
ABSTRACT
MicroRNAs (miRNAs) are important and ubiquitous regulators of gene expression in eukaryotes. However, the information about miRNAs population and their regulatory functions involving in soybean seed development remains incomplete. Base on the Dicer-like1-mediated cleavage signals during miRNA processing could be employed for novel miRNA discovery, a genome-wide search for miRNA candidates involved in seed development was carried out. As a result, 17 novel miRNAs, 14 isoforms of miRNA (isomiRs) and 31 previously validated miRNAs were discovered. These novel miRNAs and isomiRs represented tissue-specific expression and the isomiRs showed significantly higher abundance than that of their miRNA counterparts in different tissues. After target prediction and degradome sequencing data-based validation, 13 novel miRNA–target pairs were further identified. Besides, five targets of 22-nt iso-gma-miR393h were found to be triggered to produce secondary trans-acting siRNA (ta-siRNAs). Summarily, our results could expand the repertoire of miRNAs with potentially important functions in soybean.
Graphical Abstract
Identification of novel microRNAs and their targets in soybean.
MicroRNAs (miRNAs) are a class of ~ 21nt non-coding RNAs, which are well known as important regulators ubiquitously existing in both plants and animals. There are increasing evidences that they take part in a wide range of biological processes in eukaryotes, such as organogenesis [Citation1–Citation3], signal transduction [Citation4] and stress responses [Citation1,Citation5–Citation8]. In plant, a primary miRNA (pri-miRNA) is recognized and processed by DICER-LIKE1 (DCL1) to produce a miRNA precursor, which is further cropped by DCL1 to yield a single small RNA (sRNA) duplex, miRNA-miRNA*, with two-nucleotide 3ʹ overhangs [Citation9]. One strand of the duplex is the mature miRNA, it loaded onto an Argonaute (AGO) protein to form a RNA-induced silencing complex (RISC) to exert the cleavage of target transcripts. The other strand, miRNA*, is normally rapidly degraded [Citation10]. Some miRNAs have been revealed capable of triggering phased siRNA (phasiRNAs) biogenesis from their targets. The products of miRNA-guided cleavage can further serve as the substrate for RDR6, generating a dsRNA precursor which is further processed by DCL4 to produce phasiRNAs [Citation11]. Some phasiRNAs have been revealed function in trans regulation of target genes via cleavage at complementary target sites, just like many plant miRNAs. These phasiRNAs were named as trans-acting siRNAs (ta-siRNAs) [Citation12–Citation14].
Soybean is the most economically important legume in the world. It produces seeds that are enrich with proteins and oils which provide sources of energy and nutrition for both humans and animals. A lot of nutritional contents are accumulated in the soybean seeds during their development. Thus, understanding the regulation of metabolic pathways during seed development is one of the prerequisites for crop improvement [Citation15].
MiRNAs have been revealed to play an essential role in seed development [Citation15,Citation16]. In recent years, increasing investigations focused on miRNA discovery and their regulatory function analysis contributed greatly to the quality improvement of soybean seeds, such as increasing oil production by controlling fatty acid pathway [Citation17], and surviving in abiotic stress environment [Citation18,Citation19].The recent advance in next-generation small RNA sequencing technology coupled with developments of bioinformatics tools provided a powerful way for genome-wide discovery of considerable miRNAs with important regulatory functions in plants [Citation15,Citation20–Citation25]. To date, 639 mature miRNAs and 573 miRNA precursors of soybean have been registered in the miRBase (release 21). However, there are increasing evidences that the information of miRNAs population and their regulatory functions in soybean seed development remains incomplete [Citation26–Citation28].The investigation of discovering novel miRNA with potential regulatory functions during soybean seed development period is necessary to continue, which will benefits the fully understand of miRNA regulatory network, seed quality controlling and improvement of soybean.
In previous study, we found DCL1-mediated cleavage signals for the processing of the miRNA precursors could be detected by degradome sequencing. Besides, their positions are highly correlated with the boundaries of the miRNAs (−5p)/miRNAs*(−3p). So, we constructed a bio-software, miRNA Digger (), for genome-wide mining of miRNAs by using the degradome-supported Dicer cropping signals as the markers to detect potential miRNA loci [Citation29].
Figure 1. Workflow of miRNA Digger. miRNA Digger is free for download from http://www.bioinfolab.cn/miRNA_Digger/index.html [Citation29].
