
Abstract
Herein we describe the linear synthesis of a tetrasaccharyl sialoglycan found in both the Chol-1 ganglioside core and disialyl T antigen. The synthesis featured sialylation with a C5-ureido-modified sialyl donor followed by selective isolation of the desired α-sialoside via 1,5-lactamization. This methodology enables the linear synthesis of sialoglycans and provides practical access to biologically important carbohydrate molecules.
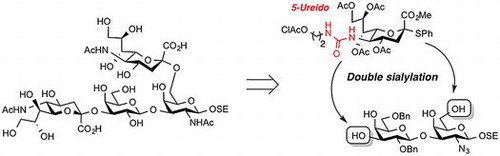
Structure of the target tetrasaccharide and key glycosylation.
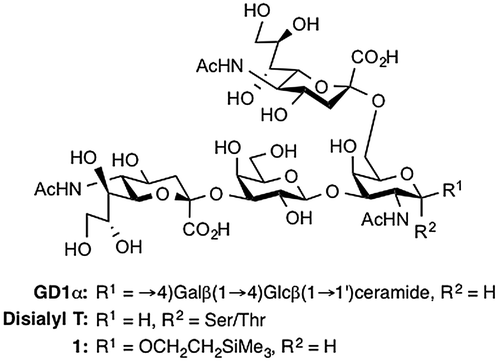
Sialoglycans, which are carbohydrate molecules containing sialic acid residues, are mainly found in vertebrates as glycoconjugates (glycoproteins and glycolipids) and have pleiotropic biological functions such as mediating cell recognition, modulating cell interactions, and serving as a receptor for neurotropic bacterial toxins and viruses.Citation1,2) Moreover, sialoglycans play a significant role in various human diseases including progressive neurodegenerative disorders.Citation3,4) Given the growing recognition of the biological importance of sialoglycans, research on the physiological and pathological implications of sialoglycans has attracted the attention of not only biologists but also chemists. Sialoglycans are an enticing target for synthetic chemists because of the difficulty of constructing both the essential α-sialoside linkages and the diverse complex structures of these molecules. In the mammalian sialoglycan biosynthetic pathway, sialic acids are enzymatically attached to the ends of the glycan chains at the last stage. In the chemical synthesis of sialoglycans, on the other hand, the introduction of sialic acid residues into preassembled glycans at the last stage has seldom been performed because of the challenging stereocontrol in the sialylation reaction and the laborious isolation of the desired α-sialosides from the reaction mixture. Thus, to date, the installation of sialic acids has generally been done early in chemical syntheses of sialoglycans, producing commonly used building blocks such as sialyl-α(2→3)/(2→6)-galactose, sialyl-α(2→6)-N-acetylgalactosamine, and sialyl-α(2→8)-sialic acid.Citation5–10) These building blocks are further modified and assembled to afford complex glycans. This strategy is called convergent synthesis. However, most mammalian sialoglycans (e.g., gangliosides) have a common asialoglycan skeleton, so the ideal synthetic strategy would be to construct a common asialoglycan unit followed by selective incorporation of sialic acids. This strategy is called linear synthesis and has the potential to be more efficient and practical. However, the linear synthesis of sialoglycans requires a high level of stereocontrol in the sialylation and careful purification of α-sialosides, both of which are difficult. Very recently, we developed a novel synthetic method for sialoglycans using a 5-ureido-modified sialyl donor.Citation11) The donor was shown to be useful for α-selective sialylation and the desired α-sialoside could be easily isolated from the reaction mixture by annulation to form a 1,5-lactam structure.Citation12) This success prompted us to apply the 5-ureido-sialyl donor to the linear synthesis of complex sialoglycans. We chose a tetrasaccharyl sialoglycan containing two sialic acid residues as our target (Figure ). This tetrasaccharyl sialoglycan is present in the Chol-1 (α-series) ganglioside core and the disialyl T antigen on O-linked glycoproteins, which play important biological roles in, for example, cholinergic synaptic transmissionCitation13) and attachment and invasion of cancer cells,Citation14) respectively. To date, the chemical synthesis of this kind of tetrasaccharide has been accomplished primarily via convergent syntheses.Citation15) Herein we describe our synthesis of sialoglycan 1, which has a 2-(trimethylsilyl)ethyl group as an aglycone, based on a linear synthetic strategy using a 5-ureido-modified sialyl donor as a key compound.
Figure 1. Structure of the target tetrasaccharides.
Results and discussion
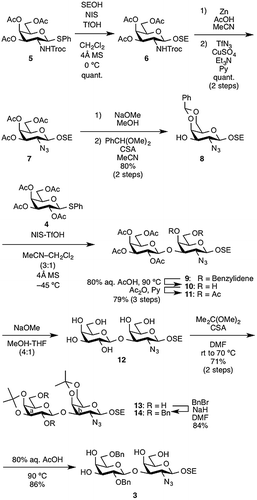
In our retrosynthetic analysis for the linear synthesis, target molecule 1 was disconnected to the C5-ureido-modified sialic acid 2 and the core 1 Galβ(1→3)GalN disaccharide unit 3 (Figure ). Disaccharide 3 was then further divided into two known compounds: galactose (Gal) derivative 4Citation16) and galactosamine (GalN) derivative 5.Citation17) The incorporation of two sialic acid residues into 3 through double sialylation was planned for the last step in the construction of the target molecule. Based on the synthetic strategy shown in Figure , we began with the synthesis of the disaccharide acceptor 3 (Scheme ). First, the known GalN thioglycoside 5 was glycosidated with 2-(trimethylsilyl)ethanol (SEOH) in the presence of NIS and TfOHCitation18,19) in CH2Cl2 at 0 °C to afford 2-(trimethylsilyl)ethyl (SE) glycoside 6 in quantitative yield. The SE group was chosen as the protecting group at the reducing end of glycan for three reasons: (1) to withstand various reaction conditions; (2) to enable selective removal followed by further elongation of the glycan as well as any necessary modifications; and (3) to increase solubility in organic solventsCitation20) Next, the N-Troc group was converted into an azide group by treatment with zinc and acetic acid followed by TfN3 in pyridine,Citation21) affording 7 in quantitative yield over two steps. Then, removal of the acetyl groups under Zemplén conditions and subsequent introduction of the benzylidene groups at the C4 and C6 positions gave the 3-OH GalN acceptor 8 in 80% yield over two steps. The glycosylation of 8 with the known Gal phenylthioglycoside donor 4 was mediated by the NIS-TfOH promoter system in MeCN–CH2Cl2 (3:1) at −45 °C.Citation22) The glycosylation reaction proceeded smoothly, but after silica gel column chromatography, the obtained disaccharide 9 was contaminated with small amounts of inseparable by-products. Thus, the mixture was used directly in the next hydrolysis reaction without additional purification. Treatment of the mixture with 80% aq. AcOH at 90 °C hydrolyzed the benzylidene acetal to give 10. However, there were small amounts of a by-product whose liberated primary hydroxyl group was acetylated, and column chromatographic purification of 10 led to the loss of the desired disaccharide derivatives. Therefore, after hydrolysis of 9, the resulting mixture was subjected directly to the acetylation without purification to obtain the per-acetylated product 11. Over the three steps of glycosylation, hydrolysis, and acetylation, 11 was obtained as the sole product in 79% yield. Next, removal of the acetyl groups from 11 and subsequent formation of two cyclic isopropylidene acetals between O-4 and O-6 of the GalN residue and between O-3 and O-4 of the Gal residue afforded 13 in 71% yield over two steps. The latter isopropylidenation generated some regioisomers as by-products. Although purification of 13 by silica gel column chromatography was troublesome, it was found that the use of both a longer column (125 g of silica gel for 0.5 g of the crude mixture was used) and ethyl acetate as the eluent made isolation of 13 easier. Subsequent benzylation with BnBr and NaH in DMF afforded the fully protected disaccharide 14 in 84% yield. Finally, hydrolysis of the isopropylidene groups by treatment with 80% aq. AcOH at 90 °C furnished the desired disaccharide acceptor 3 in 86% yield.
Figure 2. Synthetic scheme for 1 based on a linear synthetic strategy.
