
ABSTRACT
Natural products are a tremendous source of tool discovery for basic science and drug discovery for clinical uses. In contrast to the large number of compounds isolated from nature, however, the number of compounds whose target molecules have been identified so far is fairly limited. Elucidation of the mechanism of how bioactive small molecules act in cells to induce biological activity (mode of action) is an attractive but challenging field of basic biology. At the same time, this is the major bottleneck for drug development of compounds identified in cell-based and phenotype-based screening. Although researchers’ experience and inspiration have been crucial for successful target identification, recent advancements in genomics, proteomics, and chemical genomics have made this challenging task possible in a systematic fashion.
Graphical Abstract
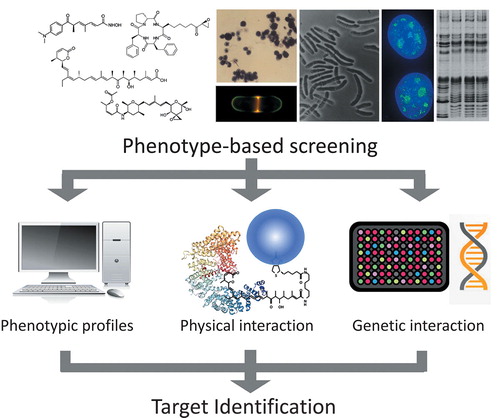
A general scheme for target identification of small molecules.
The discovery of antibiotics was one of the most socially influential achievements of biology and medicine in the 20th century. Since the accidental discovery of penicillin by Alex Fleming [Citation1], many scientists devoted themselves to developing novel antibiotics and antimicrobial natural products from numerous microorganisms, plants and animals. These efforts eventually contributed to human health by rescuing humans from various infectious diseases. Although the discovery of new antibiotics from nature has now become much less frequent, research on the biosynthesis and mode of action (MOA) of secondary metabolites has attracted much attention as the basis of future drug discovery.
Natural products including antibiotics show a large variety of structures and activities. They have played extremely important roles in not only therapy as drugs but also as tools for basic studies in various fields of chemistry and biology. For instance, FK506, which opened an innovative avenue to organ transplantation [Citation2], served as a definitive tool for dissecting the molecular mechanism underlying calcineurin-regulated immune response in T cells [Citation3] and creating artificial ligands for chemical biology [Citation4]. Thus, to elucidate the MOA of compounds that show dramatic and specific actions is a research subject that greatly attracts biologists and is also important for future drug development because successful MOA studies often identify unexpected new targets for drug discovery. However, it still remains challenging to establish truly correct target molecules, as it requires tedious trial-and-error tasks requiring both chemical and biological approaches. In this review, I will highlight three major approaches to target identification () by showing our own research as examples and discuss future directions.
Figure 1. Scheme for target identification of small molecules. It is necessary to identify targets of bioactive compounds for understanding the mode of action. There are three major approaches to target identification: comparison of phenotypic profiles with reference compounds, detection of physical interaction, and detection of genetic interaction with target molecules.
Drug-induced phenotype approach
During the “Golden Age of Antibiotics” (1950s ~ 1960s), most of the antibiotic classes used in the clinic today, as well as a large number of antibacterial natural products that were not put into the practical use, were discovered. Studies of the MOA of newly discovered antibiotics were performed, but mainly focused on macromolecular synthesis. This is because most antibiotics inhibit synthesis of the cell wall, DNA, RNA, or protein to kill bacteria. However, it became difficult to elucidate the MOA of bioactive natural products through analysis of macromolecular synthesis, as their bioactivities shifted from inhibition of bacterial cell growth to complex regulatory mechanisms of higher eukaryotes. Therefore, researchers need to predict the target or target pathway according to the phenotypic information, such as cell morphology, gene or protein expression, cell cycle progression, etc., rather than simple analysis of macromolecular synthesis, and must carefully validate the predicted target.