![Figure 1. Workflow of miRNA Digger. miRNA Digger is free for download from http://www.bioinfolab.cn/miRNA_Digger/index.html [Citation29].](/cms/asset/dd6c3ed7-6267-4486-951c-fd86b8ff5e28/tbbb_a_1536513_f0001_oc.jpg)
In this study, 12 sRNA high-throughput sequencing (HTS) data sets from six tissues of cotyledon stage soybean seed were input into miRNA Digger for miRNAs mining, followed by the expression specificity analysis and target identification. A number of novel soybean miRNAs and their targets were identified and some miRNAs registered in miRBase (Release 21) were reevaluated.
Materials and methods
Data sources
The Williams 82 soybean genome sequences, transcripts sequences and annotation information were retrieved from Phytozome. The sRNA HTS data (Supplementary Table S1a) and the degradome sequencing data libraries (GSM647200,GSM825574, GSM848963,GSM848964,GSM848965,GSM848966 and GSM848967) employed for miRNA screen were public available data sets from GEO (Gene Expression Omnibusncbi.nlm.nih.gov/geo/).
Pre-treatment of high-throughput sequencing data
In order to enable the cross-comparison between different libraries, all the HTS data sets were employed for pre-treatment. The pre-treatment procedure discarded the short sequences containing ‘N’s or with ‘0ʹ read counts, and normalized the read count of each short sequence with reads per million (PRM) as described previously [Citation29].
Genome-scale screen of miRNA-miRNA* duplexes and their corresponding precursors
miRNA Digger reports miRNA-miRNA* duplexes and their corresponding precursors based on the following five filtering procedures: (1) The genomic sequences were subdivided into 10,000-nt fragments with 500-nt overlaps. (2) The degradome tags are then mapped onto both sense and antisense strands of those 10,000-nt fragments to obtain the degradome signatures and their locations. The averaged read count (in RPM) of a degradome signature which mapped to a potential cleavage site from one library should be ‘five times or more [Citation30,Citation31] than the surrounding degradome tags, and it also should be among the top 12 most abundant degradome tags that perfectly matched to the corresponding 10,000-nt fragment in the same library [Citation32]. (3) The length of the longest pre-miRNA (the parameter “L”) was set as 700nt. And based on a single potential cleavage locus, two pre-miRNA candidates of 730 nt in length were obtained as pre-miRNA candidates, as described previously [Citation29]. (4) The pre-miRNA candidates were further employed for secondary structure prediction and followed by base complementarity analysis. Two 30nt possible “Stem” structure regions of a single miRNA precursor candidates around the cleavage locus were analyzed, and the miRNA precursor candidates with hairpin-like structures and possess complementarities higher than 70% were retained. (5) The sRNAs from HTS data sets were then mapped onto the miRNA precursor candidates and retained the potential pre-miRNAs with detectable miRNA and miRNA* clusters. The most abundant sRNA within a miRNA cluster was treated as mature miRNA, and the corresponding miRNA* was extracted based on the structurally featured 2-nt 3ʹ overhangs of the miRNA-miRNA* duplex [Citation31]. The most abundant isomiR* was treated as the counterpart of mature miRNA, if no canonical miRNA* is detected within the miRNA* cluster, which might causes by the rapid degradation of miRNA* after DCL1-mediated processing.
Expression analysis of miRNAs across different tissues
Twelve sRNA HTS data sets (Supplementary Table S1a) were employed for expression analysis. There were two duplicated data sets for every tissue sample, therefore, the average abundant (PRM) of a single miRNA was calculated and used for analysis. The log2 transformed values of abundance were displayed as heat maps. A previous reported toolkit: Heml, was utilized for heatmap profiling the differential expressions of miRNAs [Citation33].
Target prediction and identification
The potential targets of miRNAs and ta-siRNAs were predicted by using miRU algorithm, which could thoroughly search for potential complementary sites on cDNA sequences with mismatches tolerable when a miRNA/ta-siRNA sequence was given, with default parameters [Citation34]. Then, in order to get the genuine regulation of “miRNAs/ta-siRNA–target”, the degradome sequencing data (or Parallel Analysis of RNA Ends (PARE) libraries) were employed for verification of miRNA/ta-siRNA mediated cleavage sites on the predicted targets. Only when the cleavage signal resides within the middle (10 to 11nt away from the 5ʹ-end) of miRNA/ta-siRNA binding site, can the target be considered as the real target of the miRNA/ta-siRNA. Finally, target plots (T-plots) were performed to visualize the match results of the degradome data on targets [Citation35–Citation38].
