
ABSTRACT
Despite the marked progress of cancer research, cancer is the predominant cause of death in Japan, and therefore development of effective therapeutic drugs is expected.
Chemical biology is a research field utilizing small molecules to investigate biological phenomena. One of the most important aims of chemical biology is to find the small molecules, and natural products are ideal screening sources due to their structural diversity. Therefore, natural product screening based on the progress of chemical biology prompted us to find small molecules targeting cancer characteristics. Another contribution of chemical biology is to facilitate the target identification of small molecule. Therefore, among a variety of methods to uncover protein function, chemical biology is a remarkable approach in which small molecules are used as probes to elucidate protein functions related to cancer development.
Abbreviations: EGF: Epidermal growth factor; PDGF: Platelet-derived growth factor; CRPC: Castration-resistant prostate cancer; AR: Androgen receptor; FTase: Farnesyl transferase; 5-LOX: 5-Lipoxygenase; LT: Leukotriene; CysLT1: Cysteinyl leukotriene receptor 1; GPA: Glucopiericidin A; PA: Piericidin A; XN: Xanthohumol; VCP: Valosin-containing protein; ACACA: Acetyl-CoA carboxylase-α.
Graphical Abstract
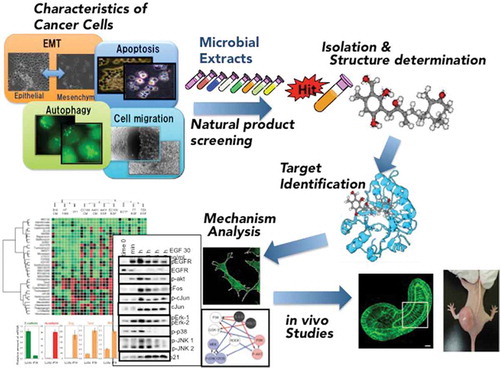
Strategy for the chemical biology studies on the small molecules targeting characteristics of cancer cells.
In the later half of 20th century, oncogenes were sequentially identified and it was shown that cancer malignant due to abnormal regulation of cellular responses by oncogenes and related molecules. After that, research on cellular response mechanisms related to the malignant transformation of oncogenes and related cancers, that is, “characteristics of cancer cells” began to develop.
On the other hand, “agricultural chemistry” traditionally studies on the isolation and structural analysis of bioactive small molecules, and also it conductes the mechanism studies on the biological response at the molecular level through analysing the mode of action of bioactive substances.
Under such background, we have challenged “The characteristics of cancer cells” research by agricultural chemistry method and have been searching for compounds targeted to it from mainly microbial culture fluid and have been analyzing and studying the cell response mechanism of cancer malignancy.
Natural product screening targeting characteristics of cancer cells
Screening for the compound targeting oncogenes and its related molecules
In 1979, the oncogene product p60src was discovered to have tyrosine kinase as a new type of protein kinase[Citation1]. Following this discovery, growth factor such as EGF (Epidermal growth factor) and PDGF (Platelet-derived growth factor) was observed to induce a rapid increase in tyrosine kinase activity of its receptor[Citation2–Citation4]. These findings suggested an important role of tyrosine phosphorylation in growth factor-mediated proliferation signals and in the src family oncogene-mediated signaling pathway for oncogenesis. These discoveries prompted me to search for an inhibitor of tyrosine kinase from the secondary metabolites of microorganisms. For this, we used the membrane fractions of EGF receptors overexpressing the human epidermoid carcinoma cell line A431 as a source of tyrosine kinase. As a result, we isolated a novel natural product named erbstatin as an inhibitor of EGF receptor tyrosine kinase from the cultured broth of Streptomyces sp. MH435-hF3[Citation5]. Erbstatin has a structure resembling tyrosine and shows a pattern of competitive inhibition with the protein to be phosphorylated[Citation6]. It is a broad spectrum tyrosine kinase inhibitor that also inhibits other tyrosine kinases, such as Her-2, Fyn and Lck. Although erbstatin is unstable in the presence of serum, it shows antitumor effects on leukemia cell line L1210 and breast cancer cell line MCF-7-bearing cancer mice by administering it in combination with a stabilizer[Citation7].
Today, many tyrosine kinase inhibitors, such as gefitinib and imatinib, are clinically used to treat patients with several types of cancer. Thus, the discovery of erbstatin has greatly contributed to the subsequent study of the development of molecularly targeted therapeutic drugs.
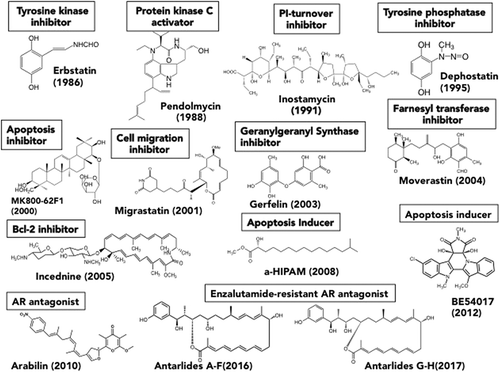
Furthermore, we searched for the compounds targeted to “characteristics of cancer cells” such as inositol phospholipid turnover[Citation8], tyrosine phosphatase[Citation9], geranylgeranyl group synthetase[Citation10,Citation11], etc., and found more than 10 novel compounds ().