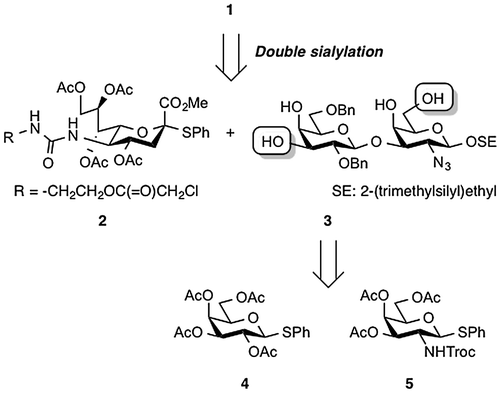
Figure 3. Model compound for studying selective deprotection of the Boc group.
Scheme 1. Synthesis of disaccharide acceptor 3.
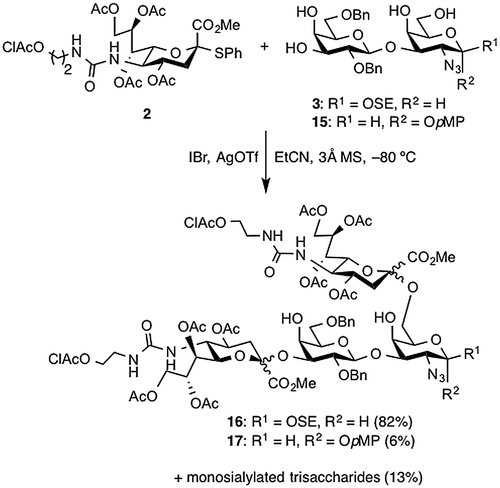
Next, we examined double sialylation of the tetraol acceptor 3 with the 5-ureido-modified sialyl donor 2 (Scheme ). We anticipated that sialylation would occur at the C3 hydroxyl group of Gal and the C6 hydroxyl group of GalN because these groups are intrinsically more reactive than those at the C4 position of Gal and GalN. In our previous study,Citation11) sialylation did not occur at the C4 position of either Gal or GalN. Using the optimized conditions from that study, sialylation was performed in the presence of IBr and AgOTfCitation23) in EtCNCitation24) at −80 °C. First, acceptor 3, which has the SE group at the reducing end, was sialylated with 4.0 equiv of donor 2, which underwent glycosylation smoothly to afford the desired disialylated tetrasaccharide 16 as a mixture of four stereoisomers in 82% yield. In this reaction, however, monosialylated products were obtained in 13% yield, even though we used an excess of donor. In contrast, sialylation of acceptor 15, which was synthesized for a comparative examination of acceptors and differed from 3 in only the protecting group at the reducing end, gave tetrasaccharide 17 in only 6% yield along with monosialylated trisaccharides in 13% yield. We attribute this result to poor solubility of the p-methoxyphenyl (pMP) glycoside in EtCN. This comparison corroborated the higher solubility of the SE glycoside and showed the usefulness of the SE group for carbohydrate synthesis.
Scheme 2. Examination of double sialylation with 2.
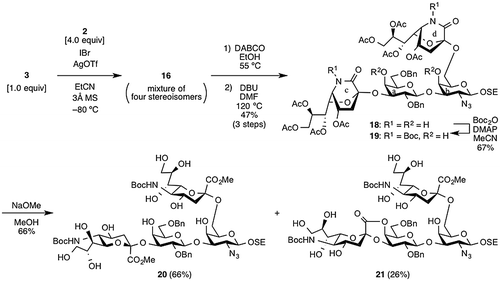
Having examined the double sialylation, we returned to the synthesis of 1 (Scheme ). The sialylation of 3 with 2 provided tetrasaccharide 16 as four stereoisomers after rough purification by gel filtration column chromatography. The product was then used in the next reaction without further purification. Treatment of 16 with DABCO in EtOH at 55 °CCitation25) followed by 1,5-lactamization with DBU in DMF at 120 °C furnished the double lactamized product 18 in 47% yield over three steps. By using the optimized lactamization conditions from our previous study,Citation11) 18 with the desired double α configuration could be easily isolated from the mixture of four stereoisomers. Subsequent introduction of Boc groups onto the two lactam rings was carried out using Boc2O and DMAP in MeCN,Citation26) giving 19 in 67% yield. Finally, by treatment with NaOMe in MeOH, lactam derivative 19 in boat form was converted into 20 in chair form as the natural conformation in 66% yield, and lactonized product 21 was obtained in 26% yield.
Scheme 3. Synthesis of tetrasaccharide derivative 20.
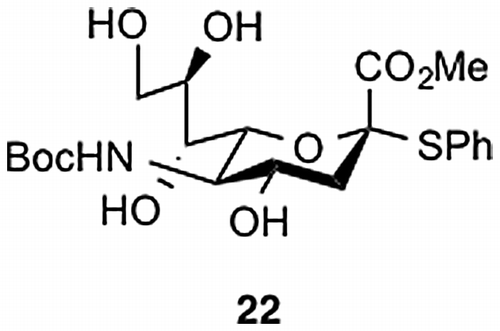
Next, we examined selective removal of the N-Boc group in 20 in the presence of the acid-labile SE group. For this, sialic acid derivative 22 was used as a model compound (Figure ). We tested several non-acidic approaches reported in the literature. However, all attempted conditions—CAN in MeCN,Citation27) NaOtBu and H2O in 2-MeTHF,Citation28) and TBAF in THFCitation29)—failed to give satisfactory results, forcing us to modify our synthetic scheme for 1. Thus, we selected the benzyloxycarbonyl (Cbz) group as an alternative to the Boc group because the Cbz can be chemoselectively removed by hydrogenolysis under neutral conditions.
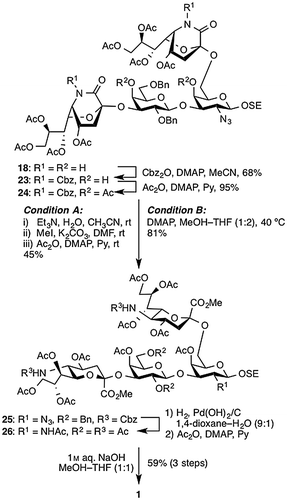
Our modified approach to 1 is shown in Scheme . The Cbz group was introduced onto the lactam ring in 18 by treatment with Cbz2O and DMAP in MeCN to afford 23 in 68% yield. Next, the hydroxyl groups at the C4 position of Gal and GalN were protected with acetyl groups to avoid lactonization between C1 of sialic acid and C4 of Gal and GalN during the subsequent lactam ring-opening reaction. Reaction of 23 with Ac2O and DMAP in pyridine gave 24 in excellent yield (95%). After acetylation at the C4 position of both Gal and GalN, the sialylated site was confirmed by a downfield shift of the C4 protons in the Citation1H NMR spectrum. Interestingly, quenching this reaction by the addition of dry MeOH produced a trace amount of the open lactam products. Next, according to our previous report,Citation12) the lactam ring-opening reaction of the N-Cbz-protected derivatives was carried out in three steps: saponification by treatment with Et3 N in aq. MeCN, methyl esterification by treatment with MeI and K2CO3 in DMF, and concomitant acetylation of the liberated amines and hydroxyl groups (condition A). As a result, 25 was obtained, but in 45% yield over the three steps. This unsatisfactory result prompted us to develop an alternative method for the lactam ring opening. Aiming for a more efficient and practical sialoglycan synthesis, we pursued a one-step operation to achieve the lactam ring opening without the need for acyl protecting groups. In considering a possible method, we recalled the by-products formed when the last acetylation was quenched. In that step, the addition of dry MeOH into the reaction mixture containing DMAP in pyridine cleaved the lactam ring, affording the corresponding methyl ester derivative with intact acetyl groups. Thus, we tried stirring 24 in pyridine–MeOH (5:1). Although no reaction was observed at room temperature, raising the reaction temperature up to 40 °C gave multiple products. Contemplating these results, we next used DMAP instead of pyridine. Treatment of 24 with 3.0 equiv of DMAP in MeOH–THF (1:2) (condition B) afforded the desired 25 in 81% yield. As anticipated, almost all acetyl groups survived under these conditions. A minor side product formed via a competing reaction that removed the Cbz group, after which the lactam could no longer open. Subsequently, hydrogenolysis-sensitive functionalities in 25 were concomitantly removed in the presence of Pearlman’s catalyst in 1,4-dioxane–H2O (9:1) under H2 atmosphere and the resulting product was then acetylated with Ac2O and DMAP in pyridine, affording 26. Finally, saponification of 26 by treatment with 1 m aq. NaOH in MeOH–THF (1:1) furnished the target 1 in 59% yield over three steps, thus completing the synthesis.
