conflict of interest was reported by the author.
Funding
This research was supported by Japan Society for the Promotion of Science [grant number 25850050], [grant number 15K18679], and [grant number 17K07747].
Acknowledgments
These studies were carried out at The University of Tokyo, University of California San Diego, and Tokyo Institute of Technology. I am very grateful to Professors Sueharu Horinouchi, Yasuo Ohnishi, Nobutaka Funa, Bradley S. Moore, Tadashi Eguchi, and Fumitaka Kudo for their support and helpful discussions. I am also grateful to Professors Hirofumi Shoun, Takayoshi Wakagi, and Shinya Fushinobu for providing me with the opportunity to study enzymology and structural biology. I thank all the co-workers for their cooperation. These studies were supported in part by the Japan Society for the Promotion Science. Finally, I would like to thank the Japan Society for Bioscience, Biotechnology and Agrochemistry for the JSBBA Awards for the Encouragement of Young Scientists.
Notes
Abbreviations: CoA, coenzyme A; ACP, acyl carrier protein; PKS, polyketide synthase; KS, ketosynthase; AT, acyltransferase; GNAT, GCN5-related N-acetyltransferase; A domain, adenylation domain; TE, thioesterase; FAD, flavin adenine dinucleotide; 3-MeAsp, (2S,3S)-3-methylaspartate; VinP1LdACP, the ACP domain of the loading module of PKS VinP1; BMOE, 1,2-bismaleimidoethane; FAS, fatty acid synthase.
References
- Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009;48:4688–4716.10.1002/anie.v48:26
- Moore BS, Hertweck C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep. 2002;19:70–99.
- Keatinge-Clay AT. The uncommon enzymology of cis-acyltransferase assembly lines. Chem Rev. 2017;117:5334–5366.10.1021/acs.chemrev.6b00683
- Gu L, Geders TW, Wang B, et al. GNAT-like strategy for polyketide chain initiation. Science. 2007;318:970–974.10.1126/science.1148790
- Kudo F, Miyanaga A, Eguchi T. Biosynthesis of natural products containingβ-amino acids. Nat Prod Rep. 2014;31:1056–1073.10.1039/C4NP00007B
- Miyanaga A, Kudo F, Eguchi T. Mechanisms of β-amino acid incorporation in polyketide macrolactam biosynthesis. Curr Opin Chem Biol. 2016;35:58–64.10.1016/j.cbpa.2016.08.030
- Ogasawara Y, Katayama K, Minami A, et al. Cloning, sequencing, and functional analysis of the biosynthetic gene cluster of macrolactam antibiotic vicenistatin in Streptomyces halstedii. Chem Biol. 2004;11:79–86.
- Ogasawara Y, Kakinuma K, Eguchi T. Involvement of glutamate mutase in the biosynthesis of the unique starter unit of the macrolactam polyketide antibiotic vicenistatin. J Antibiot. 2005;58:468–472.10.1038/ja.2005.62
- Shinohara Y, Kudo F, Eguchi T. A natural protecting group strategy to carry an amino acid starter. J Am Chem Soc. 2011;133:18134–18137.10.1021/ja208927r
- Kudo F, Kawamura K, Uchino A, et al. Genome mining of the hitachimycin biosynthetic gene cluster: involvement of a phenylalanine-2,3-aminomutase in biosynthesis. ChemBioChem. 2015;16:909–914.10.1002/cbic.201500040
- Amagai K, Takaku R, Kudo F, et al. A unique amino transfer mechanism for constructing the β-amino fatty acid starter unit in the biosynthesis of the macrolactam antibiotic cremimycin. ChemBioChem. 2013;14:1998–2006.10.1002/cbic.v14.15
- Chisuga T, Miyanaga A, Kudo F, et al. Structural analysis of the dual function thioesterase SAV606 unravels the mechanism of Michael addition of glycine to an α, β-unsaturated thioester. J Biol Chem. 2017;292:10926–10937.10.1074/jbc.M117.792549
- Hong H, Fill T, Leadlay PF. A common origin for guanidinobutanoate starter units in antifungal natural products. Angew Chem Int Ed Engl. 2013;52:13096–13099.10.1002/anie.201308136