Figure 1. Structures of the novel compounds isolated from the natural product screening targeting characteristics of cancer cells.
Natural product screening targeting androgen receptor
Prostate cancer is one of the most commonly diagnosed cancers among men worldwide[Citation12]. In recent years, the number of prostate cancer patients has been increasing rapidly in Japan. In fact, the morbidity of prostate cancer is increasing, and predicted to become the first among men in 2020 in Japan. Androgen receptor (AR) signaling plays a central role in the malignancy of prostate cancers[Citation13], and androgen deprivation by medical or surgical castration is the standard first line treatment for men with advanced prostate cancer. Over time, most men will progress to a more aggressive form of the disease called castration-resistant prostate cancer (CRPC). CRPC is treated with Androgen Receptor (AR) antagonists, but most patients experience disease progression after long-term treatment with these antagonists. A second generation AR antagonist, Enzalutamide (MDV3100), has evolved from the need for more effective and long-term AR inhibition and has been recently approved by the U.S. Food and Drug Administration[Citation14]. However, despite the initial response to enzalutamide, resistance also develops in most patients with metastatic CRPC. The recent studies have revealed that a F876L mutation in the AR confers resistance to enzalutamide[Citation15]. Therefore, new AR antagonists are required to ensure follow-up of these patients.
In the course of screening for a new type of AR antagonist, we isolated novel compounds, antarlides A-E (1–5), from the fermentation broth of Streptomyces sp. BB47[Citation16]. Antarlide A-E are mutually isomeric with regard to the double bond, having a novel macrocyclic structure of a 22-membered ring. The full stereostructure of antarlide A was established by chemical modifications including methanolysis, the Trost method, acetonide formation and the PGME method. In addition, antarlide B inhibited the transcriptional activity not only of wild-type AR but also of mutant ARs that are seen in patients with acquired resistance to clinically used enzalutamide. Interestingly, we found that this strain also produces antarlides G and H as new members of the antarlide family that have a 20-membered-ring macrocyclic structure[Citation17]. Antarlides G and H inhibited the binding of androgen to AR in vitro at concentrations similar to those observed with antarlides A-E. In addition, antarlide G inhibited the transcriptional activity of not only wild-type AR but also enzalutamide-resistant AR, suggesting that antarlides with either 22-membered- or 20 membered-rings may serve as potent third-generation AR antagonists capable of overcoming resistance to enzalutamide.
Studies of molecular mechanism of cell migration by using bioactive compounds
Cell migration inhibitors and their mode of action
Cell migration is found in embryonic morphogenesis, tissue regeneration, wound healing, leukocyte immune surveillance, which is an indispensable phenomenon for living bodies. In addition, it is closely involved in various pathological phenomena such as infiltration of cancer cells in cancer metastasis. Extensive studies have attempted to elucidate the molecular mechanisms behind cell migration. But, they still remain unclear. Bioactive compounds that regulate cell migration are promising not only as a powerful tool for studying the mechanism of cell migration but also as drug seeds for chemotherapy against tumor metastasis. Therefore, we have screened cell migration inhibitors and analyzed their mechanisms for the inhibition of cell migration.
Moverastins: Moverastin, which belongs to cylindrol family, was isolated from the culture broth of Aspergillus sp. F7720[Citation18]. However, analysis of NMR spectroscopic data raised the possibility that moverastin is a mixture of two diastereoisomers (moverastin A and B) at the C-10 secondary allylic alcohol. Prof. Watanabe at Tokyo University synthesized (10S)-moverastin and (10R)-moverastin to determine their absolute configurations. Moverastin A and moverastin B were thus identified to be (10R)-moverastin and (10S)-moverastin, respectively. Separation of the C-10 epimers of the synthetic moverastin and their bioassay revealed that both diasetereoisomers showed a same inhibitor activity of migration.
Furthermore, we demonstrated that moverastins A and B inhibited Farnesyl transferase (FTase), an enzyme that catalyzes protein prenylation, in vitro. Although farnesylation of H-Ras resulted in membrane localization and activation of H-Ras leading to activation of PI3K/Akt pathway in cultured cells, moverastins A and B inhibited membrane localization of H-Ras in EC17 cells, possibly due to the inhibition of H-Ras farnesylation. Moverastins inhibited activation of PI3K/Akt pathway, indicating that moverastins inhibited cell migration of tumor cells by inhibiting farnesylation of H-Ras, and subsequent H-Ras-dependent activation of PI3K/Akt pathway.
UTKO1: Because the inhibitory activity of moverastins for tumor cell migration was rather modest, we considered it an attractive lead compound in the search for other, more potent agents. UTKO1[Citation19] was synthesized by Prof. Watanabe at University of Tokyo as a derivative of moverastin. Although UTKO1 showed stronger inhibitory effect for cell migration than moverastin, surprisingly, it did not inhibit FTase, which is an inherent activity of moverastin.