Scheme 4. Synthesis of target 1.
Conclusion
A facile linear synthesis of the tetrasaccharyl sialoglycan found in the Chol-1 ganglioside core and disialyl T antigen was accomplished by using a 5-ureido-modified sialyl donor as a key compound. In this study, significant improvement of the lactam ring-opening reaction was achieved by using DMAP in MeOH–THF, leading to direct conversion of the lactam into the corresponding methyl ester derivative in good yield without removal of the acyl protecting groups. This method using the 5-ureido-sialyl donor streamlines the synthesis of sialoglycans and holds promise for the further development of linear syntheses of sialoglycans. We are currently investigating the application of this method to more complex sialoglycans.
Experimental
Gerenal methods
All reactions were carried out under a positive pressure of argon, unless otherwise noted. All chemicals used were purchased from commercial suppliers and used without further purification, unless otherwise noted. Molecular sieves were purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan) and dried at 300 °C for 2 h in a muffle furnace prior to use. Solvents as reaction media were purchased commercial suppliers and used without further purification, unless otherwise noted. TLC analysis was performed on Merck TLC plates (silica gel 60F254 on glass plate) or Merck HPTLC plates (silica gel 60F254 on glass plate). Compound detection was either by exposure to UV light (254 nm) or by soak in a solution of 10% H2SO4 in ethanol or 12MoO3·H3PO4-H2O/H2SO4/85% H3PO4 aq. solution or 1% aq. ninhydrin solution followed by heating. Silica gel (80 mesh, 300 mesh, and PSQ-60B) manufactured by Fuji Silysia Co. was used for flash column chromatography. Quantity of silica gel was usually estimated as 100 to 200-fold weight of sample to be charged. Sephadex LH-20 manufactured by GE Healthcare was used for gel filtration column chromatography. Solvent systems in chromatography were specified in v/v. Evaporation and concentration were carried out in vacuo. 1H-NMR and 13C-NMR spectra were recorded with Bruker AVANCE III 500 spectrometer. Chemical shifts in 1H-NMR spectra are expressed in ppm (δ) relative to the signal of Me4Si, adjusted to δ 0.00 ppm, unless otherwise noted. Data are presented as follow: Chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, dd = double of doublet, td = triple doublet, m = multiplet and/or multiple resonances), integration, coupling constant in Hertz (Hz), position of the corresponding proton. COSY methods were used to confirm the NMR signal assignments. High-resolution mass (ESI-TOF MS) spectra were acquired with a Bruker micrOTOF. MALDI-TOF mass spectra were acquired with a Bruker Autoflex operated in the positive/negative reflectron mode and α-cyano-4-hydroxycinnamic acid (CHCA) was used as the matrix. Optical rotations were measured with a “Horiba SEPA-300” high-sensitive polarimeter.
2-(Trimethylsilyl)ethyl (3,4-O-isopropylidene-β-d-galactopyranosyl)-(1→3)-2-azide-2-deoxy-4,6-O-isopropylidene-β-d-galactopyranoside (13)
To a solution of the known compound 11Citation22) (492 mg, 0.684 mmol) in THF/MeOH (1:4, 7.0 mL) was added catalytic amounts of sodium methoxide (28% solution in MeOH) at room temperature. After stirring for 24 h at room temperature as the reaction was monitored by TLC (EtOAc or CHCl3/MeOH = 20:1), the reaction was neutralized with Muromac (H+) resin. The resin was filtered out and the filtrate and washings were concentrated. The residue was exposed to high vacuum to remove solvents. The resulting residue was then dissolved in DMF (3.4 mL). To the mixture was added 2,2-dimethoxypropane (250 μL, 2.05 mmol) and 10-camphorsulfonic acid (32 mg, 0.137 mmol) at room temperature. After stirring for 19.5 h at room temperature as the reaction was monitored by TLC (EtOAc), the mixture was heated to 70 °C, and the stirring was continued for 23.5 h at the same temperature. The reaction was quenched by the addition of triethylamine. The mixture was co-evaporated with toluene. The resulting residue was purified by silica gel column chromatography (Toluene/EtOAc = 2:1) to give 13 (266 mg, 71% over two steps): [α]D = +24.2° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.43 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.26–4.24 (m, 2 H, H-4b, H-1b), 4.19 (dd, 1 H, J4,5 = 2.0 Hz, J3,4 = 5.5 Hz, H-4a), 4.11 (dd, 1 H, J2,3 = 7.5 Hz, H-3a), 4.07–4.04 (m, 2 H, H-6′b, SiCH2CH2O), 3.95–3.85 (m, 4 H, H-6b, H-5a, H-6′a, H-6a), 3.81 (dd, 1 H, J1,2 = 8.0 Hz, J2,3 = 10.5 Hz, H-2b), 3.67 (ddd, 1 H, H-2a), 3.55 (m, 1 H, SiCH2CH2O), 3.45 (dd, 1 H, J3,4 = 3.5 Hz, H-3b), 3.27 (br s, 1 H, H-5b), 2.94 (s, 1 H, 2a-OH), 2.18 (m, 1 H, 6a-OH), 1.53, 1.48, 1.46, 1.35 (4 s, 12 H, 4 Me), 1.09–1.00 (m, 2 H, SiCH2CH2O), 0.02 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 110.8, 104.7, 101.9, 99.6, 80.0, 78.8, 74.1, 74.0, 73.7, 68.3, 68.0, 66.7, 63.0, 62.8, 62.0, 29.4, 28.4, 26.6, 19.0, 18.5, -1.1; HRMS (ESI) m/z: found [M + Na]+ 570.2451, C23H41N3O10Si calcd for [M + Na]+ 570.2453.
2-(Trimethylsilyl)ethyl (2,6-di-O-benzyl-3,4-O-isopropylidene-β-d-galactopyranosyl)-(1→3)-2-azide-2-deoxy-4,6-O-isopropylidene-β-d-galactopyranoside (14)
To a solution of 13 (242 mg, 0.442 mmol) in DMF (4.4 mL) was added NaH (60% in oil; 58 mg, 1.33 mmol) at 0 °C. After stirring for 1 h at room temperature, benzyl bromide (158 μL, 1.33 mmol) was then added to the mixture at 0 °C. After stirring for 16.5 h at room temperature as the reaction was monitored by TLC (n-Hexane/EtOAc = 2:1), the reaction was quenched by the addition of MeOH at 0 °C. The mixture was co-evaporated with toluene. The resulting residue was diluted with n-Hexane/EtOAc (3:2), washed with H2O, dried over Na2SO4, concentrated. The resulting residue was purified by silica gel column chromatography (n-Hexane/EtOAc = 5:1) to give 14 (271 mg, 84%): [α]D = +45.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.44–7.23 (m, 10 H, 2 Ph), 4.88 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.81 (d, 1 H, PhCH2), 4.61 (d, 1 H, J1,2 = 7.0 Hz, H-1a), 4.57 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.54 (d, 1 H, PhCH2), 4.28 (near d, 1 H, J3,4 = 3.0 Hz, H-4b), 4.22 (d, 1 H, J1,2 = 8.0 Hz, H-1b), 4.18 (t, 1 H, J2,3 = J3,4 = 6.0 Hz, H-3a), 4.13 (dd, 1 H, J4,5 = 2.5 Hz, H-4a), 4.04 (m, 1 H, SiCH2CH2O), 3.92–3.88 (m, 3 H, H-5a, H-6′a, H-6′b), 3.81 (dd, 1 H, J2,3 = 11.0 Hz, H-2b), 3.76–3.70 (m, 2 H, H-6a, H-6b), 3.57 (m, 1 H, SiCH2CH2O), 3.48 (dd, 1 H, H-2a), 3.41 (dd, 1 H, H-3b), 3.14 (d, 1 H, J4,5 = 0.5 Hz, H-5b), 1.47, 1.41, 1.35, 1.32 (4 s, 12 H, 4 Me), 1.08–1.04 (m, 2 H, SiCH2CH2O), 0.03 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 138.7, 138.5, 128.8, 128.6, 128.5, 128.1, 127.8, 127.7, 110.3, 104.8, 102.2, 99.3, 79.2, 79.0, 78.9, 77.6, 74.0, 73.8, 73.6, 72.4, 70.2, 68.4, 67.9, 66.8, 63.0, 62.4, 29.6, 27.9, 26.6, 19.0, 18.5, -1.2; HRMS (ESI) m/z: found [M + Na]+ 750.3391, C37H53N3O10Si calcd for [M + Na]+ 750.3392.
