
Abstract
Our investigation of phenolic constituents of fruits, flower buds, and leaves of Feijoa sellowiana led to the isolation of twenty-one phenolics including three new gossypetin glycosides 1–3, and also the purification of a proanthocyanidin fraction. A high-performance liquid chromatography method for simultaneous analysis of phenolic constituents was established and then used to investigate the phenolic profiles of the parts of the plant species, to show the presence of characteristic flavonoids and ellagic acid derivatives or ellagitannins in the extracts from fruits, flower buds, and leaves. The branch extract profile also suggested the presence of alkylated ellagic acids as characteristic constituents. Inhibitory effects of feijoa flavonoids on mushroom tyrosinase were seen, although in some cases this may have resulted from direct interaction with the enzyme. Cytotoxic effect of the proanthocyanidin fraction was also shown.
New flavonoids were found in various parts of Feijoa sellowiana.
Use of polyphenol-rich plants in medicine, cosmetics, and health foods is expected [Citation1,2] based on recent findings regarding the health effects of their polyphenolic constituents, including antioxidant, antibacterial, and antitumor effects. Several distinctive plants in the family Myrtaceae are rich in polyphenolics, including flavonoids and ellagic acids [Citation3,4], as exemplified by guava (Psidium guajava) [Citation5], clove (Syzygium aromaticum) [Citation6,7], and allspice (Pimenta dioica) [Citation8].
Feijoa sellowiana Berg, also in the family Myrtaceae, is known by its edible fruits [Citation9], and this evergreen tree is widely cultivated in Oceania and the Americas. Our preliminary examination of the constituents of this plant suggested the presence of unidentified phenolics in addition to known flavonoids and related phenolics [Citation10–17]. The objective of this paper is therefore to characterize the polyphenolic constituents, especially flavonoids and ellagic acids, in this species to provide the basis for using this plant as a polyphenolic resource.
Materials and methods
General
Electrospray ionization mass spectrometry (ESI-MS) measurements were performed on a Bruker amaZon X and ETD instrument (Bruker, Billerica, MA, USA) with 50% aq. acetonitrile containing 0.1% formic acid. High-resolution (HR)-ESI-MS measurements were performed on an Agilent 6520 + 4240 Series Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) LC/MS instrument (Agilent, Santa Clara, CA, USA) with 50% aq. acetonitrile containing 0.1% formic acid. 1H and 13C NMR spectra were recorded on Varian INOVA 600AS (600 MHz for 1H, and 151 MHz for 13C) and Varian INOVA 400MR (400 MHz for 1H, and 100 MHz for 13C) instruments (Varian, Palo Alto, CA, USA). Chemical shifts were given in δ (ppm) downfield from tetramethylsilane, based on those of the solvent signals [δH 2.04 and δC 29.8 for acetone-d6, and δH 3.30 and δC 49.8 for methanol-d4]. Optical rotations were measured on a JASCO DIP-1000 digital polarimeter (JASCO, Tokyo, Japan). Electronic circular dichroism (ECD) spectra were measured on a JASCO J-720 W spectrometer. The flow rate was set to 1.0 mL/min. A YMC Pack ODS Pro C8 (4.6 mm i.d. × 150 mm) column was also used as mentioned below. Preparative HPLC was performed on a YMC ODS-A324 (10 mm i.d. × 300 mm) column (YMC, Kyoto, Japan) with H2O-CH3CN-HCOOH (85:15:5, by volume) as the eluent. A Hitachi L-2455 diode-array detector (DAD) (Hitachi, Tokyo, Japan) was used as a UV detector for HPLC. Analytical normal-phase (NP) HPLC was performed on a YMC-Pack SIL A-003 column (4.6 mm i.d. × 250 mm) with n-hexane−MeOH−tetrahydrofuran−formic acid (55:33:11:1) containing oxalic acid (450 mg/L) at room temperature. The flow rate was set at 1.5 mL/min, and UV detection at 280 nm was effected. Silica gel (YMC), Toyopearl HW-40C (TOSOH, Tokyo, Japan), YMC-gel ODS-A (75 μm) (YMC), and MCI-gel CHP-20P (Mitsubishi Chemical, Tokyo, Japan) were used for column chromatography. Apigenin, flavone, kaempferol, luteolin, morin, myricetin, naringenin, quercetagenin, quercetin, hyperoside, isoquercitrin and quercitrin were purchased from Extrasynthese (Lyon, France), cyanidin-3-O-β-glucoside were purchased from Funakoshi Co., Ltd. (Tokyo, Japan) and gossypetin was from Indofine Chemical (Hillsborough, NJ, USA). Phloroglucinol was from Ishizu Pharmaceutical (Osaka, Japan). Ellagic acid, L-tyrosine, and mushroom tyrosinase were from Sigma (USA), and kojic acid was from TCI (Tokyo, Japan).
Plant material
Fresh fruits, flower buds, leaves, and branches of a single tree of F. sellowiana (cv. “Coolidge”) were collected at the Medicinal Plant Garden, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences. Leaves and buds were collected in June, and fruits were collected in October and November. The voucher specimen No. FS-001HS was kept at the Medicinal Plant Garden.
Extraction and isolation
Fresh fruits (309 g) were homogenized in 70% acetone and the concentrated filtrate from the homogenate was extracted successively with ethyl acetate and n-butanol. The ethyl acetate extract (2.8 g) was subjected to column chromatography on Toyopearl HW-40C (2.2 cm i.d. × 40 cm) with 70% EtOH and then with 70% acetone. Combined fractions 21–25 (74.7 mg) of the 70% EtOH eluate were fractionated by column chromatography on MCI gel CHP-20P (1.1 cm i.d. × 30 cm) with increasing concentrations of MeOH in H2O, to yield phyllanthusiin E (17) (9.2 mg) from the 35% MeOH eluate, and gossypetin-3-O-l-α-arabinofuranoside (3.3 mg) (1) from the 40 and 45% MeOH eluates. The remaining part of the combined eluates with 40% MeOH and 45% MeOH, as well as the eluate with 60% MeOH from the MCI gel column were purified by preparative HPLC to yield aromadendrin-7-O-β-glucoside (1.2 mg) (19), 4-O-α-arabinofuranosylellagic acid (13) (0.4 mg), and naringenin-7-O-β-glucoside (0.8 mg) (18). Combined fractions 26–30 (81.7 mg) of the 70% EtOH eluate from the Toyopearl column were fractionated by column chromatography on MCI gel CHP-20P (1.1 cm i.d. × 20 cm) with increasing concentrations of MeOH in H2O, to yield ellagic acid (16) (3.5 mg) from the 65% MeOH eluate. The water-soluble portion (27.9 g) obtained after the n-butanol extraction was subjected to column chromatography on Diaion HP-20 (2.2 cm i.d. × 35 cm) with increasing concentrations of MeOH in H2O. The 60% MeOH eluate (396.6 mg) from the Diaion column was chromatographed on a Sephadex LH-20 column (1.1 cm i.d. × 30 cm) with the mixtures of 70% EtOH and 70% acetone, to yield PAOF-1 (22) (164.9 mg) from the eluate with a mixture of 70% EtOH–70% acetone [4:6 (v/v)].