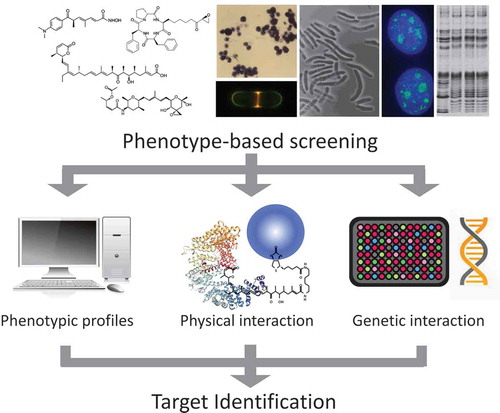
As an example, we hypothesized that trichostatin A (TSA), a Streptomyces metabolite which had been rediscovered as a potent inducer of murine erythroleukemia cell differentiation [Citation5], inhibits cyclin-dependent kinase (CDK) since it strongly inhibited the cell cycle progression of rat fibroblast 3Y1 cells in both G1 and G2 phases [Citation6]. At that time, it was shown that the cell cycle regulatory gene cdc2 was required for cell cycle progression of both G1 and G2 phases in the fission yeast Schizosaccharomyces pombe [Citation7]. We therefore studied the effect of TSA on histone H1 phosphorylation, as histone H1 was known as the major endogenous substrate of Cdc2 kinase. As a result, we found that TSA suppressed histone H1 phosphorylation in vivo but did not inhibit enzymatic activity in vitro. In this process, we happened to find an unusual increase in histone acetylation of cells treated with TSA. With this observation as a springboard, we were able to reach histone deacetylase (HDAC) as the target of TSA (). Indeed, TSA inhibited in vitro deacetylase activity at a low nanomolar concentration but the enzymatic activity of HDAC prepared from TSA-resistant mutant cells was resistant to TSA, providing the genetic evidence that HDAC is the molecular target of TSA in cells [Citation8].
Figure 2. Histone acetylation and natural inhibitors of histone deacetylase. Trichostatin A, trapoxin, and FK228 are structurally unrelated microbial metabolites that specifically inhibit histone deacetylase (HDAC). Histone acetylation is regulated by two distinct enzyme families, histone acetyltransferase (HAT) and HDAC. Acetylated histone represents a transcriptionally active state, which is recognized by acetylated histone binding proteins. HDAC inhibitors induce histone hyperacetylation, which restores expression of many tumor suppressor genes that are silenced in cancer cells.
Discovery of HDAC as the target of TSA led to the elucidation of the same target molecule for other natural products with different chemical structures one after another, based on their phenotypic similarity to TSA. Trapoxin (TPX) A and B, fungal cyclic tetrapeptides that induce morphological normalization in several oncogene-transformed cell lines, showed irreversible inhibition of HDAC activity, in contrast to reversible inhibition by TSA [Citation9] (). Structure and activity relationship (SAR) studies demonstrated that an epoxyketone moiety of TPX is responsible for the irreversible inhibition. Based on this finding, all other related natural cyclic tetrapeptides with an epoxyketone-containing side chain such as chlamydocin, HC-toxin, Cyl-1, and Cyl-2 were shown to inhibit HDACs. Furthermore, we found that the epoxyketone moiety can be replaced with a hydroxamic acid, a key structure for HDAC inhibition by TSA [Citation10], and created a series of hybrid HDAC inhibitors with anticancer activity, named Cyclic Hydroxamic Acid-containing Peptides (CHAPs) [Citation11]. The discovery of TSA and TPX as specific HDAC inhibitors opened a new area of research into the biological role of histone acetylation. Histone lysine acetylation, first described by Vincent Allfrey [Citation12], has been considered a marker for transcriptional activity, but there was no critical means to test the possibility. The emergence of TSA and TPX enabled the role of histone acetylation in various biological systems, such as cancer and immune response, to be validated because these compounds can induce histone hyperacetylation in cells, tissues, or animal bodies. Moreover, the strong affinity of TPX for the target HDAC enzyme was used for the cloning of the first HDAC gene (HDAC1) [Citation13]. HDAC inhibitors also played an important role in novel drug discovery. FK228 (also known as FR901228, romidepsin, and Istodax), which had been identified as a bicyclic depsipeptide produced by a bacterium [Citation14], showed strong anticancer activity in both animal models and clinical trials. We demonstrated that FK228 was another type of HDAC inhibitor [Citation15] (). Indeed, FK228 acts as a prodrug, of which its disulfide is reduced in the cell to become an active form that inhibits HDAC by interacting with the enzyme’s active site with its reduced sulfhydryl group [Citation16]. In the case of the synthetic compound SAHA, which had been developed as an inducer of murine erythroleukemia cell differentiation by a group in the United States, the MOA was unknown for many years, but was finally shown to inhibit HDAC by Richon et al. [Citation17] because of similarities in its structure and activity to TSA. Later, SAHA became the first HDAC inhibitor approved as an anticancer agent for treatment of lymphoma by the US Food and Drug Administration (FDA), followed by clinical application of FK228. Currently, the clinical importance of HDAC enzymes in various diseases have been established and five different HDAC inhibitors have been clinically used for treatment of cutaneous and peripheral lymphoma and multiple myeloma.