Thus, we explored the UTKO1’s mode of action by using a chemical biology approach. When cells migrate, protrusions called lamellipodia are formed at the leading edge. In A431 cells, lamellipodia formation exhibited a biphasic response to EGF with two peaks of 5 min and 12 hrs. Although UTKO1 did not affect lamellipodia formation at 5 min, it inhibited lamellipodia formation at 12 hrs. These results suggest that UTKO1 inhibits cell migration due to the inhibition of the second EGF-induced wave of lamellipodia formation. The activation of Rho family small GTPase Rac1 is reported to be required for growth factor-induced lamellipodia formation, and we found that Rac1 also exhibited a biphasic response to EGF with two peaks of 5 min and 12 hrs. UTKO1 inhibited EGF-induced Rac1 activation at 12 hrs with the same concentration to inhibit lamellipodia formation. This indicates that UTKO1 inhibits the second EGF-induced wave of lamellipodia formation via suppression of Rac1 activation.
Next, biotinyl-UTKO1 was synthesized by Prof. Watanabe to identify the molecular target of UTKO1. Among several biotinyl-UTKO1-bound proteins, 14–3-3 ζ knock down caused significant suppression on EGF-induced migration, lamellipodia formation and the activation of Rac1 as well as UTKO1. These results indicate that 14–3-3 ζ might be the molecular target of UTKO1 in A431 cells. Because 14–3-3 proteins act as adaptor or “chaperone molecules”, a binding partner of 14–3-3ζ would regulate the second EGF-induced wave of Rac1 activation, and the most likely candidate binding partner is RacGEF. Indeed, by the knock down experiments, we demonstrated that Tiam1 functions as the RacGEF responsible for the second EGF-induced wave of Rac1 activation by interacting with 14–3-3ζ. We also showed UTKO1 directly inhibits the interaction between 14–3-3ζ and Tiam1 through binding to 14–3-3ζ[Citation20].
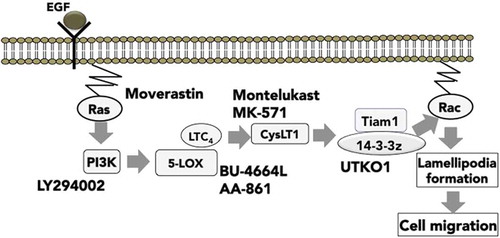
BU-4664L: In the process of further investigation into the mechanism by which EGF induces the second wave of Rac1 activation, BU-4664L[Citation21,Citation22], an inhibitor of 5-lipoxygenase (5-LOX), was found to inhibit the EGF-induced second wave but not the first wave of Rac1 activation. 5-LOX is an enzyme that is responsible for the production of leukotriene (LT)B4 and LTC4, the latter of which is further converted into stable metabolites, LTD4 and LTE4. The inhibitory activities of 5-LOX inhibitors for the EGF-induced second wave of Rac1 activation were negated by addition of LTC4 and LTD4, but not of LTB4, therefore, LTC4/D4/E4 produced by EGF stimulation act as mediators of the EGF-induced second wave of Rac1 activation. Although LTC4/D4/E4 exert their effects by binding to their receptors, cysteinyl leukotriene receptor 1 (CysLT1), CysLT1-specific antagonists MK-571 [Citation23] and montelukast [Citation24] or knockdown of CysLT1 by siRNA were found to inhibit the EGF-induced second wave, but not the first wave of Rac1 activation. These results indicate that LTC4-mediated CysLT1 signaling may regulate the EGF-induced second wave of Rac1 activation.
As described above, Tiam1 is responsible for the EGF-induced second wave of Rac1 activation, and EGF increases Tiam1 expression in terms of both mRNA and protein levels. However, the regulatory mechanisms underlying EGF-increased Tiam1 expression were still unclear. We found that the 5-LOX inhibitors BU-4664L and AA-861, as well as the CysLT1 antagonists MK-571 and montelukast, suppressed the EGF-induced increase in Tiam1 expression in terms of both mRNA and protein levels. Moreover, Tiam1 expression was increased by the addition of LTD4 into the cells where the production of LTC4/D4/E4 was inhibited by 5-LOX inhibitors. Taken together, our results indicate that the 5-LOX/LTC4/CysLT1 signaling pathway regulates EGF-induced cell migration by increasing Tiam1 expression, leading to a second wave of Rac1 activation () [Citation25].
Figure 2. Signaling mechanism for cancer cell migration elucidated by using chemical inhibitors.
Chemical genomic profiling of migration signaling
Cell migration in cancer cells is an essential step in cancer metastasis, but the regulating pathways may differ in different cancer cell types due to the variety of cellular environments and genetic mutations. Understanding the general and specific principles of the regulatory pathways of cancer cell migration is a crucial issue in metastatic cancer research. To address this issue, we conducted a chemical genomic study[Citation26]. We first set up an analytical system to detect the effect of 34 kinds of chemical inhibitors on cell migration in a wound healing assay, and subsequently performed a cluster analysis on the dataset. One significant aspect of this work is that each compound showed a characteristic cell type-specific inhibitory pattern on migration, and hierarchical clustering precisely classified the compounds according to their respective targets ().
Figure 3. Cluster analysis of the chemosensitivity profile of migration inhibition.Cells were scratched and then stimulated by EGF, serum (for HT1080, 3Y1, and EC17 cells), or conditioned medium (CM) from EC17 cells. After 16 h, wound areas were observed and photographed under microscopy.