PhasiRNA identification
Phased duplexes with 2-nt overhang at 3ʹ-end and 21–22 nt length of the target transcripts were computationally predicted by “phase processing” in double strand form from the miRNA-mediated cleavage site (termed as register 1) and other forward registers. Then each of these phased duplexes was separated into a plus siRNA and a minus siRNA, and followed by read count searching in the 12 sRNA HTS data sets. PhasiRNA searching was performed using a five-cycle sliding window, the phasiRNA production region should contain at least 5 tandem “processing” duplexes, and each of these duplexes should contain at least one phasiRNA [Citation39].
Results
Identification of novel miRNAs in soybean seeds
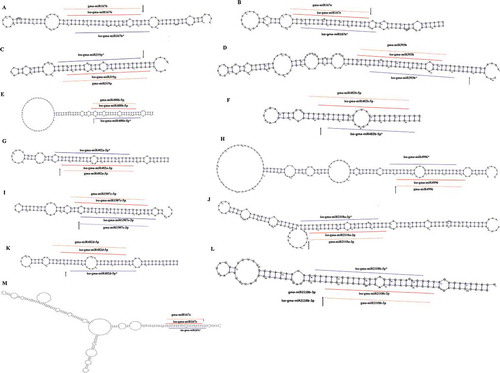
In order to discover miRNAs, the soybean genome (version 1.1) and 12 sRNA HTS libraries from six tissues: Embryo Proper (EP), Embryo Proper Cotyledon(EPC), Seed Coat Epidermis (SCE), Seed Coat Hilum (SCH), Seed Coat Inner Integument (SCII) and Seed Coat Outer Integument (SCOI) of soybean at cotelydon-stage (Supplemental Table S1a) were input into our previously constructed bio-software, miRNA Digger [Citation29]. Five filtering steps were automatically implemented to screen hairpin structure miRNA precursors containing both degradome-supported Dicer cropping sites and detectable miRNA-miRNA* duplexes (see details in Materials and methods). As a result, miRNA Digger reported totally 62 miRNA-miRNA* duplexes and their corresponding precursors (Supplemental Table S1b,c,d). Among them, 16 novel miRNAs were discovered from 16 novel precursors ( and Supplemental Table S1b). All these novel pre-miRNAs were classic “stem-loop”, and each of them possessed at least one degradome-supported DCL1 cleavage signal at the boundary of the miRNA-miRNA* duplex loci. Interestingly, another novel miRNA, gam-miR482a-5p.2, was identified from different locus on the same precursor of gam-miR482a-5p and gam-miR482a-3p which had been registered in miRBase ( and Supplemental Table S1b). Recently, Zabala et al. reported that gma-miR482a-3p represented higher expression levels than gma-miR482a-5p in various tissues of soybean [Citation40]. Besides, gma-miR482a-3p was found to be conservatively expressed in several legume and non-legume dicots, to target resistance gene receptor kinases related to defense responses and to play important role during initial stages of nodule organogenesis [Citation41,Citation42]. However, in our study, the expression analysis showed that the novel gam-miR482a-5p.2 represented a significantly high expression count in all six different tissues of soybean seed while gam-miR482a-5p and gam-miR482a-3p were almost unexpressed in all the six tissues (). Besides, 31 known miRNAs (Supplemental Table S1d) and 14 isomiRs, the variants of microRNAs,( and Supplemental Table S1c) were also identified by miRNA Digger in this study.
Figure 2. Novel miRNA-miRNA* duplexes discovered in soybean. A-P: The newly identified miRNAs and miRNA*s (marked with lines) were mapped to the pre-miRNAs. The degradome-supported DCL1-mediated cleavage signals were marked with arrows.