Fresh flower buds (105 g) were extracted with MeOH and the concentrated filtrate was then treated with n-hexane to remove the lipophilic materials. The remaining MeOH soluble part (12.2 g) was subjected to column chromatography on Diaion HP-20 (4.0 cm i.d. × 40 cm) with increasing concentrations of MeOH in H2O. The H2O eluate (8.3 g) from the column was chromatographed on a Sephadex LH-20 column (2.2 cm i.d. × 50 cm) with 70% EtOH, to yield pedunculagin (21) (254.1 mg). The eluate with 20% MeOH (1.3 g) from the Diaion column was fractionated with column chromatography on YMC-gel ODS-A (75 μm) (2.2 cm i.d. × 50 cm) with increasing concentrations of MeOH in H2O containing 5% acetic acid, to yield cyanidin-3-O-β-glucoside (20) (12.7 mg) and gossypetin-3-O-α-l-arabinofuranoside (1) (6.4 mg).
Fresh leaves (111 g) were homogenized in 70% acetone and the concentrated filtrate from the homogenate was extracted with ethyl acetate and then with n-butanol. The ethyl acetate extract (1.8 g) was subjected to column chromatography on Toyopearl HW-40C (2.2 cm i.d. × 40 cm) with 70% EtOH and then 70% acetone as the eluents. Combined fractions 21–25 (257 mg) from the 70% EtOH eluate were fractionated by column chromatography on MCI gel CHP-20P (1.1 cm i.d. × 30 cm) with increasing concentrations of MeOH in H2O, to yield hyperoside (5) (30.0 mg) from combined fractions 1–20 of the 35% MeOH eluate. Combined fractions 31–150 of the 35% MeOH eluate were further purified by preparative HPLC to yield hyperoside (5) (6.6 mg), isoquercitrin (6) (1.2 mg), reynoutrin (7) (11.6 mg), avicularin (9) (31.0 mg), and quercitrin (10) (19.6 mg). The 40% MeOH eluate from the MCI column, and the 100% MeOH eluate were respectively identified as guaijaverin (8) (19.0 mg) and flavone (11) (7.3 mg). In a separate experiment, fresh leaves (531 g) were homogenized in MeOH and, after removal of the lipophilic material from the concentrated filtrate with n-hexane, the remaining MeOH solution was evaporated to yield an extract (38.7 g). This extract was subjected to column chromatography on Diaion HP-20 (4.0 cm i.d. × 40 cm) with increasing concentrations of MeOH in H2O. The 40% MeOH eluate (3.4 g) was fractionated by column chromatography on Toyopearl HW-40C (2.2 cm i.d. × 40 cm) with 70% EtOH, and combined fractions 16–20 (274.2 mg) were purified by column chromatography on MCI gel CHP-20P (1.1 cm i.d. × 40 cm) with increasing concentrations of MeOH in H2O, to yield 4-O-α-arabinofuranosylellagic acid (13) (8.8 mg) from fractions 21–30 of the 50% MeOH eluate, and trifolin (12) (1.4 mg) from fractions 41–50 of the 50% MeOH eluate. The 80% MeOH eluate (341.5 mg) from the Diaion column was further purified by column chromatography on MCI gel CHP-20P (2.2 cm i.d. × 30 cm) with increasing concentrations of MeOH in H2O, and further purified by preparative HPLC to yield quercetin (14) (2.0 mg). Separately, dried leaves (1.78 kg) were pulverized, and extracted successively with n-hexane, ethyl acetate, MeOH, and 70% acetone. The MeOH extract (265.1 g) and the 70% acetone extract (230.7 g) were combined, and the mixture dissolved in water was extracted with n-hexane, ethyl acetate, and n-butanol, to yield the following extracts: n-hexane (14.8 g), ethyl acetate (63.0 g), n-butanol (139.7 g), and aq. extracts (265.7 g). The n-butanol extract was subjected to column chromatography on a Diaion HP-20 column (10.0 cm i.d. × 70 cm) with increasing concentrations of MeOH in H2O, and the 70% MeOH eluate was subjected to column chromatography on a YMC gel ODS-A column (75 μm) (5.0 cm i.d. × 35 cm) with increasing concentrations of MeOH in H2O. The second fraction of the 25% MeOH eluate (2.47 g) was further chromatographed on a column of MCI gel CHP-20P and preparative HPLC to give gossypetin (3.9 mg) (15). The second fraction of the 30% MeOH eluate (1.80 g) from the MCI gel column was chromatographed again on a column of MCI gel CHP-20P (2.2 cm i.d. × 35 cm) with increasing concentrations of MeOH in H2O. The 40% MeOH eluate (65.8 mg) and the first part of the 45% MeOH eluate (109.1 mg) from the MCI gel column were respectively purified by preparative HPLC to yield gossypetin-3-O-β-galactopyranoside (4) (3.4 mg), gossypetin-3-O-β-xylopyranoside (3) (4.0 mg), gossypetin-3-O-α-rhamnopyranoside (2) (7.5 mg), and gossypetin-3-O-α-l-arabinofuranoside (4.5 mg) (1).