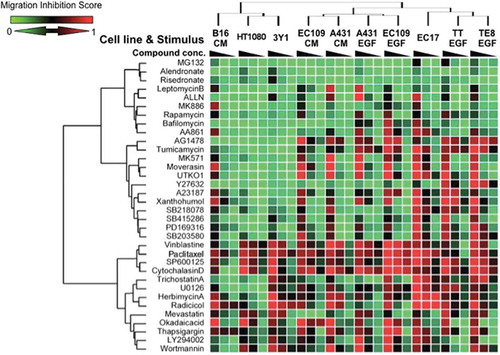
The characteristics of cell migration based on chemical inhibitor-sensitivity profiles were grouped into three clusters, and chemical inhibitors were classified into four general groups. JNK inhibitor showed a potent inhibitory effect on migration of all cell types, indicating that JNK is a common and crucial signaling molecule regulating cell migration. In contrast, some of the chemical inhibitors (AG1478, tunicamycin, the CysLT1 antagonist MK571, Moverastin and UTKO1) affected the migration of cell lines of epithelial origin, but did not affect the migration of cell lines of mesenchymal origin. This suggests that there is an essentially different regulatory mechanism of cell migration between cells of these two origins.
Moreover, we found that four cancer cell lines (TT cells, TE8 cells, A431 and EC109 cells) acquired cell motility by EGF stimulation, but chemo-sensitivity cluster analysis showed that A431 cells and EC109 cells are clustered into the same cluster, on the other hand, TT cells are TE8 cells classified into the different cluster. A431 cells and EC109 cells showed collective migration, whereas TT and TE8 cells showed an individual migration. This raises the possibility that the difference in the mode of cell migration of epithelial cells might be correlated with the differences in sensitivity to chemical inhibitors. The ROCK inhibitor, Y27632, is a representative example; it inhibited individual migration of TT cells and TE8 cells more potently than collective migration of A431 and EC109 cells. Indeed, Rho-ROCK signaling is proposed to induce actomyosin-mediated retraction at the trailing edge in individual migration[Citation27]. Moreover, the GSK-3 inhibitor (SB415286) and the p38MAPK inhibitors (PD169316 and SB203580) also inhibited the EGF-induced migration of TE8 and TT cells more potently than they did in A431 and EC109 cells. This indicates that GSK3 and p38MAPK might be involved in the Rho-ROCK signaling responsible for individual migration. Thus, our approach can be used as a tool for understanding the diversity and similarities in cancer cell migration signaling.
Target identification of bioactive compound
Metabolomic identification of the target of glucopiericidin A
Filopodia are spike-like cell membrane projections contributing to tumor metastasis; however, the molecular mechanisms controlling filopodia protrusion are complicated and unclear [Citation28,Citation29]. Finding a filopodia inhibitor in carcinoma and its molecular target that could be employed in chemical genetic studies may therefore lead to a fuller understanding of filopodia contributing to the treatment of tumor metastasis[Citation30,Citation31].
By screening the microbial broth, we found the cultured broth of one Lechevalieria sp. strain that inhibited the tumor filopodia protrusion. However, this inhibition disappeared following silica-gel chromatography. Interestingly, the inhibitory activity was almost completely recovered by re-mixing all of the silica-gel chromatography fractions, suggesting that the inhibition required the synergistic effect of two or more compounds contained within the microbial broth that eluted in different fractions. We tried to isolate the components responsible for inhibition of filopodia protrusion and found glucopiericidin A (GPA) and piericidin A (PA).
PA is a known inhibitor of mitochondrial respiration, but the mode of action of GPA has not yet been reported. We found that glycolysis inhibitor 2-deoxyglucose also showed the synergistic filopodia inhibition similarly to GPA, suggesting that GPA would be a glycolytic inhibitor. Indeed, metabolome analysis revealed that GPA decreased the cellular level of end-products in glycolysis by inhibiting either the step of glucose uptake or the subsequent step of glucose phosphorylation. Since GPA inhibited glucose transporter-mediated uptake of 3H-labeled 2-deoxyglucose, it was indicated that GPA suppressed glycolysis via inhibition of glucose transporter. Finally, we found that GPA-mediated inhibition of glycolysis dramatically decreases intracellular ATP levels only when mitochondrial respiration is inhibited, and concluded that this ATP decrease caused the synergistic filopodia inhibition by GPA and PA[Citation32].
It is widely recognized that solid tumors exposed to hypoxia can survive and grow aggressively despite low oxygen and limited nutrition. Therefore, glycolysis inhibitors are expected to be candidate drugs for tumor treatment. Many glycolytic inhibitors have been found to be inhibitors of key glycolytic enzymes, but it is not easy to evaluate whether these glycolytic enzyme inhibitors actually modulate glycolysis in living cells.
As described above, we found that glycolytic suppression resulted in the inhibition of filopodium protrusion in cancer cells only when their mitochondrial respiration was restricted. Since the test for filopodium inhibition is a quite easy, low-cost, and quick assay, we proposed it as a cell-based screening method for a new glycolytic inhibitor. In the assay, screening samples were added to A431 cells co-treated with or without the mitochondrial respiratory inhibitor rotenone and examined for their inhibitory effect on EGF-induced filopodium protrusion, which is easily judged by microscopic observation in 30 min[Citation33].