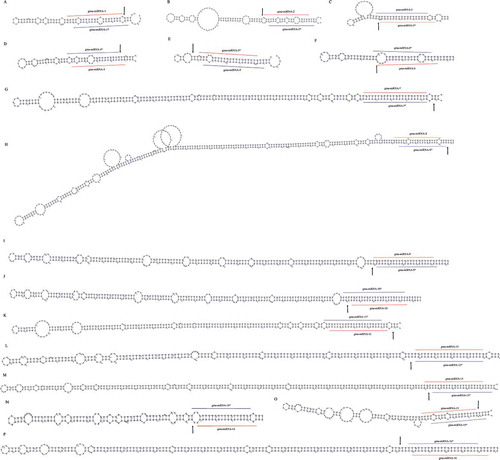
Figure 3. Novel miRNA, gam-miR482-5p.2, uncovered from known pre-miRNA. A: The newly identified gam-miR482-5p.2/gam-miR482-5p.2* (marked with lines) and miRBase registered gam-miR482-5p/gam-miR482-3p (marked with dotted lines) were mapped to the pre-miRNA. The degradome-supported DCL1-mediated cleavage signal was marked with arrow. B: Expression of gam-miR482-5p.2, gam-miR482-5p.2*, gam-miR482-5p and gam-miR482-3p in six tissues.
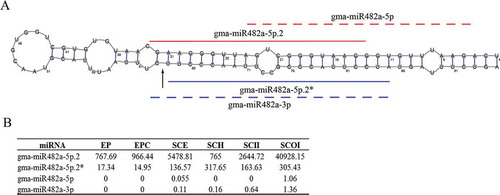
Figure 4. isomiRs uncovered in known precursors. The miRNA Digger discovered isomiRs/isomiRs* (marked with lines) and miRBase registered miRNAs counterparts (marked with dotted lines)were mapped to the pre-miRNAs. The degradome-supported DCL1-processing sites were marked with arrow.
Expression analysis of novel miRNAs across different tissues
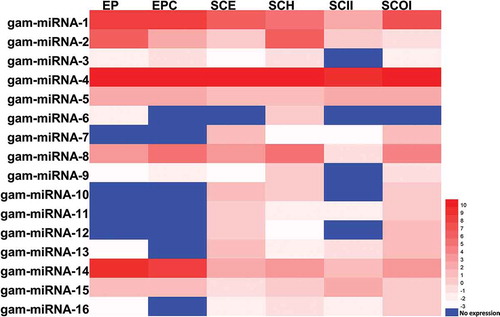
An assessment of differential expression was performed for the novel miRNAs across six tissues. The results indicated that, 16 novel miRNAs showing tissue-preferential accumulation ( and Supplemental Table S2). Most of them represent enrichment in more than two tissues; i.e., gma-miRNA-15 was observed strongly present in EP and EPC but showed low expression levels in other tissues. Gma-miRNA-5 was found strongly present in all tissues, however, it presented extremely high abundance (2900.81 RPM) in SCOI, which is 1.3 to 4.2 times of that in other tissues.
Figure 5. Expression profiling of novel miRNAs. The expression pattern of novel miRNAs in cotyledon stage across six different tissues, including: Embryo Proper (EP), Embryo Proper Cotyledon (EPC), Seed Coat Epidermis (SCE), Seed Coat Hilum (SCH), Seed Coat Inner Integument (SCII) and Seed Coat Outer Integument (SCOI) were analyzed and the log2 transformed values of abundance are displayed as a heat map.
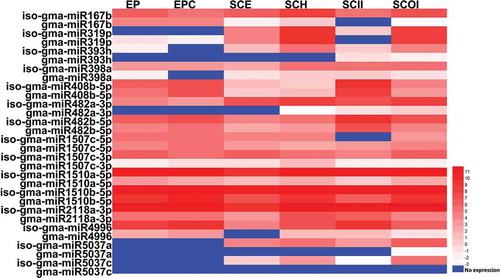
As miRNA Digger only extracted the most abundant sRNA sequence in the miRNA cluster as the mature miRNA and 14 isomiRs of known miRNAs (miRBase release 21) were reported as mature miRNAs in this study, the enrichment abundance of isomiRs and their miRNA counterparts in different tissues were also analyzed. As shown in and Supplementary Table S3, all these isomiRs represented tissue specific expression and showed significantly higher accumulation than their miRNA counterparts in different tissues, i.e., iso-gma-miR1510a-5p, iso-gma-miR1510b-5p, iso-gma-miR2118a-3p and iso-gma-miR4996 showed significantly high enrichment in all six tissues, about 6 to 1100 times of that of their miRNA counterparts (Supplementary Table S3a). Besides, in other two stages of seed development, globular and heart stage, almost all of these isomiRs present higher abundance than their miRNA counterparts (Supplementary table S3b and c). Some annotated miRNAs in miRBase showed very low enrichment in seed development stages, e.g. gma-miR393h, gma-miR482a, gma-miR1507c-3p, gma-miR5037a and gma-miR5037c ( and Supplementary Table S3). As they have high sequence similarity, isomiRs and miRNA are usually predicted to target the same mRNAs. So, the isomiRs identified in this study which had much higher expression levels may play primary regulatory roles in soybean than their miRNA counterparts.