Gossypetin-3-O-α-L-arabinofuranoside (1)
An amorphous yellow powder, [α]D − 72.8° (c 1, MeOH). ECD (c 0.01, MeOH) [θ] (nm) −4.9 × 103 (233), +5.4 × 103 (258), −1.1 × 104 (304). HR-ESI-MS found, m/z 451.0864 ([M + H]+); Calcd for C20H18O12 + H, 451.0877. ESI-MS m/z 451 ([M + H]+), 319 (MS/MS) ([M − pentose + H]+) (positive-ion mode); m/z 449 ([M−H]−), 317 (MS/MS) ([M − pentose − H]−) (negative-ion mode). 1H NMR, see Table . 13C NMR [acetone-d6–D2O (13:2, v/v), 151 MHz] δC 158.2 (C-2), 133.9 (C-3), 179.5 (C-4), 153.9 (C-5), 99.2 (C-6), 153.9 (C-7), 125.7 (C-8), 145.6 (C-9), 104.5 (C-10), 122.5 (C-1′), 116.6 (C-2′), 145.5 (C-3′), 149.1 (C-4′), 116.1 (C-5′), 122.5 (C-6′), 108.9 (C-1″), 82.3 (C-2″), 77.9 (C-3″), 87.7 (C-4″), 61.6 (C-5″). 1H NMR [methanol-d4, 600 MHz] δH 7.63 (1H d J = 2.4 Hz) (H-2′), 7.59 (1H dd J = 8.4, 2.4 Hz) (H-6′), 6.90 (1H d J = 8.4 Hz) (H-5′), 6.28 (1H s) (H-6), 5.46 (1H br s) (H-1″), 4.33 (1H dd J = 3.0, 0.6 Hz) (H-2″), 3.91 (1H dd J = 5.4, 3.0 Hz) (H-3″), 3.91 (1H dd J = 8.4, 3.6 Hz) (H-4″), 3.51 (1H dd J = 12.0, 3.6 Hz) (H-5″a), 3.48 (1H dd J = 12.0, 8.4 Hz) (H-5″b). 13C NMR [methanol-d4, 151 MHz] δC 160.1 (C-2), 135.4 (C-3), 181.2 (C-4), 155.7 (C-5), 100.4 (C-6), 155.8 (C-7), 127.0 (C-8), 147.5 (C-9), 106.1 (C-10), 124.0 (C-1′), 117.9 (C-2′), 147.1 (C-3′), 150.7 (C-4′), 114.2 (C-5′), 124.1 (C-6′), 110.3 (C-1″), 84.1 (C-2″), 79.5 (C-3″), 88.9 (C-4″), 63.4 (C-5″).
Table 1. 1H-NMR assignment of compound 1–3 and correspond known compounds 7, 9, and 10 [1H-NMR 600 MHz, Acetone-d6–D2O (13:2), 27 °C] (J in Hz).
Gossypetin-3-O-α-rhamnopyranoside (2)
An amorphous yellow powder, [α]D −98.3° (c 1, MeOH). ECD (c 0.01, MeOH) [θ] (nm) −7.2 × 103 (231),+8.0 × 103 (255), −7.2 × 103 (322). HR-ESI-MS Found, m/z 463.0889 ([M−H]−); Calcd for C21H20O12−H, 463.0871. ESI-MS m/z 465 ([M + H]+), 319 (MS/MS) ([M − deoxyhexose + H]+) (positive-ion mode); m/z 463 ([M−H]−), 317 (MS/MS) ([M − deoxyhexose − H]−) (negative-ion mode). 1H NMR, see Table . 13C NMR [acetone-d6–D2O (13:2, v/v), 151 MHz] δC 158.4 (C-2), 135.3 (C-3), 179.3 (C-4), 153.5 (C-5), 99.1 (C-6), 154.1 (C-7), 125.7 (C-8), 145.6 (C-9), 104.9 (C-10), 122.4 (C-1′), 116.8 (C-2′), 145.5 (C-3′), 149.0 (C-4′), 115.9 (C-5′), 122.5 (C-6′), 102.7 (C-1″), 71.3 (C-2″), 72.4 (C-3″), 71.5 (C-4″), 71.1 (C-5″), 17.5 (C-6″).
Gossypetin-3-O-β-xylopyranoside (3)
An amorphous yellow powder, [α]D −139.0° (c 1, MeOH). ECD (c 0.01, MeOH) [θ] (nm) −7.8 × 103 (207), +1.1 × 104 (221), +9.1 × 103 (239). HR-ESI-MS Found, m/z 449.0723 ([M−H]−); Calcd for C20H18O12−H, 449.0715. ESI-MS m/z 451 ([M + H]+), 319 (MS/MS) ([M − pentose + H]+) (positive-ion mode); m/z 449 ([M−H]-), 317 (MS/MS) ([M − pentose − H]−) (negative-ion mode). 1H NMR, see Table . 13C NMR [acetone-d6–D2O (13:2, v/v), 151 MHz] δC 157.9 (C-2), 134.5 (C-3), 179.0 (C-4), 153.7 (C-5), 99.1 (C-6), 153.9 (C-7), 125.7 (C-8), 145.5 (C-9), 104.7 (C-10), 122.5 (C-1′), 117.1 (C-2′), 145.3 (C-3′), 149.2 (C-4′), 115.7 (C-5′), 122.8 (C-6′), 103.7 (C-1″), 74.3 (C-2″), 76.5 (C-3″), 70.0 (C-4″), 66.4 (C-5″).
Hydrolysis of compounds 1–3
Compound 1 (100 μg) in 1 M HCl (200 μL) was heated for 4 h at 100 °C in water bath. The solution was then concentrated to dryness, and analyzed by RP-HPLC, to show the presence of gossypetin in the reaction mixture based on the comparison of the authentic one. Compounds 2 and 3 were treated analogously, to show the formation of gossypetin.
Proanthocyanidin PAOF-1 (22)
13C NMR [acetone-d6–D2O (13:2, v/v), 151 MHz] δC 169.3 (galloyl), 156.0, 154.3 (C-5, 7, 8a), 145.8, 145.1 (C-3′, 4′), 131.9 (C-1′), 118.7 (C-6′), 115.6 (C-5′), 114.5 (C-2′), 110.1 (C-6, 8), 107.1 (C-4a), 101.6 (C-4a), 97.1 (C-6, 8), 76.4 (C-2), 72.1 (C-3 extension), 67.8 (C-3 terminal), 36.9 (C-4). ECD (c 0.01, MeOH) [θ] (nm) −5.3 × 105 (206), +1.1 × 106 (238), −3.5 × 104 (277).
Constituent analysis of PAOF-1 (22)
The constituent units of PAOF-1 (22) were identified by phloroglucinolysis as follows [Citation18]: PAOF-1 (30.1 mg) was dissolved in 1% HCl-EtOH containing 0.5% (w/v) phloroglucinol, and left to stand for 3 days at room temperature, and the reaction mixture (38.6 mg) was subjected to column chromatography on MCI gel CHP-20P (1.1 cm i.d. × 20 cm), to give (−)- epicatechin-(4β-2)-phloroglucinol (22a) (2.1 mg) (from the eluate with 25% + 30% MeOH) and (−)-3-O-galloylepicatechin-(4β-2)-phloroglucinol (22b) (2.8 mg) (from the eluate with 40% MeOH eluate. The presence of catechin from the terminal unit in the reaction mixture was also observed in the NP- and RP-HPLC analyses.