2-(Trimethylsilyl)ethyl (2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-2-azide-2-deoxy-β-d-galactopyranoside (3)
To a solution of 14 (50 mg, 77.2 μmol) in acetic acid (1.2 mL) was added H2O (0.3 mL) at room temperature. After stirring for 7 h at 90 °C as the reaction was monitored by TLC (EtOAc), the mixture was diluted with toluene and then concentrated. The resulting residue was purified by silica gel column chromatography (Toluene/EtOAc = 1:10) to give 3 (43 mg, 86%): [α]D = +9.1° (c 1.0, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.37–7.28 (m, 10 H, 2 Ph), 5.07 (d, 1 H, Jgem = 11.5 Hz, PhCH2), 4.66 (d, 1 H, PhCH2), 4.50–4.49 (m, 3 H, 2 PhCH2, H-1a), 4.28 (d, 1 H, J1,2 = 8.0 Hz, H-1b), 4.04–3.98 (m, 2 H, H-4b, SiCH2CH2O), 3.93 (m, 1 H, H-4a), 3.87 (m, 1 H, H-6′b), 3.73 (m, 1 H, H-6′a), 3.70–3.64 (m, 4 H, H-6a, H-2b, H-6b, H-5a), 3.62–3.53 (m, 3 H, SiCH2CH2O, H-3a, H-2a), 3.45–3.41 (m, 2 H, H-3b, H-5b), 3.02 (s, 1 H, 4b-OH), 2.48 (d, 1 H, J4,OH = 2.0 Hz, 4a-OH), 2.44 (d, 1 H, J3,OH = 3.0 Hz, 3a-OH), 3.96 (dd, 1 H, J6′b,OH = 3.5 Hz, J6b,OH = 9.0 Hz, 6b-OH), 1.02 (m, 2 H, SiCH2CH2O), 0.00 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 138.5, 137.9, 129.0, 128.9, 128.5, 128.4, 128.3, 128.2, 104.2, 102.2, 81.7, 78.7, 75.0, 74.1, 74.1, 74.0, 73.3, 69.9, 69.0, 68.0, 67.9, 62.8, 62.7, 18.6, -1.1; HRMS (ESI) m/z: found [M + Na]+ 670.2767, C31H45N3O10Si calcd for [M + Na]+ 670.2766.
2-(Trimethylsilyl)ethyl (4,7,8,9-tetra-O-acetyl-5-amino-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(4,7,8,9-tetra-O-acetyl-5-amino-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→6)]-2-azide-2-deoxy-β-d-galactopyranoside (18)
A mixture of 2 (229 mg, 0.326 mmol) and 3 (53 mg, 81.6 μmol) in propionitrile (8.2 mL), which was distilled prior to use, was transferred into a two-neck round bottom flask, in which 3Å molecular sieves (820 mg) were placed, using a cannula and the suspension was stirred for 30 min at rt. The mixture was cooled to −80 °C and the stirring was continued for 30 min. Subsequently, IBr (1 m soln in CH2Cl2, 0.49 mL, 0.489 mmol) and AgOTf (38 mg, 0.147 mmol) were added to the mixture at −80 °C. After stirring for 2.5 h at the same temperature as the reaction was monitored by HPTLC (CHCl3/MeOH = 30:1, developed twice), additional portion of AgOTf (12 mg, 46.7 μmol) was added to the mixture and the stirring was continued. After stirring for total 5 h, the reaction was quenched by the addition of satd aq Na2CO3. The precipitate was filtered through Celite pad and the pad was washed with CHCl3. The combined filtrate and washings were washed with satd aq Na2S2O3, dried over Na2SO4, and concentrated. The resulting residue was purified by gel filtration column chromatography (LH-20, CHCl3/MeOH = 1:1) to give 16 containing four stereoisomers. The crude mixture was then dissolved in EtOH (3.7 mL). To the solution was added 1,4-diazabicyclo[2.2.2]octane (DABCO) (124 mg, 1.11 mmol) at room temperature. After stirring for 3 h at 55 °C as the reaction was monitored by TLC (CHCl3/MeOH = 15:1), the mixture was evaporated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH = 30:1 → 15:1) to give the mixture of dechloroacetylated compounds. The mixture was co-evaporated with toluene and then exposed to high vacuum overnight. The resulting residue was dissolved in DMF (12 mL). To the solution was added 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (14 μL, 96.1 μmol) at room temperature. After stirring for 50 min at 120 °C as the reaction was monitored by TLC (CHCl3/MeOH = 10:1), the reaction was quenched by the addition of acetic acid at room temperature. The mixture was co-evaporated with toluene. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH = 40:1) to give 18 (55 mg, 47%): [α]D = +19.1° (c 2.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (m, 2 H, 2 NH of Neu), 7.33–7.16 (m, 10 H, 2 Ph), 5.82–5.77 (m, 2 H, 2 H-7 of Neu), 5.22–5.17 (m, 2 H, 2 H-8 of Neu), 4.96–4.94 (m, 3 H, 2 H-4 of Neu, PhCH2), 4.68–4.66 (m, 2 H, PhCH2, H-1a), 4.51 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.48 (d, 1 H, PhCH2), 4.32 (dd, 1 H, J8,9′ = 2.5 Hz, Jgem = 12.0 Hz, H-9′ of Neu), 4.29 (dd, 1 H, J8,9′ = 2.5 Hz, 12.0 Hz, H-9′ of Neu), 4.24 (d, 1 H, J1,2 = 8.0 Hz, H-1b), 4.17–4.11 (m, 5 H, H-4b, 2 H-9 of Neu, 2 H-6 of Neu), 4.03 (dd, 1 H, J5,6′ = Jgem = 9.0 Hz, H-6′a), 3.99–3.89 (m, 4 H, SiCH2CH2O, H-2a, 2 H-5 of Neu), 3.80–3.71 (m, 5 H, H-6a, H-6′b, H-3a, H-4a, H-2b), 3.65 (dd, 1 H, J5,6 = 6.0 Hz, Jgem = 9.0 Hz, H-6b), 3.61–3.52 (m, 3 H, H-5b, SiCH2CH2O, H-5a), 3.48 (dd, 1 H, J3,4 = 3.0 Hz, J2,3 = 10.5 Hz, H-3b), 2.38 (dd, 1 H, J3 eq,4 = 5.5 Hz, Jgem = 15.0 Hz, H-3 eq of Neu), 2.26–2.03 (m, 24 H, H-3 eq of Neu, 2 H-3ax of Neu, 7 Ac), 1.89 (s, 3 H, Ac), 1.06–0.97 (m, 2 H, SiCH2CH2O), 0.03 (s, 9 H, SiMe3); Citation13C NMR (125 MHz, CDCl3) δ 171.3, 171.1, 170.4, 170.3, 170.3, 170.2, 170.2, 170.1, 170.0, 169.8, 139.1, 138.5, 128.7, 128.6, 128.5, 128.3, 127.9, 127.7, 104.6, 102.3, 96.2, 96.0, 79.0, 78.3, 78.0, 78.0, 77.9, 76.1, 75.5, 73.9, 73.6, 73.3, 70.8, 70.5, 70.4, 69.9, 69.6, 69.5, 69.3, 68.9, 67.6, 67.5, 63.1, 62.4, 62.3, 49.6, 49.4, 36.4, 35.0, 21.2, 21.2, 21.1, 21.1, 21.0, 21.0, 18.5, 0.3, -1.1; HRMS (ESI) m/z: found [M + Na]+ 1468.5096, C65H87N5O30Si calcd for [M + Na]+ 1468.5097.