Thus, if there is a reference compound that has a specific target, the MOA can be easily predicted according to its phenotypic similarity. Creation of a database for the profiles of each reference compound would be useful for facilitating the process of searching for similar MOA. Recently, various databases for the actions of compounds such as transcriptome [Citation18], proteome [Citation19], cancer cell panel [Citation20], cell morphology [Citation21] have been established, which greatly accelerate MOA studies based on phenotypic changes induced by target-unknown compounds. In particular, the expanded connectivity map (Cmap), a transcriptome dataset covering more than one million expression profiles, can be used for discovering the MOA of small molecules [Citation22].
Physical interaction-based approach
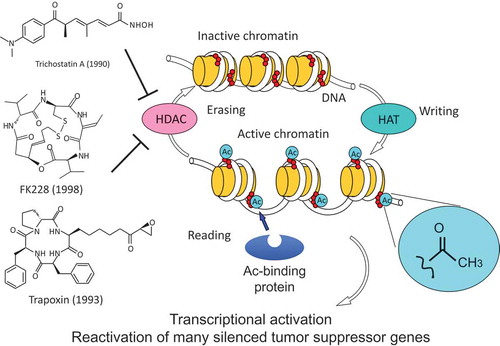
If there is existing information about the MOA of a known compound that is structurally or phenotypically similar to a new compound of interest, it is possible to infer its target, but it is not easy to do so if the MOA of the compound of interest is completely new. In the case of TSA, we happened to discover HDAC as an important target molecule by chance in the course of biochemical experiments. However, such a biochemical approach toward elucidation of a new target totally depends on the experience and intuition of the researcher, and cannot be considered general methodology. In 1990s, a group led by Stuart L. Schreiber changed this situation completely. They first synthesized a bioactive compound and then modified it to develop affinity probes by immobilizing on resin, which allowed for identification of binding proteins from cell lysates (). The representative examples were calcineurin for the FK506-FKBP complex [Citation3], HDAC1 for trapoxin [Citation13], and the proteasome for lactacystin, a differentiation inducer for neuroblastoma cells [Citation23]. As such, if the physical interaction is sufficiently strong, specific and detectable in the in vitro cell-free system, then screening for the binding protein using the affinity probe is the most powerful and straightforward way to identify the target molecule. Our group also succeeded in the identification of a surprising molecule as the target of the potent anticancer natural product FR901464 by employing the physical interaction approach in collaboration with the organic chemistry team at the University of Tokyo led by Takeshi Kitahara and Hidenori Watanabe. Based on the SAR study, a chemically stable methyl ketal derivative of FR901464 named spliceostatin A (SSA) was created and its biotinylated probe was used for screening the binding protein. The target molecule identified from the cell lysate was the SF3b complex, which is an essential component of the spliceosome, a large ribonucleoprotein complex responsible for eukaryotic splicing (). As a result of specific binding to SF3b, SSA inhibited the splicing reaction in vitro and in the cell, resulting in the accumulation of precursor mRNA (pre-mRNA) [Citation24].
Figure 3. Identification of the splicing factor SF3b as the target of FR901464/spliceostatin A (SSA). SSA is a methyl ketal derivative of the natural product FR901464, which retains potent antitumor activity with better stability than FR901464 (a). SSA was used for synthesizing the biotinylated probe (b). Using the biotinylated probe (b-SSA), a protein complex containing SAP155, SAP145, SAP130, and SAP49 was identified as the SSA-binding protein (c). The SF3b complex, which recognizes the branch point sequence of introns in pre-mRNA, is an essential component of U2 snRNP in the spliceosome (d).