Molecular weight distribution analysis of proanthocyanidins in PAOF-1 (22) [Citation19]
The molecular weight distribution was estimated by size exclusion chromatography (SEC) on a TSK-gel SuperAW3000 column (6.0 mm i.d. × 150 mm) (TOSOH, Tokyo, Japan) using N,N-dimethylformamide (DMF) containing 3 M aq. ammonium formate [0.5% (v/v)] as the eluent at 40 °C in a column oven. The flow rate was set at 0.5 mL/min. The calibration line for the molecular weight distribution was prepared using (−)-epicatechin (monomer), procyanidin B2 (dimer), procyanidin C1 (trimer) and cinnamtannin A2 (tetramer), isolated from bark of Cinnamomum cassia following a method applied to Cynomorium songaricum [Citation20].
The HPLC-DAD (diode array detector) analyses of the extracts and quantitation of the major constituents
Extracts for the quantitative HPLC analyses (other than the MeOH extracts of fresh leaves and flower buds) were prepared as follows. Fresh fruits (127.7 g) were homogenized in MeOH (500 mL), and the concentrated filtrate (100 mL) was extracted with n-hexane (100 mL × 3) to remove the lipophilic material. The MeOH-soluble portion was evaporated to yield the extract (13.8 g) used for the HPLC analysis. Fresh branches (570.0 g) were homogenized in MeOH (300 mL), and the concentrated filtrate (120 mL) was treated with n-hexane (120 mL × 3) to remove the lipophilic material. The remaining MeOH solution was evaporated to yield the extract (7.5 g) for the HPLC analysis.
Each of the extracts (5 mg) thus obtained was dissolved in MeOH (100 μL), and 3 μL each of the solutions was injected into a YMC Pack ODS Pro C8 (4.6 mm i.d. × 150 mm) column (YMC, Kyoto, Japan) with the following gradient profile of solvents A [H2O–CH3CN–AcOH (95:5:1, v/v/v)] and B [H2O–CH3CN–AcOH (60:40:1, v/v/v)] using a Hitachi L-2130 gradient system (Hitachi, Tokyo, Japan): 0–40 min, 0% → 30%, linearly; 40–60 min, 30% → 100% (%B in A + B) for the simultaneous detection of the phenolics. The column temperature was set to 40 °C in a column oven. Flow rate was set to 1.0 mL/min. Detection was accomplished using UV absorption in the 200–400 nm range on the L-2155 photodiode-array instrument (Hitachi), and monitored at 280 and 320 nm for flavonoids and ellagic acids, respectively.
The following isocratic conditions were used for quantitation purposes. Each solution (2 μL) of the extracts (5 mg) in MeOH (100 μL) was injected into a YMC Pack ODS Pro C8 (4.6 mm i.d. × 150 mm) column (YMC, Kyoto, Japan) with a mixture of H2O and CH3CN (85:15, by volume) containing 1% acetic acid for flavonoid glycosides and ellagic acids and a mixture of H2O and CH3CN (7:3, by volume) containing 1% acetic acid for flavone (11) as the eluents. Solutions of the authentic samples with concentrations of 100, 50, and 25 μg/100 μL were prepared and injected onto the column, and the quantitation of the constituents was based on their peak areas, and on triplicate experiments.
Tyrosinase inhibitory assay
A tyrosinase inhibitory assay was performed based on a method reported previously [Citation21] with slight modification using 96-well microplates. A mixture of phosphate buffer (pH 6.8) (160 μL) and MeOH (20 μL) containing the followings additives were prepared: mushroom tyrosinase (62.5 mU) (A), without any additives (B), mushroom tyrosinase and the tested compound (C), and the tested compound without the enzyme (D). The mushroom tyrosinase was added as the buffered solution and the tested compounds were added as the MeOH solution. After pre-incubation of these solutions for 10 min at room temperature, L-tyrosine in 20 μM phosphate buffer (1 μM, 20 μL) was added to each solution and the mixtures were incubated for 20 min at room temperature. Based on the absorbance at 475 nm before (λbefore) and after (λafter) the incubation for each solution, the inhibition (%) for the tested compound was calculated using the equation
where A, B, C, and D are the differences between λafter and λbefore for the respective solutions mentioned above. The inhibition % was expressed as the average from triplicate experiments. Kojic acid and quercetin were used as the positive controls.
Antitumor assay
The antitumor assay was performed according to the method reported previously [Citation22], using human oral squamous cell carcinoma (OSCC) cell lines (gingival tissue-derived Ca9-22, and tongue tissue-derived HSC-2, HSC-3 and HSC-4) and human normal oral cells [gingival fibroblast (HGF), pulp cell (HPC), periodontal ligament fibroblast (HPLF)]. The tumor selectivity index (TS) was calculated by dividing the mean CC50 for four OSCC cell lines to mean CC50 for three normal oral cells [(D/B) in Table ]. Since both Ca9-22 and HGF cells were derived from the gingival tissue, the relative sensitivity of these cells was also compared [(C/A) in Table ].
Results and discussion
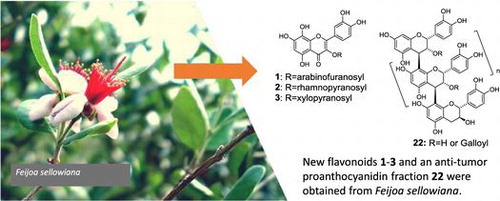
Twenty-one phenolic compounds (1–21), including three new ones (1, 2, and 3; Figure ), and a proanthocyanidin oligomer fraction named PAOF-1 (22), were obtained from the fruits, flower buds, and leaves of an F. sellowiana tree. Among them, seven compounds (1, 13, 16–19) and PAOF-1 (22) were from the fruits, three (1, 20, and 21) were from the flower buds, and 1–15 were from the leaves. The known compounds among those isolated were identified based on the comparisons of the NMR and MS data with those in the literature (4 [Citation23], 7 [Citation24], 8 [Citation25], 9 [Citation25], 12 [Citation26], 13 [Citation27], 17 [Citation28], 18 [Citation29], 19 [Citation30]), and direct comparisons with the authentic samples on HPLC (5, 6, 10, 11, 14–16, 20, 21, 23). Although compounds 5, 9, 10, 13, 14, 16 and 20, 21, 23 were reported as the constituents of this plant species, the isolation of compounds 4, 6–8 12, 15, 17–19, 22 was not reported previously.
Figure 1. Phenolic constituents of Feijoa sellowiana. Arrows in structure 1 indicate key HMBC correlations.