2-(Trimethylsilyl)ethyl (4,7,8,9-tetra-O-acetyl-5-tert-butoxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(4,7,8,9-tetra-O-acetyl-5-tert-butoxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→6)]-2-azide-2-deoxy-β-d-galactopyranoside (19)
To a solution of 18 (55 mg, 38.0 μmol) in MeCN (1.8 mL) were added di-tert-butyl dicarbonate (25 mg, 11.4 μmol) and DMAP (0.1 m soln in MeCN; 19 μL, 1.9 μmol) at room temperature. After stirring for 30 min at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 15:1), the reaction was evaporated. The resulting residue was purified by silica gel column chromatography (CHCl3) to give 19 (42 mg, 67%): [α]D = +14.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.24 (m, 10 H, 2 Ph), 5.87 (dd, 1 H, J7,8 = 5.5 Hz, J6,7 = 9.5 Hz, H-7c), 5.80 (dd, 1 H, J7,8 = 5.0 Hz, J6,7 = 10.0 Hz, H-7d), 5.27–5.24 (m. 2 H, H-8c, H-8d), 5.01–4.97 (m, 2 H, H-4c, H-4d), 4.94 (d, 1 H, Jgem = 11.0 Hz, PhCH2), 4.91 (m, 1 H, H-5c), 4.87 (m, 1 H, H-5d), 4.68 (d, 1 H, PhCH2), 4.56 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.54 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.49 (d, 1 H, PhCH2), 4.28–4.18 (m, 6 H, 4 H-9 of Neu, H-1b, H-6d), 4.15 (m, 1 H, H-6c), 4.03 (d, 1 H, J3,4 = 3.0 Hz, H-4b), 4.01–3.97 (m, 3 H, H-5a, SiCH2CH2O, H-6ʹb), 3.95 (dd, 1 H, J3,4 = 3.0 Hz, J2,3 = 9.5 Hz, H-3a), 3.90 (dd, 1 H, J5,6 = 6.5 Hz, Jgem = 10.0 Hz, H-6b), 3.76 (d, 1 H, H-4a), 3.72–3.57 (m, 6 H, H-6ʹa, H-2a, H-6a, H-2b, SiCH2CH2O, H-5b), 3.43 (dd, 1 H, J2,3 = 10.0 Hz, H-3b), 2.97 (br s, 1 H, 4b-OH), 2.93 (br s, 1 H, 4a-OH), 2.43–2.37 (m, 3 H, H-3d ax, H-3d eq, H-3c eq), 2.19 (dd, 1 H, J3ax,4 = 5.5 Hz, Jgem = 14.5 Hz, H-3c ax), 2.14, 2.12, 2.11, 2.09, 2.08, 2.05, 2.04, 1.92 (8 s, 24 H, 8 Ac), 1.56, 1.53 (2 s, 18 H, 2 t-Bu), 1.05–1.01 (m, 2 H, SiCH2CH2O), 0.02 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.7, 170.7, 170.3, 170.1, 170.1, 169.9, 169.9, 169.8, 165.7, 165.0, 149.6, 149.5, 138.7, 138.3, 128.7, 128.6, 128.5, 128.3, 128.1, 127.9, 104.2, 102.0, 96.6, 96.5, 85.8, 85.7, 80.8, 77.9, 77.2, 76.9, 76.3, 75.4, 74.0, 73.6, 73.3, 70.8, 70.3, 70.1, 69.6, 69.1, 68.9, 67.7, 67.7, 67.6, 64.3, 62.8, 61.9, 61.8, 51.4, 51.2, 36.2, 35.3, 28.3, 21.2, 21.1, 21.1, 21.1, 21.1, 21.0, 21.0, 20.9, 18.5, 0.3; HRMS (ESI) m/z: found [M + Na]+ 1668.6145, C75H103N5O34Si calcd for [M + Na]+ 1668.6146.
2-(Trimethylsilyl)ethyl (methyl 5-tert-butoxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)-(2→3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(methyl 5-tert-butoxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)-(2→6)]-2-azide-2-deoxy-β-d-galactopyranoside (20)
To a solution of 19 (42 mg, 25.5 μmol) in MeOH (2.6 mL) was added sodium methoxide (28% solution in MeOH, 14 mg, 0.255 mmol) at room temperature. After stirring for 3 h at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 10:1), the reaction was neutralized with Muromac (H+) resin. The resin was filtered out and washed with MeOH. The filtrate and washings were concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3/MeOH = 20:1 → 10:1) to give 20 (23 mg, 66%) and lactonized product 21 (9 mg, 26%): 20: [α]D = −5.5° (c 1.0, MeOH); 1H NMR (500 MHz, CD3OD) δ 7.47–7.23 (m, 10 H, 2 Ph), 4.93 (d, 1 H, Jgem = 11.0 Hz, PhCH2), 4.74 (d, 1 H, PhCH2), 4.66 (d, 1 H, J1,2 = 7.5 Hz, H-1a), 4.57 (s, 2 H, 2 PhCH2), 4.38 (m, 1 H, H-1b), 4.13 (dd, 1 H, J3,4 = 3.0 Hz, J2,3 = 10.0 Hz, H-3a), 4.09 (br s, 1 H, H-4a), 4.01 (m, 1 H, SiCH2CH2O), 3.93–3.45 (m, 31 H, ring H, 2 OMe, SiCH2CH2O), 2.66–2.61 (m, 2 H, 2 H-3 eq of Neu), 1.95 (t, 1 H, J3ax,4 = Jgem = 12.5 Hz, H-3ax of Neu), 1.72 (t, 1 H, J3ax,4 = Jgem = 12.5 Hz, H-3ax of Neu), 1.44 (s, 18 H, 2 t-Bu), 1.07–0.96 (m, 2 H, SiCH2CH2O), 0.05 (s, 9 H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 171.1, 170.7, 159.5, 140.0, 139.6, 129.5, 129.4, 129.2, 128.9, 128.8, 128.5, 106.0, 103.3, 101.0, 100.3, 81.5, 80.8, 78.5, 76.4, 76.0, 75.7, 75.4, 74.4, 74.2, 74.1, 72.7, 72.5, 71.6, 70.4, 70.3, 70.2, 68.8, 68.6, 68.4, 64.8, 64.5, 63.6, 54.6, 54.5, 53.5, 53.4, 49.8, 49.7, 49.6, 49.5, 49.3, 41.6, 40.1, 30.7, 19.2, -0.0; HRMS (ESI) m/z: found [M + Na]+ 1396.5824, C61H95N5O28Si calcd for [M + Na]+ 1396.5825. 21: [α]D = +7.7° (c 1.1, MeOH); 1H NMR (500 MHz, CD3OD) δ 7.40–7.24 (m, 10 H, 2 Ph), 5.33 (d, 1 H, J3,4 = 4.0 Hz, H-4a), 4.92 (d, 1 H, Jgem = 12.5 Hz, PhCH2), 4.77 (d, 1 H, PhCH2), 4.71 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.65 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.62 (d, 1 H, PhCH2), 4.35 (d, 1 H, J1,2 = 7.5 Hz, H-1b), 4.25 (dd, 1 H, J2,3 = 9.0 Hz, H-3a), 4.09 (m, 1 H, H-4 of Neu), 4.03–3.42 (m, 28 H, ring H, OMe, SiCH2CH2O), 3.39 (dd, 1 H, H-2a), 2.64 (dd, 1 H, J3 eq,4 = 4.5 Hz, Jgem = 13.0 Hz, H-3 eq of Neu), 2.25 (dd, 1 H, J3 eq,4 = 5.5 Hz, Jgem = 13.0 Hz, H-3 eq of Neu), 1.71 (t, 1 H, J3ax,4 = 13.0 Hz, H-3ax of Neu), 1.64 (t, 1 H, J3ax,4 = 13.0 Hz, H-3ax of Neu), 1.44 (s, 18 H, 2 t-Bu), 1.06–0.96 (m, 2 H, SiCH2CH2O), 0.05 (s, 9 H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 170.7, 166.9, 159.8, 159.5, 139.6, 139.1, 129.6, 129.5, 129.4, 129.2, 129.2, 129.1, 128.7, 105.1, 103.2, 100.3, 97.0, 81.5, 81.0, 80.8, 79.5, 79.4, 79.3, 79.2, 79.0, 75.5, 75.4, 75.1, 74.9, 74.6, 74.2, 73.4, 72.5, 72.2, 71.9, 70.3, 69.6, 68.7, 68.5, 68.4, 65.2, 64.8, 64.6, 63.4, 54.7, 54.6, 53.5, 41.9, 41.5, 28.7, 19.2, -1.3; HRMS (ESI) m/z: found [M + Na]+ 1364.5564, C60H91N5O27Si calcd for [M + Na]+ 1364.5563.