Identification of the target of autophagy inhibitor, xanthohumol
Autophagy is a bulk, nonspecific protein degradation pathway that is involved in the pathogenesis of cancer and neurodegenerative disease[Citation34,Citation35]. But its molecular mechanism is still unclear.
So we searched for novel modulators of autophagy by screening small compounds, in-house natural product library. As a result, we identified xanthohumol (XN) as an autophagy modulator. Therefore, we attempted to identify the target protein of XN, which regulates autophagy[Citation36]. By using XN-immobilized beads, valosin-containing protein (VCP) was identified as an XN-binding protein. VCP, also known as p97, is one of the well-characterized type II AAA ATPase (ATPases associated with diverse cellular activities)[Citation37]. VCP consists of a substrate, a cofactor-binding N domain, and two AAA ATPase domains, D1 and D2, and forms a hexameric double-ring structure. XN bound directly to the N domain, which is known to mediate co-factor and substrate binding to VCP. VCP has been reported to be an essential protein for autophagosome maturation. These results suggested that XN inhibited the function of VCP and resulted in impairment of autophagosome maturation.
Next, we examined the antitumor activity of XN targeting VCP. Several human tumor cell lines exhibited high sensitivity to XN both in vitro and in vivo. Overexpression of VCP occurs in many cancers and clinical studies have reported a correlation between elevated VCP expression and its progression. However, because significant differences in the expression levels of VCP were not found among the cell lines we tested, XN-sensitivity is not dependent on VCP expression levels. However, XN was found to induce a decrease in the expression levels of survivin, a member of the inhibitor of apoptosis (IAP) protein family that inhibits caspases and blocks cell death, in XN-sensitive cells. On the contrary, XN weakly suppressed the expression levels of survivin in XN-insensitive tumor cells. Therefore, it is likely that effect of XN on survivin expression may be related to the sensitivity of tumor cells to XN.
We next performed genome-wide shRNA screening and identified the adenylate cyclase (AC) pathway as genes relating to the antitumor activity of XN against human tumor cells. This pathway regulates various cellular functions via activating PKA-dependent phosphorylation. These results raised the possibility that the AC/PKA pathway could contribute to preventing apoptosis induced by VCP inhibition and that the activity of AC/PKA pathway in tumor cells could determine sensitivity to XN. We also found that PKA inhibition induced a decrease in expression levels of survivin in XN-sensitive and XN-insensitive tumor cells. In addition, suppressed expression of survivin by PKA inhibitor was further enhanced in the presence of XN. Therefore, decreased expression of survivin by both PKA inhibition and VCP inhibition may be responsible for the synergistically enhancement of apoptosis by VCP inhibition and PKA inhibition[Citation38].
Target protein identification combining in silico screening and experimental verification
Anti-apoptotic oncoproteins Bcl-2 and Bcl-xL are overexpressed in many cancers[Citation39,Citation40], resulting in the expansion of a transformed population and the advancement of the multidrug-resistant stage. Consequently, Bcl-2/Bcl-xL have stood out among molecular targets in oncology and the functional blockade of these proteins will be an aid to novel anti-tumor therapies. Therefore, we constructed a cell-based chemical-genetic screening system to discover small molecules that induce apoptosis in Bcl-xL-overexpressing human small cell lung carcinoma Ms-1 cells when combined with anti-tumor drugs.
In the course of our screening, we isolated a structurally and functionally unique compound, named Incednine, from the culture broth of Streptomyces sp. ML694-90F3[Citation41]. Incednine was isolated as a pale yellow powder from the cultured broth of producing strain by Centrifugal liquid-liquid Partition Chromatography, because this compound decomposed easily under acidic conditions or when exposed to light and the isolation using solid carrier such as silica gel was inefficient.
Incednine was tested for its suppressive activity against the anti-apoptotic function of Bcl-2/Bcl-xL. Bcl-xL or Bcl-2-overexpressing Ms-1 cells displayed resistance to various types of anti-tumor agents, such as adriamycin. However, this resistance was overcome by the sequential combination of anti-tumor agents and incednine. Bcl-2/Bcl-xL are known to show an anti-apoptotic effect partly through forming a heterodimer with pro-apoptotic Bcl-2 members, such as Bax and Bak, however, incednine did not inhibit the binding capacity of Bcl-xL to Bax.
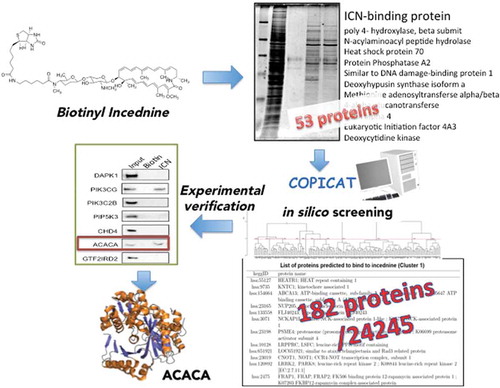
Because incednine inhibits the anti-apoptotic function of Bcl-2/Bcl-xL without affecting its binding to pro-apoptotic Bcl-2 family proteins, it may target other proteins associated with the Bcl-2/Bcl-xL-regulated apoptotic pathway. To address the mode of action of incednine underlying its unique function, we first synthesized affinity-tagged incednine, and proteins bound to incednine were separated by SDS-PAGE, and fifty-three proteins were identified. However, these proteins tested did not appear to be the target responsible for modulating Bcl-2/Bcl-xL anti-apoptotic function.