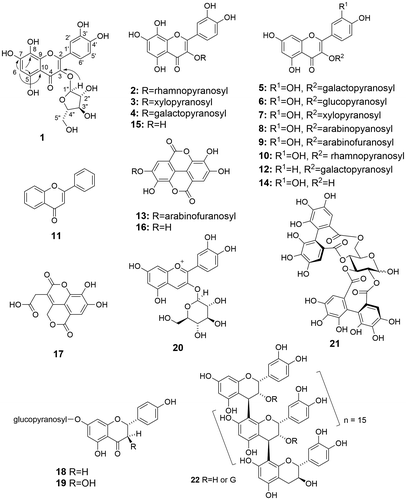
Structures of new flavonoid glycosides
Compound 1 was obtained as an amorphous yellow powder. The molecular formula of this compound was determined to be C20H18O12 based on the [M + H]+ ion peak from positive-ion mode of the HR-ESI-MS. The low-resolution ESI-MS also showed the ions at m/z 451 in the positive-ion mode and m/z 449 in the negative-ion mode, corresponding to the [M + H]+ and the [M − H]− ions, respectively, and the MS/MS of these ions showed the ions at m/z 319 [M − pentose + H]+ (positive-ion mode) and m/z 317 [M − pentose–H]− (negative-ion mode), corresponding to the aglycone of mass 318. The 1H NMR spectrum (Table ) in acetone-d6 containing D2O showed a singlet at δH 6.33, and signals of three protons forming an ABX system at δH 7.70, 7.59, and 6.95. The former singlet was assigned to an A-ring proton of its flavonol skeleton, and the latter ABX signals were attributed to the B-ring protons of the skeleton. The spectrum also showed proton signals corresponding to the pentose moiety. The chemical shifts and the coupling constants of the 1H-NMR signals of the pentose are similar to those of avicularin (9) (= quercetin-3-O-α-l-arabinofuranoside) (see Table ), suggesting the presence of arabinose as the pentose in 1. Combined with the molecular formula of 1 shown above, the structure where an A-ring proton in 9 is replaced with a hydroxyl group was assigned to compound 1. The 13C NMR spectrum of 1 substantiated this structural assignment as shown in the “Materials and methods” section). An A-ring carbon signal in the spectrum of 1 shifted downfield noticeably (δC 125.7, C-8), relative to the corresponding carbon signal (δC 94.6) of 9, and three A-ring carbon signals shifted upfield [δC 153.9 (C-5), 153.9 (C-7), 145.6 (C-9)], relative to the corresponding carbon signals of 9 [δC 162.2 (C-5), 165.4 (C-7), 158.3 (C-9)]. These shifts in the A-ring carbons indicated that the flavonol aglycone in 1 is gossypetin. The assignments of the A-ring carbon signals were further substantiated by the correlations of H-6 to C-5, 7, 8, and 10 in the heteronuclear multiple-bond correlation (HMBC) spectrum of 1 (Figure S4 in the Supplementary data) in methanol-d4. The HMBC spectrum also showed a correlation of H-1″ to C-3 (Figure S3 in the Supplementary data) in acetone-d6–D2O, substantiating the location O-3 of the arabinofuranose residue on the flavonol structure in 1. The electronic circular dichroism (ECD) spectrum of compound 1 showed negative and positive maxima at 304 and 258 nm ([θ]304 −1.1 × 104, [θ]258 + 0.5 × 104), forming a pattern similar to that of 9 ([θ]295 − 0.8 × 104, [θ]254 + 1.1 × 104). Because the l-configuration of the arabinofuranose moiety in 9 found in the present study was shown by its specific rotation ([α]D −152°; lit. [Citation25], [α]D −152°), the arabinofuranose residue in compound 1 was determined to have the l-configuration. The structure of compound 1 was thus considered to be gossypetin- 3-O-α-l-arabinofuranoside (Figure ).
Compound 2 was obtained as an amorphous yellow powder. The molecular formula C21H20O12, was indicated by the [M − H]− ion peak in the HR-ESI-MS. The low-resolution ESI-MS also showed the [M + H]+ ion peak at m/z 465 and the [M − deoxyhexose + H]+ ion peak at m/z 319 (MS/MS of the ion) (positive-ion mode), and the [M − H]− ion peak at m/z 463 and the [M–deoxyhexose–H]− ion peak at m/z 317 (MS/MS of the ion) (negative-ion mode). Thus, the aglycone with mass 318 was consistent with the gossypetin structure. The 1H NMR spectrum of compound 2 also showed the presence of gossypetin as the aglycone moiety (Table ). The sugar proton signals corresponding to the deoxyhexose moiety in 2 forms a pattern similar to that of the rhamnose moiety of quercitrin (= quercetin-3-O-α-rhamnopyranoside) (10). The 13C NMR spectrum also showed the signals attributed to structure 2 as shown in the “Materials and methods” section. The structure of gossypetin-3-O-α-rhamnopyranoside was thus assigned to compound 2.
Compound 3 was also obtained as an amorphous yellow powder. Its molecular formula C20H18O12 was indicated by the [M − H]− ion peak in the HR-ESI-MS. The low-resolution ESI-MS showed the [M + H]+ ion peak at m/z 451 and the ion peak at [M − pentose + H]+ ion peak at m/z 319 (MS/MS) (positive-ion mode), and the [M − H]− ion peaks at m/z 449 and the [M − pentose − H]− ion peak at m/z 317 (MS/MS) (negative-ion mode). The ions for the mass-318 aglycone are also consistent with the gossypetin structure. The 1H NMR spectrum (Table ) showed the signals of the aglycone gossypetin, too. The sugar proton signals showed a pattern similar to the corresponding one of reynoutrin (= quercetin-3-O-β-xylopyranoside) (7), indicating that the pentose in 3 is β-xylopyranose. The 13C NMR spectrum also showed the signals assignable to structure 3 (See the “Materials and methods” section). The structure of gossypetin-3-O-β-xylopyranoside was thus assigned to compound 3.
The aglycone structure in 1–3 was further confirmed by acid hydrolysis of these compounds, to give gossypetin.
Characterization of PAOF-1
PAOF-1 (22) was obtained from the water-soluble residue of the feijoa fruit extract as a brown amorphous powder. The 13C NMR spectrum indicated the signals assignable to the galloyl moieties in addition to those of the procyanidin skeleton (see the Materials and Method section), and the galloylated procyanidin structure was further substantiated by acid degradation in the presence of phloroglucinol, to show that it is composed of (−)-epicatechin and (−)-3-O-galloylepicatechin as the extension units and (+)-catechin as the terminal unit. These constituent units were identified based on their comparisons on RP-/NP-HPLC with the authentic samples. The relative molar ratio of (−)-epicatechin-(4β-2)-phloroglucinol and (−)-3-O-galloylepicatechin-(4β-2)-phloroglucinol in the reaction mixture was estimated to be 5:7. The 2R stereochemistry [Citation31] of the respective flavan units were indicated by the negative Cotton effect at 273 nm in the ECD spectrum. The number-average (Mn) and weight-average (Mw) molecular weights of the procyanidin were estimated to be 6400 and 7600, respectively, based on the SEC analysis [Citation19]. The polymerization degree (PD) 17 was also calculated based on the Mn data. Although these values are based on the extrapolation of the standard procyanidins and epicatechin, this value is not so far from that calculated from the phloroglucinol treatment (PD, 14).