2-(Trimethylsilyl)ethyl (4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→3)-(2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→6)]-2-azide-2-deoxy-β-d-galactopyranoside (23)
To a solution of 18 (159 mg, 0.110 mmol) in MeCN (4.4 mL) were added dibenzyl dicarbonate (94 mg, 0.330 mmol) and DMAP (0.1 m soln in MeCN; 55 μL, 5.5 μmol) at room temperature. After stirring for 40 min at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 30:1), the reaction was quenched by the addition of dry MeOH at 0 °C and concentrated. The resulting residue was purified by silica gel column chromatography (CHCl3 → CHCl3/MeOH = 300:1 → 200:1) to give 23 (129 mg, 68%): [α]D = +12.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.46–7.24 (m, 20 H, 4 Ph), 5.88 (dd, 1 H, J7,8 = 6.0 Hz, J6,7 = 9.5 Hz, H-7c), 5.81 (dd, 1 H, J7,8 = 6.0 Hz, J6,7 = 10.0 Hz, H-7d), 5.36 (s, 2 H, PhCH2), 5.34 (d, 1 H, Jgem = 12.5 Hz, PhCH2), 5.31 (d, 1 H, PhCH2), 5.25–5.22 (m. 2 H, H-8c, H-8d), 5.03–4.99 (m, 3 H, H-4c, H-4d, H-5d), 4.96–4.94 (m, 2 H, H-5c, PhCH2), 4.68 (d, 1 H, Jgem = 11.5 Hz, PhCH2), 4.57 (d, 1 H, J1,2 = 8.0 Hz, H-1a), 4.52 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 4.47 (d, 1 H, PhCH2), 4.25–4.17 (m, 6 H, H-1b, 4 H-9 of Neu, H-6d), 4.14 (m, 1 H, H-6c), 4.04–3.99 (m, 3 H, H-4b, H-6ʹb, SiCH2CH2O), 3.95 (dd, 1 H, J3,4 = 3.5 Hz, J2,3 = 9.5 Hz, H-3a), 3.91 (dd, 1 H, J5,6 = 6.5 Hz, Jgem = 10.0 Hz, H-6b), 3.76 (near s, 1 H, H-4a), 3.73–3.58 (m, 7 H, H-2a, H-6ʹa, H-6a, H-5a, H-2b, H-5b, SiCH2CH2O), 3.44 (dd, 1 H, J3,4 = 3.0 Hz, J2,3 = 10.5 Hz, H-3b), 2.89 (br s, 2 H, 2 OH), 2.45–2.34 (m, 3 H, H-3d eq, H-3c eq, H-3d ax), 2.26 (dd, 1 H, J3ax,4 = 6.0 Hz, Jgem = 14.5 Hz, H-3c ax), 2.15, 2.12, 2.11, 2.03, 2.03, 1.99, 1.99, 1.92 (8 s, 24 H, 8 Ac), 1.05–1.02 (m, 2 H, SiCH2CH2O), 0.03 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.7, 170.7, 170.2, 170.0, 169.9, 169.8, 169.7, 165.4, 164.7, 151.3, 151.1, 138.5, 138.2, 134.7, 134.7, 129.0, 129.0, 129.0, 128.7, 128.6, 128.5, 128.5, 128.4, 128.2, 128.0, 127.9, 104.1, 102.0, 96.6, 96.5, 80.9, 77.2, 76.9, 76.4, 75.5, 73.9, 73.5, 73.2, 70.3, 70.1, 70.0, 69.9, 69.8, 69.7, 69.0, 68.9, 67.7, 67.5, 67.5, 67.4, 64.4, 62.8, 61.9, 61.8, 51.9, 51.7, 35.9, 35.1, 21.1, 21.1, 21.0, 20.9, 20.9, 18.4, -1.1; HRMS (ESI) m/z: found [M + Na]+ 1736.5832, C81H99N5O34Si calcd for [M + Na]+ 1736.5833.
2-(Trimethylsilyl)ethyl (4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→3)-(4-O-acetyl-2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylono-1,5-lactam)-(2→6)]-4-O-acetyl-2-azide-2-deoxy-β-d-galactopyranoside (24)
To a solution of 23 (96 mg, 56.0 μmol) in pyridine (1.8 mL) were excess amounts of Ac2O (0.9 mL) and DMAP (0.1 m soln in CH2Cl2; 28 μL, 2.8 μmol) at room temperature. After stirring for 20 h at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 45:1), the mixture was co-evaporated with toluene. The resulting residue was purified by silica gel column chromatography (CHCl3) to give 24 (96 mg, 95%): [α]D = +32.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.46–7.23 (m, 20 H, 4 Ph), 5.85 (dd, 1 H, J7,8 = 6.0 Hz, J6,7 = 9.5 Hz, H-7c), 5.74 (dd, 1 H, J7,8 = 5.0 Hz, J6,7 = 9.5 Hz, H-7d), 5.38–5.29 (m, 5 H, PhCH2, H-4b), 5.28–5.23 (m, 2 H, H-8c, H-8d), 5.20 (d, 1 H, J3,4 = 3.0 Hz, H-4a), 5.03 (m, 1 H, H-4c), 4.97–4.94 (m, 3 H, H-5c, H-5d, H-4d), 4.87 (d, 1 H, Jgem = 11.0 Hz, PhCH2), 4.70 (d, 1 H, J1,2 = 7.5 Hz, H-1a), 4.61 (d, 1 H, PhCH2), 4.48 (d, 1 H, Jgem = 11.5 Hz, PhCH2), 4.39 (d, 1 H, PhCH2), 4.29 (m, 1 H, H-1b), 4.25–4.17 (m, 6 H, 4 H-9 of Neu, H-6d, H-6c), 3.99–3.95 (m, 2 H, SiCH2CH2O, H-3a), 3.83 (dd, 1 H, J5,6ʹ = 3.5 Hz, Jgem = 10.0 Hz, H-6b), 3.75–3.57 (m, 7 H, H-5b, H-5a, H-6b, H-2b, H-3b, SiCH2CH2O, H-2a), 3.51 (dd, 1 H, J5,6ʹ = 5.5 Hz, Jgem = 9.0 Hz, H-6ʹa), 3.38 (dd, 1 H, J5,6 = 7.0 Hz, H-6a), 2.47–2.38 (m, 2 H, H-3c eq, H-3d eq), 2.31–2.22 (m, 2 H, H-3d ax, H-3c ax), 2.14, 2.11, 2.10, 2.07, 2.06, 2.04, 2.01, 2.00, 1.98, 1.91 (10 s, 30 H, 10 Ac), 1.08–1.04 (m, 2 H, SiCH2CH2O), 0.03 (s, 9 H, SiMe3); 13C NMR (125 MHz, CDCl3) δ 170.9, 170.8, 170.6, 170.1, 170.0, 169.9, 169.8, 169.7, 165.0, 163.6, 151.5, 151.2, 138.5, 138.0, 134.9, 134.8, 129.0, 128.9, 128.8, 128.7, 128.7, 128.5, 128.6, 128.4, 128.3, 128.1, 128.0, 103.2, 102.2, 96.4, 96.3, 78.5, 76.9, 76.1, 75.6, 74.0, 73.7, 73.2, 72.4, 71.3, 70.3, 70.1, 70.0, 69.9, 69.8, 69.3, 69.1, 68.3, 68.3, 67.7, 67.4, 63.9, 63.5, 61.9, 61.8, 51.9, 51.7, 35.7, 34.8, 21.2, 21.2, 21.1, 21.1, 21.0, 20.9, 20.9, 18.6, -1.1; HRMS (ESI) m/z: found [M + Na]+ 1820.6045, C85H103N5O36Si calcd for [M + Na]+ 1820.6044.