Figure 6. Expression profiling of isomiRs and their miRNA counterparts. The expression pattern of newly discovered isomiRs and their miRBase-registered mature miRNA counterparts in cotyledon stage across six different tissues, including: Embryo Proper (EP), Embryo Proper Cotyledon (EPC), Seed Coat Epidermis (SCE), Seed Coat Hilum (SCH), Seed Coat Inner Integument (SCII) and Seed Coat Outer Integument (SCOI) were analyzed and the log2 transformed values of abundance are displayed as a heat map.
miRNA target verification using PARE libraries
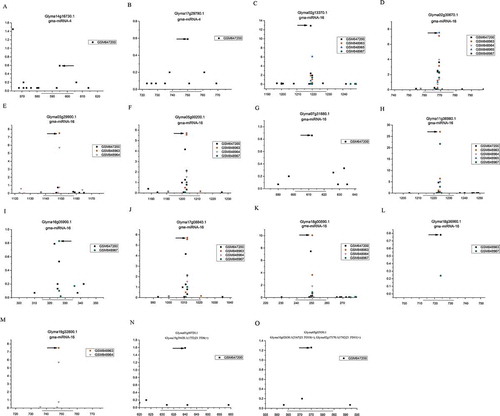
All the novel miRNAs were employed for target prediction using miRU algorithm, followed by degradome-based verification using public PARE data and T-plot profiling. The principle of target identification were described in the “materials and methods”. As a result, 2 novel miRNAs involving in 13 novel miRNA-target interaction were identified. The novel miRNAs cleave their targets all in the position of 10-11nt, which fits the characteristic of canonical miRNA very well.
As shown in and , gma-miRNA-1 targets three SQUAMOSA PROMOTER BINDING PROTEIN-LIKE6 (SPL6) genes (Glyma02g13370.1, Glyma05g00200.1 and Glyma17g08840.1), four SPL9 genes (Glyma02g30670.1, Glyma03g29900.1, Glyma18g36960.1 and Glyma19g32800.1), one SPL2 gene (Glyma11g36980.1) and one SPL3 gene (Glyma07g31880.1). SPL2 has been revealed controlling proper development of lateral organs in association with shoot maturation in the reproductive phase [Citation43], SPL3 functions in the control of flowering time and phase change [Citation44], SPL6 has been revealed essential for the resistance of pathogens and virus in Arabidopsis and Tobacco, respectively [Citation45] and SPL9 has strong effects on leaf initiation rate [Citation44]. These results imply that gma-miRNA-1 is involved in the regulation of soybean development and pathogen resistance. Gma-miRNA-7 was detected targeting two genes (Glyma14g16730.1 and Glyma17g29790.1) encoding DUF581 containing proteins. DUF581 domain has been revealed encoding plant specific FCS-Like Zinc Fingers and involving in protein-protein interaction. Many DUF581 family proteins have been identified in A. thaliana, and one of them has been revealed involving in ABA-mediated seed dormancy and induced during senescence [Citation46–Citation49].
Table 1. Targets of novel miRNAs.
Figure 7. Degradome sequencing data-based validation of the miRNA-target interactions in soybean. Five libraries of degradome sequencing data libraries (GSM647200,GSM848963,GSM848964,GSM848965 and GSM848967) were recruited for T-plot profiling. The IDs of the target transcripts and the corresponding miRNAs are listed on the top. The y axes measure the normalized reads (in RMP, reads per million) of the degradome signals, and the x axes represent the position of the cleavage signals on the target transcripts. The binding sites of the phased siRNA on their target transcripts were denoted by horizontal lines, and the dominant cleavage signals were marked by arrows.