FR901464/SSA inhibits the cell cycle progression of a variety of cancer cells at G1 phase. In order to clarify the mechanism, expression of known cell cycle regulators was analyzed, which revealed that the C-terminal truncated form of p27 (p27*), one of the CDK inhibitory proteins, specifically accumulated in cells treated with FR901464. p27* was found to lack the C-terminal part of p27 encoded by exon 2, suggesting that translation of p27 pre-mRNA occurs, resulting in the production of a protein consisting of the sequences of exon 1 and intron 1 up to the in-frame termination codon. Therefore, we introduced a p27 minigene containing an HA tag prior to the termination codon within intron 1 and a FLAG tag prior to the original termination codon in exon 2 to see which termination codon is used in cells treated with SSA. The result clearly showed that the intron 1 sequence was translated in the presence of SSA [Citation24]. This finding indicates that SSA has surprising activity that induces not only pre-mRNA accumulation through splicing inhibition but also translation of intron sequences in at least a subset of pre-mRNAs including p27, probably through the leakage of the nuclear pre-mRNA into the cytoplasm where translation occurs [Citation25].
Another screening project for physically interacting proteins successfully elucidated the long-awaited target molecule of the clinically important drug thalidomide, which is famous for showing teratogenicity and therapeutic activity toward multiple myeloma. Handa and colleagues identified the ubiquitin ligase called cereblon (CRBN) as the target molecule of thalidomide, using ferrite-glycidyl methacrylate beads bound to a carboxylic thalidomide derivative FR259625 [Citation26]. Thalidomide suppresses the degradation of the original substrates of CRBN, which is responsible for its teratogenicity. In addition, thalidomide binding to CRBN changes its target proteins for ubiquitination, and causes degradation of new protein regardless of the physiological function of CRBN, which may be involved in its anticancer activity [Citation27,Citation28].
Thus, the detection of physical interactions between a compound and its binding protein is widely established as the most direct method for target identification. In some cases, however, it is difficult to find a functional group required for synthesizing affinity probes. It is generally necessary to perform extensive, time-consuming SAR studies for determining the functional moiety to link to the resin. The Osada group at RIKEN developed an almost universal method for making affinity probes for bioactive small molecules using an aryl diazirine group [Citation29]. Because of its highly active, nonspecific photo-crosslinking reaction, a compound of interest can be immobilized on the linker of the beads in a random orientation after UV irradiation. Therefore, the photo-crosslinked product should always contain compound that remains biologically active even after covalent coupling, which can be used for affinity purification of target molecules.
Genetic interaction-based approach
Although the physical interaction approach is a truly straightforward way to identify target molecules, it is sometimes difficult to detect binding proteins because of low affinity to a probe, low abundance in the cell, high background levels that disturb detection, or unstable binding intolerable to washing. Indeed, the actual success rate for drug target identification by the physical interaction approach is low. In such cases, detecting genetic interaction is useful as an alternative method. A genetic interaction between a compound and target protein represents a change in the cell’s sensitivity to the compound (compound-induced phenotype) when the function of the target molecule is modulated by genetic alterations. One of the most famous examples in which a target molecule was identified in this way is rapamycin. The target molecule TOR (Target of Rapamycin) was identified from a rapamycin-resistant mutation in Saccharomyces cerevisiae [Citation30].
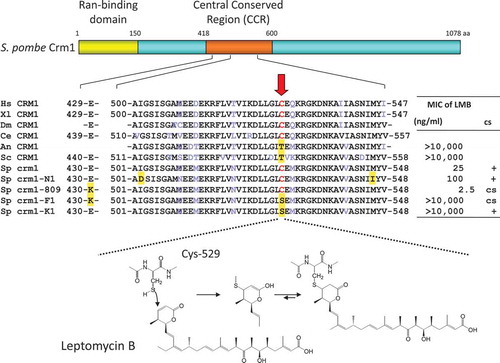
We applied this method to understand the MOA of the antifungal antibiotic leptomycin B (LMB) produced by a Streptomyces strain [Citation31]. LMB induces morphological changes in a limited class of fungi such as Mucor and Schizosaccharomyces at low nanomolar concentrations but shows no obvious effect on other fungal species such as Aspergillus and Saccharomyces. Cloning of the LMB resistance gene in the fission yeast Schizosaccharomyces pombe identified a mutation in the crm1 gene that is responsible for LMB resistance [Citation32]. Since the phenotypes induced by LMB and the crm1 deletion mutation in S. pombe were identical, we concluded that CRM1 is the target of LMB, but at that time the physiological function of CRM1 was not known. CRM1 is a highly conserved protein that localizes to both the nucleus and cytoplasm and accumulates in the nuclear envelope. We cloned a human homologue of CRM1 and analyzed its function [Citation33]. During the course of the analysis, we found that LMB inhibits nuclear export of proteins bearing the nuclear export signal (NES). This finding led to the discovery of the long-awaited receptor for the NES [Citation34,Citation35]. Indeed, CRM1 was proven to be the nuclear export factor essential for protein nuclear export in all eukaryotes. Importantly, further genetic and biochemical analyses demonstrated that LMB specifically binds via a Michael-type addition to a specific cysteine residue on CRM1 that is conserved in all LMB-sensitive organisms but not in LMB-insensitive organisms [Citation36] (). Recent X-ray crystallographic studies showed that the cysteine residue resides in the NES binding cleft [Citation37]. As CRM1 has been considered an important target for cancer therapy, the development of synthetic CRM1 inhibitors and their clinical studies are underway [Citation38].