2-(Trimethylsilyl)ethyl (methyl 4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)-(2→3)-(4-O-acetyl-2,6-di-O-benzyl-β-d-galactopyranosyl)-(1→3)-[(methyl 4,7,8,9-tetra-O-acetyl-5-benzyloxycarbamoyl-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonate)-(2→6)]-4-O-acetyl-2-azide-2-deoxy-β-d-galactopyranoside (25)
To a solution of 24 (30 mg, 16.7 μmol) in THF/MeOH (2:1, 1.8 mL) was added DMAP (6 mg, 50.1 μmol) at room temperature. After stirring for 68.5 h at 40 °C as the reaction was monitored by TLC (CHCl3/MeOH = 30:1), the reaction mixture was evapo column chromatography (CHCl3/MeOH = 200:1) to give 25 (25 mg, 81%): [α]D = +5.0° (c 1.0, CHCl3); 1H NMR (500 MHz, CD3OD) δ 7.45–7.30 (m, 20 H, 4 Ph), 5.58 (m, 1 H, H-8c), 5.42–5.38 (m, 3 H, H-7c, H-7d, H-4b), 5.35 (m, 1 H, H-8d), 5.13 (d, 1 H, Jgem = 12.5 Hz, PhCH2), 5.11 (d, 1 H, Jgem = 12.0 Hz, PhCH2), 5.01 (d, 1 H, J3,4 = 3.0 Hz, H-4a), 4.97–4.84 (m, 5 H, H-1a, 3 PhCH2, H-4c), 4.81 (td, 1 H, J3 eq,4 = 4.5 Hz, J3ax,4 = 12.5 Hz, H-4d), 4.75 (d, 1 H, Jgem = 13.0 Hz, PhCH2), 4.50–4.36 (m, 6 H, 2 PhCH2, 2 5-NH, H-3a, H-9ʹc), 4.27 (dd, 1 H, J8,9ʹ = 2.5 Hz, Jgem = 12.5 Hz, H-9ʹd), 4.21 (d, 1 H, J1,2 = 8.0 Hz, H-1b), 4.09 (dd, 1 H, J8,9 = 5.5 Hz, H-9d), 4.04–3.99 (m, 2 H, SiCH2CH2O, H-6d), 3.98 (dd, 1 H, J8,9 = 5.0 Hz, Jgem = 13.0 Hz, H-9c), 3.82–3.68 (m, 12 H, 2 OMe, H-6ʹb, H-5a, H-3b, H-5c, H-5d, H-6c), 3.65–3.56 (m, 3 H, H-5b, SiCH2CH2O, H-2b), 3.48 (dd, 1 H, J5,6ʹ = 6.0 Hz, Jgem = 9.5 Hz, H-6ʹa), 3.41 (m, 2 H, H-2a, H-6a), 3.28 (dd, 1 H, J5,6 = 6.0 Hz, Jgem = 9.5 Hz, H-6b), 2.59 (dd, 1 H, J3 eq,4 = 4.5 Hz, Jgem = 12.5 Hz, H-3c eq), 2.55 (dd, 1 H, Jgem = 12.5 Hz, H-3d eq), 2.15, 2.12, 2.10, 2.05, 2.03, 2.021.98 (7 s, 21 H, 7 Ac), 1.84 (dd, 1 H, H-3d ax), 1.82 (s, 6 H, 2 Ac), 1.77–1.72 (m, 4 H, Ac, H-3c ax), 1.07–1.02 (m, 2 H, SiCH2CH2O), 0.04 (s, 9 H, SiMe3); 13C NMR (125 MHz, CD3OD) δ 171.1, 171.0, 170.9, 170.6, 170.5, 170.3, 169.9, 169.8, 168.3, 168.0, 156.2, 156.1, 140.1, 138.5, 136.7, 128.8, 128.7, 128.6, 128.5, 128.4, 128.0, 127.9, 127.3, 127.2, 102.4, 102.1, 99.0, 97.3, 79.1, 77.9, 75.5, 74.9, 73.6, 73.5, 72.9, 72.6, 72.5, 72.2, 69.8, 69.2, 69.0, 68.9, 68.5, 68.4, 67.7, 67.6, 67.4, 64.1, 63.4, 62.6, 62.5, 53.4, 53.2, 51.7, 51.5, 38.2, 37.9, 21.7, 21.3, 21.2, 21.1, 21.0, 20.9, 20.7, 18.6, -1.1; HRMS (ESI) m/z: found [M + Na]+ 1884.6568, C87H111N5O38Si calcd for [M + Na]+ 1884.6569.
2-(Trimethylsilyl)ethyl (5-acetamide-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→3)-β-d-galactopyranosyl-(1→3)-[(5-acetamide-3,5-dideoxy-d-glycero-α-d-galacto-2-nonulopyranosylonic acid)-(2→6)]-2-acetamide-2-deoxy-β-d-galactopyranoside (1)
To a solution of 25 (47 mg, 25.2 μmol) in 1,4-dioxane/H2O (9:1, 5.0 mL) was added palladium hydroxide (20 wt % Pd (dry basis) on carbon, wet; 47 mg) at room temperature. After stirring under H2 atmosphere for 29 h at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 15:1) and MALDI-TOF MS, the reaction mixture was filtered through membrane filter and washed with MeOH. The combined filtrate and washings were concentrated and exposed to high vacuum. The residue was then dissolved in pyridine (5.0 mL). To the mixture were added excess amounts of Ac2O (0.5 mL) and a catalytic amount of DMAP at room temperature. After stirring for 37 h at room temperature as the reaction was monitored by TLC (CHCl3/MeOH = 15:1) and MALDI-TOF MS, the reaction mixture was concentrated. The residue was then diluted with CHCl3, washed with 2 m HCl, H2O, and satd aq Na2CO3, dried over Na2SO4, concentrated. The resulting residue was roughly purified by gel filtration column chromatography (LH-20, CHCl3/MeOH = 1:1) to give per-acetylated compound 26 along with small amounts of contaminants. The crude mixture was dissolved in THF/MeOH (1:1, 5.0 mL). To the solution was added 1 m aq NaOH (500 μl, 0.504 mmol) at room temperature. After stirring for 9 h at room temperature as the reaction was monitored by TLC (CHCl3/MeOH/5% aq. CaCl2 = 5:4:1), the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography (PSQ-60B, CHCl3/MeOH/H2O = 5:4:0.8 → 5:4:1) to give 1 (16 mg, 59% over three steps); [α]D = −5.5° (c 1.0, MeOH/H2O = 1:1); 1H NMR (500 MHz, D2O, Chemical shifts are expressed in ppm (δ) relative to the signal of Me3Si in the SE group, adjusted to 0.00 ppm) δ 4.51 (d, 1 H, J1,2 = 8.5 Hz, H-1b), 4.47 (d, 1 H, J1,2 = 7.5 Hz, H-1a), 4.17 (d, 1 H, J3,4 = 3.0 Hz, H-4b), 4.05 (dd, 1 H, J3,4 = 3.0 Hz, J2,3 = 9.5 Hz, H-3a), 4.02 (m, 1 H, SiCH2CH2O), 3.97 (dd, 1 H, J2,3 = 10.5 Hz, H-2b), 3.95–3.55 (m, 23 H, ring H, SiCH2CH2O), 3.52 (dd, 1 H, H-2a), 2.75–2.69 (m, 2 H, 2 H-3 eq of Neu), 2.02, 2.01, 1.99 (3 s, 9 H, 3 Ac), 1.78 (t, 1 H, J3ax,4 = Jgem = 12.0 Hz, H-3ax of Neu), 1.67 (t, 1 H, J3ax,4 = Jgem = 12.0 Hz, H-3ax of Neu), 1.01–0.83 (m, 2 H, SiCH2CH2O), 0.00(s, 9 H, SiMe3); 13C NMR (125 MHz, D2O, Chemical shifts are expressed in ppm (δ) relative to the signal of Me3Si in the SE group, adjusted to 0.00 ppm) δ 177.4, 177.3, 177.0, 176.3, 175.7, 107.1, 103.1, 102.8, 102.1, 82.9, 78.0, 77.1, 75.6, 75.2, 75.0, 74.2, 74.0, 71.4, 70.8, 70.7, 70.6, 70.6, 70.5, 70.3, 69.8, 65.8, 65.0, 64.9, 63.4, 54.2, 54.1, 53.4, 51.3, 42.6, 42.1, 24.8, 24.4, 19.6, 0.0; HRMS (ESI) m/z: found [M + Na]− 1086.3790, C41H69N3O27Si calcd for [M + Na]− 1086.3791.