Comparison of composition profiles of the extracts from various parts of F. sellowiana
Because differences were seen among the phenolic constituents isolated from the parts obtained from the single tree, we compared the composition profiles of the extracts from the fruits, flower buds, leaves, and branches using an HPLC system equipped with a photodiode-array detector. An HPLC condition with a gradient elution system using (A) H2O–CH3CN (95:5) containing 1% acetic acid and (B) H2O–CH3CN (60:40) containing 1% acetic acid was established for simultaneous analyses of the phenolics isolated from three of the four parts. The chromatographic profiles of the extracts from the four parts, monitored at 280 and 320 nm, are shown in Figure ; this wavelength combination was adjusted to detect the phenolics, including flavonoids and ellagic acids, in the extracts.
Figure 2. RP-HPLC profiles of the crude methanol extracts from (a) fruits, (b) buds, (c) leaves, and (d) branches of Feijoa sellowiana. Red and blue lines indicate profiles monitored at 280 and 320 nm, respectively.
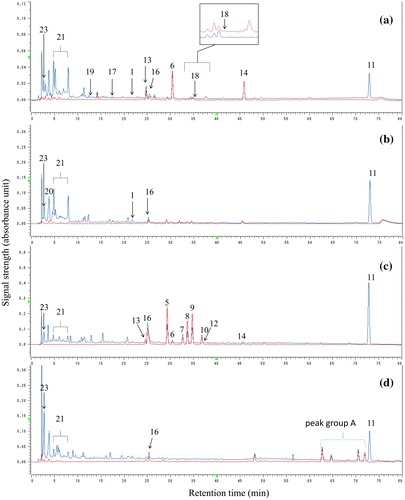
The fruit extract exhibited the HPLC profile of Figure (a). A pair of characteristic peaks of pedunculagin (21) forming an anomer mixture was observed there, and the remaining four prominent peaks were assigned to those of isoquercitrin (6), 4-O-α-arabinofuranosylellagic acid (13), quercetin (14), and flavone (11). Compound 11 was reported as one of the characteristic constituents with anticancer activity in a feijoa fruit extract [Citation32]. Furthermore, the peaks corresponding to those of gallic acid (23) and pedunculagin (21) were also assigned based on their DAD patterns in addition to their retention times (data not shown). Although some other phenolic constituents (16–19) were also isolated from the fruit extract in the present study, their HPLC peaks were much smaller.
The flower bud extract showed the profile of Figure (b). The peaks of pedunculagin (21) and gallic acid (23) were also seen there, as described above. The HPLC peak of an anthocyanin constituent, cyanidin glucoside (20), which is ascribed to the red color of feijoa flowers, also appeared. There were also HPLC peaks due to flavone (11), ellagic acid (16), and the new flavonoid, gossypetin arabinofuranoside (1).
The leaf extract showed the HPLC peaks of the phenolic compounds 5–14 isolated from the leaf extract, as shown by Figure (c). Although the peaks due to the quercetin glycosides (5–10) were distinctive, the HPLC peak of their aglycone, quercetin (14), was much smaller than those of the corresponding glycosides. Flavone (11) was one of the major constituents in the leaf extract. Peaks due to ellagic acid (16), pedunculagin (21), and gallic acid (23) were also observed.
The branch extract showed remarkable HPLC peaks in the region of retention time 60–72 min, as shown by peak group A in Figure (d). The UV (DAD) spectra of the HPLC peaks of group A (except for the peak at retention time 70.5 min) had characteristic absorption maxima in the UV region 350–365 nm (Figure S15 in the Supplementary data), which are similar to that of ellagic acid (16), shown in Figure (d). The similarity of the DAD spectra suggested the presence of alkylated derivatives of ellagic acids [Citation27] such as O-methyl ethers. Although the presence of the alkylated derivatives was reported for some myrtaceous plants [Citation3], further confirmation of the presence of those types of compounds in this source plant is still awaited.
The amounts of the flavonoids (5–10 and 11) and ellagic acids (13 and 16) in the extracts were quantitated and the results are summarized in Table . Isoquercitrin (6) was the most abundant flavonoid in the fruit extract, while hyperoside (5) was the most abundant flavonoid in the leaf extract (Table ). It should be noted that considerable contents of flavone (11) were seen in all the extracts from the tree. The content of 16 in the fruit extract, in addition to that of arabinofuranosylellagic acid (13), was also noteworthy. Ellagic acid (16) was one of the most abundant phenolics in the extracts from the leaves, flower buds, and branches of the tree (Table ).
Table 2. Contents of flavonoids and ellagic acids in the leaves, flower buds, branches, and fruits of Feijoa sellowianaa.
Ellagic acid was found in edible fruits of some plants such as strawberry, and the plants containing ellagic acid or its derivatives have been expected to be beneficial for health [Citation33], since various pharmacological effects, such as anticancer, anti-inflammatory, anti-mutagenic, antioxidant, and antimicrobial effects, have been reported for ellagic acid. The presence of ellagic acid also can be correlated with the presence of ellagitannins because ellagic acid is known as the product formed upon hydrolysis of ellagitannins. Ellagitannins have been found in plant species of the family Myrtaceae, and an ellagitannin pedunculagin (21) was obtained from flower buds in the present study. Additional studies to identify hydrolysable tannins of this plant species are in progress.
The accumulation of these antioxidant flavonoids and phenolics in plants can be explained by the roles in the plant survival in tropical and subtropical regions [Citation34]. The present study indicated that all four parts of feijoa trees, including leaves, can be used as resources for antioxidant phenolics.