So, we proposed a novel protocol combining in silico screening and experimental verification for the identification of target proteins (). The training dataset of protein-chemical interactions to construct the SVM-based statistical learning model[Citation6,Citation42,Citation43] was collected from the approved DrugCards data in the DrugBank database [Citation44], and 53 interactions with incednine. This method solves the problem of the low sensitivity of the traditional method. Among 24,245 human proteins in the KEGG repository, 182 proteins target protein candidates were computationally predicted to bind to incednine by the statistical prediction method and 16 proteins among the predictions were experimentally verified. As a result, 40% accuracy of the computational predictions was achieved successfully, and ACACA (acetyl-CoA carboxylase-α) were revealed to bind to incednine by applying our novel protocol to identify potential target proteins of incednine. ACACA is the rate-limiting enzyme for long-chain fatty acid synthesis that catalyzes the ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA, playing a critical role in cellular energy storage and lipid synthesis[Citation45]. There is strong evidence that cancer cell proliferation and survival are dependent on de novo fatty acid synthesis. Indeed, it suggested that chemical inhibition of ACACA results in the induction of apoptosis in Bcl-xL-overexpressing Ms-1 cells when combined with anti-tumor drugs, supposing that ACACA might be a molecular target of incednine.
Figure 4. Proposed protocol of predicting target protein combining in silico screening and experimental verification.
Conclusion
Despite common recognition of the importance of natural compounds in drug discovery development, natural product screening tends to be avoided in recent years due to difficulty in identifying target molecules. However, natural screening is one of the art of agricultural chemistry, and it can also be said to be the culture of our country. Searching for natural products having various activities and diverse structures is exactly a “Treasure finding”. On the other hand, analysis of the mechanism of action through identification of target molecules of natural products is exactly a “Mystery solving”. “Natural product chemical biology” challenging both “Treasure finding” and “Mystery solving” should be actively deployed.
Glossary
PI3K: Phosphoinositide 3-kinase
PI3K is a kinase that catalyzes the phosphorylation of the hydroxyl group at the 3-position of the inositol ring of the inositol phospholipid. Activation of PI3K is known to cause diverse biological activities such as cell differentiation/proliferation and metabolism, cell migration, reconstruction of cytoskeleton.
Akt:
Akt is a serine/threonine kinase having a PH (Plekstrin Homology) domain at its N-terminus.
PI (3, 4, 5) P3 produced by PI3 kinase binds to the PH domain of Akt to collect Akt near the membrane, and Akt is phosphorylated by other protein kinases and activated. It controls many cellular response mechanisms such as cell proliferation and survival, glucose metabolism, tissue invasion and angiogenesis.
Rho:
Rho is one of the small G proteins and functions as a molecular switch within the cell by reciprocating between activated GTP binding form and inactivated GDP binding form. Rho regulates cellular responses such as cell motility and cell adhesion, and cytokinesis.
Rac1: RAS-related C3 botulinus toxin substrate 1
Rac1 is a member of the Rac subfamily, a Rho family small G protein. Rac1 controls a variety of intracellular events such as cell proliferation and cytoskeleton remodeling, and activation of protein kinase.
14-3-3:
14-3-3 is an adapter protein of about 29 kDa, and seven isoforms of β, γ, ε, η, θ, σ, and ζ are known in mammals. 14-3-3 forms homodimers or heterodimers in cells, usually recognizes and binds phosphorylated Ser/Thr of the substrate protein and controls the activity of the protein .
Tiam1: T-lymphoma invasion and metastasis-inducing protein 1
Tiam1 is a GDP/GTP exchange factor acting on Rac, which activates Rac.
JNK: c-Jun NH2-terminal kinase
JNK is a kinase belonging to the MAP kinase family that phosphorylates the N-terminus of the transcription factor c-Jun, and is not only involved in stress response but also is involved in regulating various cell functions such as cell proliferation, cell polarity, cell migration, metabolism.
ROCK:Rho-associated kinase
ROCK is a protein kinase, which is activated by specifically binding with activated Rho. ROCK functions downstream of Rho and was found to contribute to actin cytoskeleton rearrangement by Rho.
GSK: glycogen synthase kinase
GSK is a kind of serine/threonine protein kinase. GSK is contained in various intracellular signaling pathways such as cell proliferation, cell migration, glucose regulation and apoptosis.
PKA: Protein kinase A
PKA is serine/threonine protein kinase activated by cAMP. It is included in various cellular response mechanisms.