Figure 4. Schematic representation of fission yeast CRM1 and the binding mode of leptomycin B (LMB). A single cysteine residue (Cys-529) in the central conserved region is the binding site of LMB, which is conserved in LMB-sensitive organisms such as human, but not in LMB-insensitive organisms such as the budding yeast S. cerevisiae. Other mutations such as crm1-809 and crm1-N1 were also mapped to this region. It is now known that the central conserved region is responsible for recognizing the NES. The unsaturated delta lactone ring mediates covalent bond formation with the cysteine residue by a Michael-type addition.
Yeast chemical genetics has proven highly useful for identifying chemical-genetic interactions, as illustrated not only by TOR targeted by rapamycin, but also CRM1. Both TOR and CRM1 are key regulatory molecules conserved in all eukaryotes and are considered important therapeutic targets, indicating that model organisms such as yeast that are amenable to genetic manipulation are important tools for target identification.
Chemical genomics
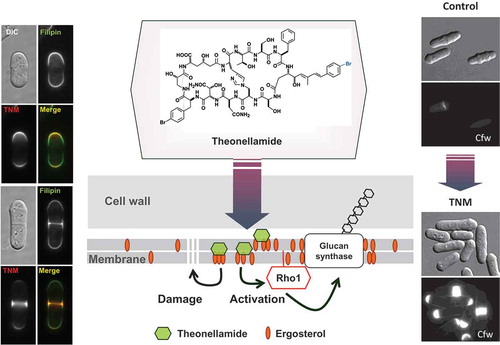
In recent years, techniques for analyzing genetic interactions between all genes and compounds at once have been developed by utilizing genomic information, with post-genomic tools such as gene-disrupted or gene over-expressing strains for all genes in an organism of interest’s genome. For example, we succeeded in cloning and forcibly expressing all open reading frames (ORFs) encoded by the genome of fission yeast. We used this library to test drug susceptibility upon gene overexpression and to create the protein localization database “Localizome” [Citation39]. In general, overexpression of a target molecule for a given compound of interest or protein involved in the target pathway often confers resistance to the compound in cells. If the localization of the compound in cells can be visualized using a fluorescently labeled derivative, then global information about the protein subcellular localization can be utilized to narrow down the potential interacting proteins, since the compound and its target should colocalize with the compound-target complex. Using such information, we succeeded in identifying the target molecule of the anti-fungal marine natural product theonellamide, whose MOA had been completely unknown for many years. Theonellamide was found to serve as a new type of antifungal compound that binds to ergosterol in the fungal cell membrane and derails signal transduction, thereby inducing abnormal beta-glucan synthesis and cell death [Citation40] ().
Figure 5. Mechanism of action of theonellamide (TNM) that binds ergosterol in fungi. A fluorescently labeled TNM was colocalized with filipin, a fluorescent probe for sterol, in fission yeast. On the other hand, TNM induces accumulation of 1,3-beta-D-glucan, which is visualized with calcofluor white (Cfw). Genetic analysis revealed that TNM binding to ergosterol causes activation of glucan synthase, which is mediated by Rho1, a small GTPase.