Tyrosinase inhibitory assay
Tyrosinase inhibitors are expected to have suppressing effects on diet browning via affecting production of melanin or melanin-like substances [Citation35], and the inhibitory effects of various flavonoids on this enzyme including the structure-activity relationship have been revealed [Citation35]. Therefore, we examined the effects of flavonoids found in F. sellowiana and related phenolics on the tyrosinase activity. The results are summarized in Table . The flavonoid glycosides examined did not affect the tyrosinase activity at the concentration used, as observed in previous reports [Citation36–38]. However, gossypetin (15), the aglycone of the new compounds, 1–3, had an inhibitory effect, along with the other unglycosylated flavonoids examined in the present study. The inhibitory effect of gossypetin on tyrosinase has not yet been reported, and the strength of the inhibition was almost the same as that observed for one of the positive controls, kojic acid, and somewhat weaker than that of quercetin (14) (Table ). A hydrolysable tannin pedunculagin (21), and the proanthocyanidin (condensed tannin) PAOF-1 (22) did not show the inhibitory activity (< 10% inhibition). The inhibitory effects seen in the present study could be attributable to their antioxidant effects, including those via trapping copper ions in the enzyme. Because the tyrosinase used in this study was from mushrooms, the effects may be different from the findings using the human enzyme; further experiments using that type are expected.
Table 3. Tyrosinase inhibitory activity of flavonoids.
On the other hand, ellagic acid (16) and myricetin (26) showed unanticipated inhibitory values (−47.0 ± 7.5%, and 139.8 ± 6.9% inhibition of tyrosinase, respectively). These results suggest that ellagic acid acts as a substrate or cofactor of tyrosinase, and myricetin binds to the products of the enzyme reaction. Previous papers suggested that polyphenols might work as substrates and/or cofactors of tyrosinase, or as an enzyme binding agent [Citation38]. Although these explanations might rationalize the unusual effects of these two compounds on tyrosinase activity, further studies are needed to clarify them.
Antitumor assay
Although various antitumor drugs have been developed, most of them had potent cytotoxicity on the normal cells [Citation31]. On the other hand, various tannins with large molecular weights, especially oligomeric hydrolysable tannins, showed remarkable tumor selectivity (TS) in the cytotoxic experiments [Citation39]. Therefore, we examined cytotoxicity of the galloylated proanthocyanidin oligomer PAOF-1 (22) using OSCC lines (Ca9–22, HSC-2, HSC-3, HSC-4) and normal oral cells (HGF, HPC, HPLF) in the present study. As a result, CC50 of PAOF-1 against human oral squamous cell carcinoma cell lines were 151 μM, while CC50 for normal human oral cells showed over 477 μM (Table ). The TS index was over 3.2 based on these values [(D)/(B) in Table ]. When tumor selectivity was calculated using cells both derived from gingival tissue (Ca9-22 vs. HGF), PAOF-1 had slightly higher TS (>4.6), comparable with that of 5-FU (TS > 5.1) and doxorubicin (>5), except for docetaxel (TS > 128) [(C)/(A) in Table ].
Table 4. Antitumor activity of PAOF-1 and antitumor agents.
Since the cytotoxic property of an acetonic extract from feijoa fruit peels was reported previously [Citation40], this result suggested that one of the active constituents may be regarded to be the oligomeric proanthocyanidin in this plant.
Conclusions
In the present study, we isolated new gossypetin glycosides (1–3), along with eighteen known compounds, including another gossypetin glycoside 4 and its aglycone gossypetin (15), quercetin (14) and its glycosides (5–10), a kaempferol glycoside (12), a naringenin glycoside (18), an aromadendrin glycoside (19), a cyanidin glycoside (20), flavone (11), ellagic acid (16) and its derivatives (13 and 17), pedunculagin (21) and proanthocyanidin oligomer PAOF-1 (22) as the phenolic constituents of the leaves, fruits, and flower buds of F. sellowiana. This is the first report of the isolation of gossypetin-3-O-β-galactopyranoside (4), isoquercitrin (6), reynoutrin (7), guaijaverin (8) trifolin (12), phyllanthusiin E (17), naringenin-7-O-β-glucoside (18), and aromdendrin-7-O-β-glucoside (19) from feijoa. The presence of the alkylated ellagic acid derivatives was also suggested in the branch extract.
The contents of nine of the phenolic compounds in the extracts from various parts were quantitated (Table ), to reveal that feijoa can be regarded as a resource for those polyphenolic compounds. Unglycosylated flavonoids examined in this study showed inhibitory effects on tyrosinase, comparable to that of kojic acid (Table ). The proanthocyanidin oligomer fraction (PAOF-1) (22) derived from F. sellowiana fruits showed selective cytotoxity against OSCC lines, comparable to that of 5-FU (Table ). Previous studies on feijoa constituents generally focused on its edible fruits; however, the findings in the present study suggested not only fruits, but also the other parts, especially leaves, of this plant species might be worth studying to find bioactive polyphenolics or flavonoids.
Author contributions
H. A. performed isolation, identification, structure elucidation, tyrosinase inhibitory assay of the constituents, and prepared the manuscript; H. S. contributed to antitumor assay and improved the manuscript; T. H. designed the study and reviewed manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by a Grant-in-Aid from JSPS [grant number 25450170].
Supplemental data
Supplementary material for this paper is available online at https://doi.org/10.1080/09168451.2017.1412246.
FS-SI-All-11252017.pdf
Download PDF (1.5 MB)Acknowledgements
The Varian INOVA 600AS NMR and Varian INOVA 400-MR NMR instruments used in this study are the property of the SC-NMR laboratory of Okayama University.
Related Research Data
References
- Manach C, Williamson G, Morand C, et al. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81:230S–242S.
- Gharras HE. Polyphenols: food sources, properties and applications – a review. Int J Food Sci Technol. 2009;44:2512–2518.10.1111/j.1365-2621.2009.02077.x
- Lowry JB. The distribution and potential taxonomic value of alkylated ellagic acid. Phytochemistry. 1968;7:1803–1813.10.1016/S0031-9422(00)86652-2
- Reynertson KA, Yang H, Jiang B, et al. Quantitative analysis of antiradical phenolic constituents from fourteen edible Myrtaceae fruits. Food Chem. 2008;109:883–890.10.1016/j.foodchem.2008.01.021
- Gutiérrez RMP, Mitchell S, Solis RV. Psidium guajava: a review of its traditional uses, phytochemistry and pharmacology. J Ethnopharmacol. 2008;117:1–27.10.1016/j.jep.2008.01.025
- Cai L, Wu CD. Compounds from Syzygium aromaticum possessing growth inhibitory activity against oral pathogens. J Nat Prod. 1996;59:987–990.10.1021/np960451q
- Nassa MI. Flavonoid triglycosides from the seeds of Syzygium aromaticum. Carbohydr Res. 2006;341:160–163.10.1016/j.carres.2005.10.012
- Kikuzaki H, Miyajima Y, Nakatani N. Phenolic glycosides from berries of pimenta dioica. J Nat Prod. 2008;71:861–865.10.1021/np0705615
- Weston RJ. Bioactive products from fruit of the feijoa (Feijoa sellowiana, Myrtaceae): a review. Food Chem. 2010;121:923–926.10.1016/j.foodchem.2010.01.047
- Okuda T, Yoshida T, Hatano T, et al. Ellagitannins of the casuarinaceae, Stachyuraceae and Myrtaceae. Phytochemistry. 1982;21:2871–2874.