Acknowledgments
I am grateful to the late Dr. Hamao Umezawa (Former Director of Institute of Microbial Chemistry) and Professor Kazuo Umezawa (Professor Emeritus of Keio University) for their continuous guidance and encouragement. I offer my heartfelt thank all collaborators for their support and advice. I would also like to thank the graduates and current students of the graduate and undergraduate schools of Keio University. Lastly, I would like to express my sincere gratitude to Professor Emeritus Akikazu Hatanaka (Yamaguchi University) and Professor Hiroyuki Osada (RIKEN) for their recommendation to this award.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Collett MS, Purchio AF, Erikson RL. Avian sarcoma virus-transforming protein, pp60src shows protein kinase activity specific for tyrosine. Nature. 1980 May 15;285(5761):167–169. PubMed PMID: 6246443.
- Ushiro H, Cohen S. Identification of phosphotyrosine as a product of epidermal growth factor-activated protein kinase in A-431 cell membranes. J Biol Chem. 1980 Sep 25;255(18):8363–8365. PubMed PMID: 6157683.
- Pike LJ, Bowen-Pope DF, Ross R, et al. Characterization of platelet-derived growth factor-stimulated phosphorylation in cell membranes. J Biol Chem. 1983 Aug 10;258(15):9383–9390. PubMed PMID: 6192129.
- Heldin CH, Ek B, Ronnstrand L. Characterization of the receptor for platelet-derived growth factor on human fibroblasts. Demonstration of an intimate relationship with a 185,000-Dalton substrate for the platelet-derived growth factor-stimulated kinase. J Biol Chem. 1983 Aug 25;258(16):10054–10061. PubMed PMID: 6309764.
- Umezawa H, Imoto M, Sawa T, et al. Studies on a new epidermal growth factor-receptor kinase inhibitor, erbstatin, produced by MH435-hF3. J Antibiot (Tokyo). 1986 Jan;39(1):170–173. PubMed PMID: 3005217.
- Nakamura H, Iitaka Y, Imoto M, et al. The structure of an epidermal growth factor-receptor kinase inhibitor, erbstatin. J Antibiot (Tokyo). 1986 Feb;39(2):314–315. PubMed PMID: 3007417.
- Imoto M, Umezawa K, Komuro K, et al. Antitumor activity of erbstatin, a tyrosine protein kinase inhibitor. Jpn J Cancer Res. 1987 Apr;78(4):329–332. PubMed PMID: 3108212.
- Imoto M, Morii T, Deguchi A, et al. Involvement of phosphatidylinositol synthesis in the regulation of S phase induction. Exp Cell Res. 1994 Nov;215(1):228–233. PubMed PMID: 7957673.
- Imoto M, Kakeya H, Sawa T, et al. Dephostatin, a novel protein tyrosine phosphatase inhibitor produced by Streptomyces. I. Taxonomy, isolation, and characterization. J Antibiot (Tokyo). 1993 Sep;46(9):1342–1346. PubMed PMID: 8226312.
- Zenitani S, Tashiro S, Shindo K, et al. Gerfelin, a novel inhibitor of geranylgeranyl diphosphate synthase from Beauveria felina QN22047. I. Taxonomy, fermentation, isolation, and biological activities. J Antibiot (Tokyo). 2003 Jul;56(7):617–621. PubMed PMID: 14513904.
- Islam MS, Kitagawa M, Imoto M, et al. Synthesis of gerfelin and related analogous compounds. Biosci Biotechnol Biochem. 2006 Oct;70(10):2523–2528. PubMed PMID: 17031062.
- Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008 Mar–Apr;58(2):71–96. PubMed PMID: 18287387.
- Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004 Apr;25(2):276–308. . PubMed PMID: 15082523.
- Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009 May 8;324(5928):787–790. PubMed PMID: 19359544; PubMed Central PMCID: PMC2981508.
- Korpal M, Korn JM, Gao X, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013 Sep;3(9):1030–1043. PubMed PMID: 23842682.
- Saito S, Fujimaki T, Panbangred W, et al. Antarlides: a new type of Androgen Receptor (AR) antagonist that overcomes resistance to AR-targeted therapy. Angew Chem Int Ed Engl. 2016 Feb 18;55(8):2728–2732. PubMed PMID: 26805525.
- Saito S, Fujimaki T, Panbangred W, et al. Antarlides F-H, new members of the antarlide family produced by Streptomyces sp. BB47. J Antibiot (Tokyo). 2017 May;70(5):595–600. PubMed PMID: 28174422.
- Takemoto Y, Watanabe H, Uchida K, et al. Chemistry and biology of moverastins, inhibitors of cancer cell migration, produced by Aspergillus. Chem Biol. 2005 Dec;12(12):1337–1347. PubMed PMID: 16356851.
- Sawada M, Kubo S, Matsumura K, et al. Synthesis and anti-migrative evaluation of moverastin derivatives. Bioorg Med Chem Lett. 2011 Mar 1;21(5):1385–1389. PubMed PMID: 21306898.
- Kobayashi H, Ogura Y, Sawada M, et al. Involvement of 14-3-3 proteins in the second epidermal growth factor-induced wave of Rac1 activation in the process of cell migration. J Biol Chem. 2011 Nov 11;286(45):39259–39268. PubMed PMID: 21868386; PubMed Central PMCID: PMC3234750.