Boone and colleagues utilized synthetic lethality in budding yeast to predict the gene that encodes the drug target. Synthetic lethality is a phenomenon wherein a combination of deficiencies of two or more genes causes cell death, whereas a deficiency in only one of the genes does not. When a compound of interest inhibits the function of one gene in a synthetic lethal pair, the compound does not inhibit the growth of a wild-type strain, but does inhibit the growth of the particular mutant strain in which the other synthetic lethal partner gene is disrupted, because the function of both the synthetic lethal genes is simultaneously impaired [Citation41]. In this case, the compound of interest and the gene corresponding to the growth-inhibited mutant shares a chemical-genetic interaction, and this is analogous to the genetic interaction shared between the synthetic lethal genes. To find every synthetic lethal gene pair in budding yeast, they quantified the proliferation of double-knockout mutants by comparing the degree of fitness between double- and single-knockout strains, and mapped the resulting global genetic interaction network in yeast [Citation42]. This genome-wide genetic interaction map provided a key for interpreting chemical-genetic interactions. They established a high-throughput assay to detect chemical-genetic interactions between mutant strains and compounds, using barcode sequencing of genomic DNA prepared from a pooled culture of gene disrupted mutants, each marked with different DNA barcodes. Treating the DNA-barcoded pools with compounds in a microculture format has allowed the rapid generation of chemical-genetic profiles, and has enabled the large-scale analysis of compounds from whole chemical libraries [Citation43].
Meanwhile, Ohya et al. established a method called CalMorph, which allows for quantification of yeast morphological phenotypes using numerical parameters, and statistically extracts and profiles the phenotypes of each viable gene disruptant [Citation44]. This dataset is also useful for predicting the target or target pathway of a compound of interest by comparing the drug-induced phenotype and the disruptant phenotype. Such analytical techniques for genome-wide chemical genetic interactions are progressing year by year, and have now expanded to not only yeast but also nematodes and animal cells. Our group also utilizes a barcoded shRNA library that enables global knockdown of every single gene in human cells in order to determine the genes that affect sensitivity to small molecules [Citation45]. With the advent of genome editing technology, this field is expected to accelerate more and more [Citation46].
Perspectives
On the frontline of drug discovery, in vitro target-based assay systems developed for promising therapeutic targets obtained from clinical genome projects have been prevailing over the in vivo cell-based or phenotype-based assays. In recent years, however, the success rate for drug development from target-based assays has rapidly declined, while the cost of screening is increasing. This is probably because straightforward therapeutic targets have been exhausted and it has become difficult to find new targets. On the other hand, HDAC, CRM1, and SF3b, which were identified as the targets of TSA, LMB, and SSA, respectively, are currently drawing attention as novel clinical targets for cancer therapy. Indeed, some HDAC inhibitors have already been used clinically, and new inhibitors of CRM1 and SF3b are being tested clinically. These target molecules are seemingly difficult to envision as drug targets because of their fundamental functions in life. However, if a specific inhibitor actually exists, the potential as a therapeutic target can actually be tested. In this way, HDAC and TOR have emerged as important drug targets. In the future, it is expected that new disease model cells will be created using iPS and genome editing technologies [Citation47,Citation48], and that phenotypic screening using those model cells will become mainstream again. In order to realize iPS cell-based drug discovery, it will be necessary to strengthen the methodology for target identification and determining the MOA of hit compounds, which will greatly accelerate drug development from disease models.
Acknowledgments
The author thanks S. Li and Y. Yashiroda for critical reading, E. Bradshaw for proofreading, and H. Kobayashi for assistance in figure preparation.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Fleming A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenza. Br J Exp Pathol. 1929;10:226–236.
- Kino T1, Hatanaka H, Hashimoto M, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physic-chemical and biological characteristics. J Antibiot (Tokyo). 1987;40(9):1249–1255.
- Liu J, Farmer JD Jr, Lane WS, et al. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66(4):807–815.
- Spencer DM, Wandless TJ, Schreiber SL, et al. Controlling signal transduction with synthetic ligands. Science. 1993;262(5136):1019–1024.
- Yoshida M, Nomura S, Beppu T. Effects of trichostatins on differentiation of murine erythroleukemia cells. Cancer Res. 1987;47:3688–3691.
- Yoshida M, Beppu T. Reversible arrest of proliferation of rat 3Y1 fibroblasts in both the G1 and G2 phases by trichostatin A. Exp Cell Res. 1988;177:122–131.