- Lowry JB. Anthocyanins of the melastomataceae, Myrtaceae and some allied families. Phytochemistry. 1976;15:513–516.
- Hardy PJ, Michael BJ.Volatile components of feijoa fruits. Phytochemisty. 1970;9:1355–1357.
- Shaw GJ, Allen JM, Yates MK. Volatile flavour constituents of the skin oil from Feijoa sellowiana. Phytochemistry. 1989;28:1529–1530.10.1016/S0031-9422(00)97781-1
- Ruberto G, Tringali C. Secondary metabolites from the leaves of Feijoa sellowiana berg. Phytochemistry. 2004;65:2947–2951.10.1016/j.phytochem.2004.06.038
- Vanidze MR, Shalashvili AG, Chkhikvishvili ID, et al. Catechins of feijoa leaves. Subtropicheskie Kul’tury. 1991;6:87–90.
- ElShenawy SM, Marzouk MS, Dib RAE, Elyazed HEA, et al. A polyphenols and biological activities of Feijoa sellowiana leaves and twigs. Rev Latinoamer Quím. 2008;36:104–120.
- Oldrich L, Borivoj K, Ladislav K, et al. Identification of isoflavones in Acca sellowiana and two Psidium spesies (Myrtaceae). Biochem Syst Ecol. 2005;33:983–992.
- Foo LY, Karchesy JJ. Procyanidin tetramers and pentamers from Douglas fir bark. Phytochemistry. 1991;30:667–670.
- Kusuda M, Inada K, Ogawa T, et al. Polyphenolic constituent structures of Zanthoxylum piperitum fruit and the antibacterial effects of its polymeric procyanidin on methicillin-resistant Staphylococcus aureus. Biosci Biotechnol Biochem. 2006;70:1423–1431.10.1271/bbb.50669
- Jin S, Eerdunbayaer, Doi A, et al. Polyphenolic constituents of Cynomorium songaricum Rupr. and antibacterial effect of polymeric proanthocyanidin on methicillin-resistant Staphylococcus aureus. J Agric Food Chem. 2012;60:7297–7305.10.1021/jf301621e
- Likhitwitayawuid K, Srutularak B. A new dimeric stilbene with tyrosinase inhibitiory activity from Artocarpus gomezianus. J Nat Prod. 2001;64:1457–1459.10.1021/np0101806
- Orabi MAA, Taniguchi S, Sakagami H, et al. Hydrolyzable tannins of tamaricaceous plants. v. structures of monomeric–trimeric tannins and cytotoxicity of macrocyclic-type tannins isolated from Tamarix nilotica. J Nat Prod. 2013;76:947–956.10.1021/np4001625
- Suzuki H, Sasaki R, Ogata Y, et al. Metabolic profiling of flavonoids in Lotus japonicus using liquid chromatography Fourier transform ion cyclotron resonance mass spectrometry. Phytochemistry. 2008;69:99–111.10.1016/j.phytochem.2007.06.017
- Olszawska M, Wolbiś M. Further flavonoids from the flowers of Prunus spinosa L. Drug Res. 2007;64:241–245.
- Andres S, Petra S, Jurgen C, et al. Elution order of quercetin glycosides from apple pomace extracts on a new HPLC stationary phase with hydrophilic endcapping. J Sep Sci. 2002;23:361–364.
- Wolbiś M, Nowak S, Kicel A. Polyphenolic compounds in Scopolia caucasica Kolesn. ex Kreyer (Solanaceae). Acta Pol Pharm. 2007;64:241–246.
- Zafrilla P, Ferreres F, Tomás-Barberán FA. Effect of processing and storage on the antioxidant ellagic acid derivatives and flavonoids of red raspberry (Rubus idaeus) jams. J Agric Food Chem. 2001;49:3651–3655.10.1021/jf010192x
- Yoshida T, Ito H, Matsunaga S, et al. Tannins and related polyphenols of euphorbiaceous plants. IX. Hydrolyzable tannins with 1C4 glucose core form Phyllanthus flexuosus Muell. Arg Chem Pharm Bull. 1992;40:53–60.10.1248/cpb.40.53
- Moco S, Tseng LH, Spraul M, et al. Building-up a comprehensive database of flavonoids based on nuclear magnetic resonance data. Chromatographia. 2006;64:503–508.10.1365/s10337-006-0077-6
- Slimestad R, Andersen OM, Francis GW. Ampelopsin 7-glucoside and other dihydroflavonol 7-glucosides from needles of Picea Abies. Phytochemistry. 1984;35:550–552.
- Korver O, Wilkins CK. Circular dichroism spectra of flavonols. Tetarhedron. 1971;27:5459–5465.
- Bontempo P, Mita L, Miceli M, et al. Feijoa sellowiana derived natural flavone exerts anti-cancer action displaying HDAC inhibitory activities. Int J Biochem Cell Biol. 2007;39:1902–1914.10.1016/j.biocel.2007.05.010
- Vattem DA, Shetty K. Biological functionality of ellagic acid: review. J Food Biochem. 2005;29:234–266.10.1111/jfbc.2005.29.issue-3
- Casati P, Walbot V. Gene expression profiling in response to ultraviolet radiation in maize genotypes with varying flavonoid content. Plant Physiol. 2003;132:1739–1754.10.1104/pp.103.022871
- Chang T. An updated review of tyrosinase inhibitor. Int J Mol Sci. 2009;10:2440–2475.10.3390/ijms10062440
- Nugroho A, Choi J, Park J, et al. two new flavonol glycosides from Lamium amplexicaule L. and their in vitro free radical scavenging and tyrosinase inhibitory activities. Planta Med. 2009;75:364–366.10.1055/s-0028-1112216
- Kubo I, Kinst-Hori I. Flavonols from saffron flower: tyrosinase inhibitory activity and inhibition mechanism. J Agric Food Chem. 1999;47:4121–4125.10.1021/jf990201q
- Kim Y, Uyama H. Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cell Mol Life Sci. 2005;62:1707–1723.10.1007/s00018-005-5054-y
- Sakagami H. Biological activities and possible dental application of three major groups of polyphenols. J Pharmacol Sci. 2014;126:92–106.10.1254/jphs.14R04CR
- Motohashi N, Kawase M, Shirataki Y, et al. Biological activity of feijoa peel extracts. Anticancer Res. 2000;20:4323–4329.