- Igarashi Y, Miyanaga S, Onaka H, et al. Revision of the structure assigned to the antibiotic BU-4664L from Micromonopora. J Antibiot (Tokyo). 2005 May;58(5):350–352. PubMed PMID: 16060388.
- Peppelenbosch MP, Tertoolen LG, den Hertog J, et al. Epidermal growth factor activates calcium channels by phospholipase A2/5-lipoxygenase-mediated leukotriene C4 production. Cell. 1992 Apr 17;69(2):295–303. PubMed PMID: 1314702.
- Jones TR, Zamboni R, Belley M, et al. Pharmacology of L-660,711 (MK-571): a novel potent and selective leukotriene D4 receptor antagonist. Can J Physiol Pharmacol. 1989 Jan;67(1):17–28. PubMed PMID: 2540892.
- Tate KM, Lee C, Edelman S, et al. Interactions among polymorphic and conserved residues in MHC class II proteins affect MHC-peptide conformation and T cell recognition. Int Immunol. 1995 May;7(5):747–761. PubMed PMID: 7547702.
- Magi S, Takemoto Y, Kobayashi H, et al. 5-lipoxygenase and cysteinyl leukotriene receptor 1 regulate epidermal growth factor-induced cell migration through Tiam1 upregulation and Rac1 activation. Cancer Sci. 2014 Mar;105(3):290–296. PubMed PMID: 24350867; PubMed Central PMCID: PMC4317946.
- Magi S, Tashiro E, Imoto M. A chemical genomic study identifying diversity in cell migration signaling in cancer cells. Sci Rep. 2012;2:823. PubMed PMID: 23139868; PubMed Central PMCID: PMC3492869.
- Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008 Oct 31;135(3):510–523. PubMed PMID: 18984162.
- Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008 Jun 1;9(6):446–454. . PubMed PMID: 18464790; eng.
- Faix J, Rottner K. The making of filopodia. Curr Opin Cell Biol. 2006 Feb 1;18(1):18–25. . PubMed PMID: 16337369; eng.
- Bacon C, Lakics V, Machesky L, et al. N-WASP regulates extension of filopodia and processes by oligodendrocyte progenitors, oligodendrocytes, and Schwann cells-implications for axon ensheathment at myelination. Glia. 2007 Jun 1;55(8):844–858. PubMed PMID: 17405146; eng.
- Shulman Z, Shinder V, Klein E, et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. 2009 Mar 20;30(3):384–396. PubMed PMID: 19268609; eng.
- Kitagawa M, Ikeda S, Tashiro E, et al. Metabolomic identification of the target of the filopodia protrusion inhibitor glucopiericidin A. Chem Biol. 2010 Sep 24;17(9):989–998. PubMed PMID: 20851348.
- Kitagawa M, Misawa M, Ogawa S, et al. A new, convenient cell-based screening method for small-molecule glycolytic inhibitors. Biosci Biotechnol Biochem. 2011;75(2):367–369. PubMed PMID: 21307586.
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001 Mar;2(3):211–216. PubMed PMID: 11265251.
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004 Jan 9;313(2):453–458. PubMed PMID: 14684184.
- Sasazawa Y, Kanagaki S, Tashiro E, et al. Xanthohumol impairs autophagosome maturation through direct inhibition of valosin-containing protein. ACS Chem Biol. 2012 May 18;7(5):892–900. PubMed PMID: 22360440.
- Tresse E, Salomons FA, Vesa J, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010 Feb;6(2):217–227. PubMed PMID: 20104022; PubMed Central PMCID: PMC2929010.
- Shikata Y, Yoshimaru T, Komatsu M, et al. Protein kinase A inhibition facilitates the antitumor activity of xanthohumol, a valosin-containing protein inhibitor. Cancer Sci. 2017 Apr;108(4):785–794. PubMed PMID: 28122154; PubMed Central PMCID: PMC5406609.
- Tsujimoto Y, Cossman J, Jaffe E, et al. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985 Jun 21;228(4706):1440–1443. PubMed PMID: 3874430.
- Wang S, Yang D, Lippman ME. Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Semin Oncol. 2003 Oct;30(5 Suppl 16):133–142. PubMed PMID: 14613034.
- Futamura Y, Sawa R, Umezawa Y, et al. Discovery of incednine as a potent modulator of the anti-apoptotic function of Bcl-xL from microbial origin. J Am Chem Soc. 2008 Feb 13;130(6):1822–1823. PubMed PMID: 18205364.
- Nagamine N, Shirakawa T, Minato Y, et al. Integrating statistical predictions and experimental verifications for enhancing protein-chemical interaction predictions in virtual screening. PLoS Comput Biol. 2009 Jun;5(6):e1000397. PubMed PMID: 19503826; PubMed Central PMCID: PMC2685987.
- Nagamine N, Sakakibara Y. Statistical prediction of protein chemical interactions based on chemical structure and mass spectrometry data. Bioinformatics. 2007 Aug 1;23(15):2004–2012. . PubMed PMID: 17510168.
- Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration [research support, non-U.S. gov’t]. Nucleic Acids Res. 2006 Jan 1;34(Database issue):D668–72. PubMed PMID: 16381955; PubMed Central PMCID: PMC1347430. eng.
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007 Oct;7(10):763–777. . PubMed PMID: 17882277.