- Simanis V, Nurse P. The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell. 1986;45(2):261–268.
- Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179.
- Kijima M, Yoshida M, Sugita K, et al. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–22435.
- Furumai R, Komatsu Y, Nishino N, et al. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci U S A. 2001;98:87–92.
- Komatsu Y, Tomizaki KY, Tsukamoto M, et al. Cyclic hydroxamic-acid-containing peptide 31, a potent synthetic histone deacetylase inhibitor with antitumor activity. Cancer Res. 2001;61(11):4459–4466.
- Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794.
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272(5260):408–411.
- Ueda H, Nakajima H, Hori Y, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J Antibiot (Tokyo). 1994;47(3):301–310.
- Nakajima H, Kim YB, Terano H, et al. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res. 1998;241(1):126–133.
- Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62(17):4916–4921.
- Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci U S A. 1998;95(6):3003–3007.
- Lamb J, Crawford ED, Peck D, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935.
- Muroi M, Kazami S, Noda K, et al. Application of proteomic profiling based on 2D-DIGE for classification of compounds according to the mechanism of action. Chem Biol. 2011;17(5):460–470.
- Yamori T. Panel of human cancer cell lines provides valuable database for drug discovery and bioinformatics. Cancer Chemother Pharmacol. 2003;52(Suppl 1):S74–79.
- Futamura Y, Kawatani M, Kazami S, et al. Morphobase, an encyclopedic cell morphology database, and its use for drug target identification. Chem Biol. 2012;19(12):1620–1630.
- Subramanian A, Narayan R, Corsello SM, et al. 4. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437–1452.e17.
- Fenteany G, Standaert RF, Lane WS, et al. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268(5211):726–731.
- Kaida D, Motoyoshi H, Tashiro E, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3(9):576–583.
- Yoshimoto R, Kaida D, Furuno M, et al. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA. 2017;23(1):47–57.
- Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350.
- Krönke J1, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305.
- Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–309.
- Kanoh N, Honda K, Simizu S, et al. Photo-cross-linked small-molecule affinity matrix for facilitating forward and reverse chemical genetics. Angew Chem Int Ed Engl. 2005;44(23):3559–3562.
- Kunz J, Henriquez R, Schneider U, et al. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73(3):585–596.
- Hamamoto T, Gunji S, Tsuji H, et al. Leptomycins A and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J Antibiot (Tokyo). 1983;36(6):639–645.
- Nishi K, Yoshida M, Fujiwara D, et al. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269(9):6320–6324.
- Kudo N, Khochbin S, Nishi K, et al. Molecular cloning and cell cycle-dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J Biol Chem. 1997;272(47):29742–29751.
- Fornerod M, Ohno M, Yoshida M, et al. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90(6):1051–1060.
- Kudo N, Wolff B, Sekimoto T, et al. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res. 1998;242(2):540–547.
- Kudo N, Matsumori N, Taoka H, et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96(16):9112–9117.
- Dong X, Biswas A, Süel KE, et al. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458(7242):1136–1141.
- Mendonca J, Sharma A, Kim HS, et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget. 2014;5(15):6102–6112.
- Matsuyama A, Arai R, Yashiroda Y, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2006;24(7):841–847.
- Nishimura S, Arita Y, Honda M, et al. Marine antifungal theonellamides target 3beta-hydroxysterol to activate Rho1 signaling. Nat Chem Biol. 2010;6(7):519–526.
- Parsons AB, Brost RL, Ding H, et al. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22(1):62–69.
- Costanzo M, Baryshnikova A, Bellay J, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–431.
- Piotrowski JS, Li SC, Deshpande R, et al. Functional annotation of chemical libraries across diverse biological processes. Nat Chem Biol. 2017;13(9):982–993.
- Ohya Y, Sese J, Yukawa M, et al. High-dimensional and large-scale phenotyping of yeast mutants. Proc Natl Acad Sci U S A. 2005;102(52):19015–19020.
- Takase S, Kurokawa R, Arai D, et al. A quantitative shRNA screen identifies ATP1A1 as a gene that regulates cytotoxicity by aurilide B. Sci Rep. 2017;7(1):2002.
- Hart T, Chandrashekhar M, Aregger M, et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163(6):1515–1526.
- Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17(3):170–182.
- Shi Y, Inoue H, Wu JC, et al. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16(2):115–130.