Author contributions
All authors participated in planning the research. Maiko Tanase and Akihiro Imamura designed and synthesized new compounds. Akihiro Imamura wrote the manuscript with the assistance of Hiromune Ando, Makoto Kiso, and Hideharu Ishida. Hideharu Ishida supervised the research. All authors approved the final version of the manuscript for submission.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the JSPS KAKENHI [grant number JP16K18694], [grant number JP15K07409].
Acknowledgements
This article is reported as synthetic studies on sialoglycoconjugates 166. Part 165: Ref. [Citation30].
References
- Varki A, Cummings RD, Esko JD, et al., editors. Essentials of glycobiology. 2nd ed. New York (NY): Cold Spring Harbor Laboratory Press; 2008.
- Inoue Y, Lee YC, Troy FA II, editors. Sialobiology and other novel forms of glycosylation. Osaka: Gakushin Publishing Company; 1999.
- Schnaar RL, Gerardy-Schahn R, Hildebrandt H. Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev. 2014;94:461–518.
- Schengrund CL. Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem Sci. 2015;40(7):397–406.
- Hasegawa A. Synthesis of sialoglycoconjugates. In: Khan SH, O’Neill RA, editors. Modern methods in carbohydrate synthesis. Amsterdam: Harwood Academic Publishers; 1996. p. 277–301.
- Hasegawa A, Kiso M. Chemical synthesis of sialyl glycosides. In: Hanessian S, editor. Preparative carbohydrate chemistry. New York (NY): Marcel Dekker, Inc.; 1997. p. 357–379.
- Ando H, Kiso M. Selective α-sialylation. In: Fraser-Reid B, Tatsuta K, Thiem J, editors. Glycoscience. vol. 2. New York (NY): Springer-Verlag, Berlin Heidelberg; 2008. p. 1315–1359.
- Navuluri C, Crich D. Stereocontrolled synthesis of sialosides. In: Hung SC, Zulueta MML, editors. Glycochemical synthesis. Hoboken (NJ): John Wiley & Sons, Inc.; 2016. p. 131–154.
- Halcomb RL, Chappell MD. Recent developments in technology for glycosylation with sialic acid. In: Wang PG, Bertozzi CR, editors. Glycochemistry. New York (NY): Marcel Dekker, Inc.; 2001. p. 177–220.
- De Meo C, Boons GJ, Demchenko AV. Synthesis of glycosides of sialic acid. In: Kamerling JP, Boons GJ, Lee YC, et al., editors. Comprehensive glycoscience. Vol. 1, Introduction to glycoscience, Synthesis of carbohydrates. Oxford (UK): Elsevier; 2007. p. 583–604.
- Tanase M, Imamura A, Ando H, et al. A 5-ureido-modified sialyl donor: a tool for the synthesis of α-sialosides. Org Lett. 2016;18(6):1454–1457.
- Tanaka H, Ando H, Ishida H, et al. Synthetic study on α(2→8)-linked oligosialic acid employing 1,5-lactamization as a key step. Tetrahedron Lett. 2009;50(31):4478–4481.
- Ando S, Tanaka Y, Kobayashi S, et al. Synaptic function of cholinergic-specific Chol-1α ganglioside. Neurochem Res. 2004;29(4):857–867.
- Kudelka MR, Ju T, Heimburg-Molinaro J, et al. Simple sugars to complex disease–Mucin-type O-glycans in cancer. In: Drake RR, Ball LE, editors. Advanced in cancer research. Vol. 126, Glycosylation and cancer. Oxford (UK): Elsevier; 2015. p. 53–135.
- Ito H, Ishida H, Collins BE, et al. Systematic synthesis and MAG-binding activity of novel sulfated GM1b analogues as mimics of Chol-1 (α-series) gangliosides: highly active ligands for neural siglecs. Carbohydr Res. 2003;338(16):1621–1639.
- Capon B, Collins PM, Levy AA, et al. Reactions at position 1 of carbohydrates. V. Nucleophilic displacement reactions of acetylglycosyl halides. J Chem Soc. 1964;3242–3254.
- Nitz M, Bundle DR. Efficient synthesis of 3,6-dideoxy-β-d-arabino-hexopyranosyl-terminated LacdiNac glycan chains of the Trichinella spiralis parasite. J Org Chem. 2000;65(10):3064–3073.
- Konradsson P, Udodong UE, Fraser-Reid B. Iodonium promoted reactions of disarmed thioglycosides. Tetrahedron Lett. 1990;31(30):4313–4316.
- Veeneman GH, van Leeuwen SH, van Boom JH. Iodonium ion promoted reactions at the anomeric centre. II An efficient thioglycoside mediated approach toward the formation of 1,2-trans linked glycosides and glycosidic esters. Tetrahedron Lett. 1990;31(9):1331–1334.
- Magnusson G. 2-(Trimethylsilyl)ethyl(TMSEt) glycosides; anomeric blocking, deblocking and activation in the synthesis of oligosaccharides. Trends Glycosci Glycotech. 1992;4(18):358–367.
- Yan RB, Yang F, Wu Y, et al. An efficient and improved procedure for preparation of triflyl azide and application in catalytic diazotransfer reaction. Tetrahedron Lett. 2005;46(52):8993–8995.
- Wilstermann M, Magnusson G. Synthesis of disaccharide glycosyl donors suitable for introduction of the β-d-Galp-(1→3)-α- and -β-d-GalpNAc groups. Carbohydr Res. 1995;272(1):1–7.
- Meijer A, Ellervik U. Study of interhalogens/silver trifluoromethanesulfonate as promoter systems for high-yielding sialylations. J Org Chem. 2002;67(21):7407–7412.
- Hasegawa A, Nagahama T, Ohki H, et al. Reactivity of glycosyl promoters in α-glycosylation of N-acetyl-neuraminic acid with the primary and secondary hydroxyl groups in the suitably protected galactose and lactose derivatives. J Carbohydr Chem. 1991;10(3):493–498.
- Lefeber DJ, Kamerling JP. Vliegenthart JFG. The use of diazabicyclo[2.2.2]octane as a novel highly selective dechloroacetylation reagent. Org Lett. 2000;2(5):701–703.
- Burk MJ, Allen JG. A mild amide to carbamate transformation. J Org Chem. 1997;62(20):7054–7057.
- Hwu JR, Jain ML, Tsay SC, et al. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Lett. 1996;37(12):2035–2038.
- Tom NJ, Simon WM, Frost HN, et al. Deprotection of a primary Boc group under basic conditions. Tetrahedron Lett. 2004;45(5):905–906.
- Routier S, Saugé L, Ayerbe N, et al. A mild and selective method for N-Boc deprotection. Tetrahedron Lett. 2002;43(4):589–591.
- Goto K, Sawa M, Tamai H, et al. The total synthesis of starfish ganglioside GP3 bearing a unique sialyl glycan architecture. Chem Eur J. 2016;22(24):8323–8331.