- Miyanaga A, Iwasawa S, Shinohara Y, et al. Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc Natl Acad Sci USA. 2016;113:1802–1807.10.1073/pnas.1520042113
- Oefner C, Schulz H, D’Arcy A, et al. Mapping the active site of Escherichia coli malonyl-CoA-acyl carrier protein transacylase (FabD) by protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 2006;62:613–618.10.1107/S0907444906009474
- Wang F, Wang Y, Ji J, et al. Structural and functional analysis of the loading acyltransferase from avermectin modular polyketide synthase. ACS Chem Biol. 2015;10:1017–1025.10.1021/cb500873 k
- Shinohara Y, Miyanaga A, Kudo F, et al. The crystal structure of the amidohydrolase VinJ shows a unique hydrophobic tunnel for its interaction with polyketide substrates. FEBS Lett. 2014;588:995–1000.10.1016/j.febslet.2014.01.060
- Miyanaga A, Hayakawa Y, Numakura M, et al. Identification of the fluvirucin B2 (Sch 38518) biosynthetic gene cluster from Actinomadura fulva subsp. indica ATCC 53714: substrate specificity of β-amino acid selective adenylating enzyme FlvN. Biosci Biotechnol Biochem. 2016;80:935–941.10.1080/09168451.2015.1132155
- Takaishi M, Kudo F, Eguchi T. Identification of incednine biosynthetic gene cluster: characterization of novel β-glutamate-β-decarboxylase IdnL3. J Antibiot. 2013;66:691–699.10.1038/ja.2013.76
- Miyanaga A, Cieślak J, Shinohara Y, et al. The crystal structure of adenylation enzyme VinN reveals a unique β-amino acid recognition mechanism. J Biol Chem. 2014;289:31448–31457.10.1074/jbc.M114.602326
- Cieślak J, Miyanaga A, Takaku R, et al. Biochemical characterization and structural insight into aliphatic β-amino acid adenylation enzymes IdnL1 and CmiS6. Proteins. 2017;85:1238–1247.10.1002/prot.v85.7
- Dunn BJ, Khosla C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J Roy Soc Interface. 2013;10:20130297.10.1098/rsif.2013.0297
- Liou GF, Khosla C. Building-block selectivity of polyketide synthases. Curr Opin Chem Biol. 2003;7:279–284.10.1016/S1367-5931(03)00016-4
- Wong FT, Chen AY, Cane DE, et al. Protein−protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry. 2010;49:95–102.10.1021/bi901826 g
- Dunn BJ, Watts KR, Robbins T, et al. Comparative analysis of the substrate specificity of trans versus cis- acyltransferases of assembly line polyketide synthases. Biochemistry. 2014;53:3796–3806.10.1021/bi5004316
- Crosby J, Crump MP. The structural role of the carrier protein — active controller or passive carrier. Nat Prod Rep. 2012;29:1111–1137.10.1039/c2np20062 g
- Tripathi S, Li H, Poulos TL. Structural basis for effector control and redox partner recognition in cytochrome P450. Science. 2013;340:1227–1230.10.1126/science.1235797
- Park H, Kevany BM, Dyer DH, et al. A polyketide synthase acyltransferase domain structure suggests a recognition mechanism for its hydroxymalonyl-acyl carrier protein substrate. PLoS One. 2014;9:e110965.10.1371/journal.pone.0110965
- Zha L, Wilson MR, Brotherton CA, et al. Characterization of polyketide synthase machinery from the pks Island facilitates isolation of a candidate precolibactin. ACS Chem Biol. 2016;11:1287–1295.10.1021/acschembio.6b00014
- Hertweck C, Luzhetskyy A, Rebets Y, et al. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep. 2007;24:162–190.10.1039/B507395 M
- Seto H, Sato¯ T, Urano S, et al. Utilization of 13C–13C coupling in structural and biosynthetic studies. VII. The structure and biosynthesis of vulgamycin. Tetrahedron Lett. 1976;17:4367–4370.10.1016/0040-4039(76)80117-7
- Piel J, Hertweck C, Shipley PR, et al. Cloning, sequencing and analysis of the enterocin biosynthesis gene cluster from the marine isolate ‘Streptomyces maritimus’: evidence for the derailment of an aromatic polyketide synthase. Chem Biol. 2000;7:943–955.10.1016/S1074-5521(00)00044-2
- Xiang L, Kalaitzis JA, Moore BS. EncM, a versatile enterocin biosynthetic enzyme involved in Favorskii oxidative rearrangement, aldol condensation, and heterocycle-forming reactions. Proc Natl Acad Sci USA. 2004;101:15609–15614.10.1073/pnas.0405508101
- Cheng Q, Xiang L, Izumikawa M, et al. Enzymatic total synthesis of enterocin polyketides. Nat Chem Biol. 2007;3:557–558.10.1038/nchembio.2007.22
- Hertweck C, Xiang L, Kalaitzis JA, et al. Context-dependent behavior of the enterocin iterative polyketide synthase: a new model for ketoreduction. Chem Biol. 2004;11:461–468.10.1016/j.chembiol.2004.03.018
- Teufel R, Miyanaga A, Michaudel Q, et al. Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature. 2013;503:552–556.10.1038/nature12643
- Piano V, Palfey BA, Mattevi A. Flavins as covalent catalysts: new mechanisms emerge. Trends Biochem Sci. 2017;42:457–469.10.1016/j.tibs.2017.02.005
- Teufel R, Stull F, Meehan MJ, et al. Biochemical establishment and characterization of EncM’s flavin-N5-oxide cofactor. J Am Chem Soc. 2015;137:8078–8085.10.1021/jacs.5b03983
- Adak S, Begley TP. Dibenzothiophene catabolism proceeds via a flavin-N5-oxide intermediate. J Am Chem Soc. 2016;138:6424–6426.10.1021/jacs.6b00583
- Adak S, Begley TP. RutA-catalyzed oxidative cleavage of the uracil amide involves formation of a flavin-N5-oxide. Biochemistry. 2017;56:3708–3709.10.1021/acs.biochem.7b00493
- Austin MB, Noel JP. The chalcone synthase superfamily of type III polyketide synthases. Nat Prod Rep. 2003;20:79–110.10.1039/b100917f
- Lim YP, Go MK, Yew WS. Exploiting the biosynthetic potential of type III polyketide synthases. Molecules. 2016;21:806.10.3390/molecules21060806
- Reusch RN, Sadoff HL. Novel lipid components of the Azotobacter vinelandii cyst membrane. Nature. 1983;302:268–270.10.1038/302268a0
- Miyanaga A, Ohnishi Y. Biosynthesis of phenolic lipid by type III polyketide synthases. In: Geiger O., editor. Biogenesis of fatty acids, lipids and membranes in handbook of hydrocarbon and lipid microbiology. Basel: Springer; 2017. DOI:10.1007/978-3-319-43676-0_14-3.
- Miyanaga A, Ohnishi Y. Type III polyketide synthases responsible for phenolic lipid synthesis. In: Geiger O., editor. Biogenesis of fatty acids, lipids and membranes in handbook of hydrocarbon and lipid microbiology. Basel: Springer; 2017. DOI:10.1007/978-3-319-43676-0_28-2.
- Funa N, Ozawa H, Hirata A, et al. Phenolic lipid synthesis by type III polyketide synthases is essential for cyst formation in Azotobacter vinelandii. Proc Natl Acad Sci USA. 2006;103:6356–6361.10.1073/pnas.0511227103
- Miyanaga A, Funa N, Awakawa T, et al. Direct transfer of starter substrates from type I fatty acid synthase to type III polyketide synthases in phenolic lipid synthesis. Proc Natl Acad Sci USA. 2008;105:871–876.10.1073/pnas.0709819105
- Miyanaga A, Horinouchi S. Enzymatic synthesis of bis-5-alkylresorcinols by resorcinol-producing type III polyketide synthases. J Antibiot. 2009;62:371–376.10.1038/ja.2009.44
- Satou R, Miyanaga A, Ozawa H, et al. Structural basis for cyclization specificity of two Azotobacter type III polyketide synthases. A single amino acid substitution reverses their cyclization specificity. J Biol Chem. 2013;288:34146–34157.10.1074/jbc.M113.487272