According to the previous reports, 22-nt miRNAs have the ability to trigger the ta-siRNAs production from the cleaved targets [Citation50]. So the twelve sRNA HTS data sets were employed for 22-nt novel miRNAs/isomiRs triggered secondary ta-siRNAs identification. The results showed that only five targets of iso-gma-miR393h generated secondary phasiRNAs. Based on the modified ta-siRNA biogenesis model which we have reported previously, 135 novel phasiRNAs were discovered (Supplementary Table S4). All these phasiRNAs were then employed for target identification, and 3 novel ta-siRNA-target interactions were identified ( and ).
Table 2. Targets of miRNA-triggered secondary ta-siRNAs.
The ta-siRNA, Glyma19g39420.1 (1752)21 3ʹD8(+), was found targeting a gene (Glyma01g00720.1) which encoding a regulator of Vps4 activity in the Multivesicular Body (MVB) pathway. In recent years, the MVBs have been revealed playing important roles in mediating protein traffic and plant immune responses in plants [Citation51]. Although the regulatory mechanism of Vps4 and MVBs in soybean are not well studied, however, Shahriari et al. revealed that the homologous of Vps4 in Arabidopsis, AtSKD1, related to the cell surviving during seed development [Citation52]. Therefore, we speculated that Glyma19g39420.1(1752)21 3ʹD8(+) might involving in regulation of mediating protein traffic, pathogen response and cell surviving during seed development of soybean.
Another two ta-siRNAs, Glyma10g02630.1 (2167)21 3ʹD10(+) and Glyma02g17170.1(1742)21 3ʹD11(+), were found targeting a Class I peptide chain release factor gene (Glyma05g03930.1), indicating that they are both play important roles for cell viability [Citation53].
Discussion
Soybean is an important economic crop. In recent years, a number of miRNAs in soybean have been revealed playing crucial regulatory roles in organ development [Citation53], nutrient signaling [Citation45], biotic and abiotic stress responses. In this study, a previously constructed bio-software, miRNA Digger, was used for miRNA mining from 12 public available sRNA data sets. As a result, 17 novel miRNAs were discovered. Two of them were further identified involving in 13 novel miRNA-target interactions and might play important roles in regulation of protein–protein interaction, stress response, plant development, plant proliferation and differentiation.
In addition, 14 isomiRs rather than their miRNA counterparts were discovered in this study. Recently, literatures revealed that, a single pre-miRNA can generate a series of miRNA variants (termed as isomiRs) with various length and 5ʹ or 3ʹ ends [Citation54,Citation55], and different variants of the same miRNA accumulated divergently in different tissues and development stages. However, the isomiRs uncovered in this study represented significantly higher abundance than those registered in miRBase (Release 21) in all three stages of seed development (Supplementary Table S3), which indicated that these isomiRs might play primary regulatory roles than their miRNA counterparts in soybean. Although numerous miRNAs from variant species have been discovered and registered in miRBase, some incorrect annotation of miRNAs in miRBase have been reported [Citation1,Citation56,Citation57]. So, using different methods to reevaluate the annotated miRNAs is necessary and important for fully understanding of the miRNA family and their functions.
The 22-nt size class miRNAs have been drawn a great attention in recent years because of their potential ability of triggering the production of secondary ta-siRNAs which have multiple biological functions [Citation12,Citation58,Citation59]. Therefore, all the target transcripts of 22-nt miRNAs/isomiRs were employed for secondary ta-siRNAs identification. However, except those of iso-gma-miR393h, the other target transcripts could not enter into the synthesis pathway for secondary ta-siRNA generation. This anomaly indicates that the current ta-siRNA triggering models are imperfect and needs to be further investigated.
Author contributions
L.Y. accomplished the identification of novel miRNAs and phasiRNAs; R.G. accomplished miRNAs expression analysis and miRNAs/ta-siRNAs target verification. L.Y. and R.G. wrote the main manuscript. L.Y, R.G, Y.M. and C.S. designed the experiments. Y.J., X.Y. and Z.Y. contributed to collection and pre-treatment of the data, and preparation of supplemental files. Y.M. and C.S. revised the manuscript. All of the authors reviewed the manuscript.
Table_S4.pdf
Download PDF (47.6 KB)Table_S3c.pdf
Download PDF (30.6 KB)Table_S3b.pdf
Download PDF (30.5 KB)Table_S3a_.pdf
Download PDF (33.7 KB)Table_S2.pdf
Download PDF (24.4 KB)Table_S1_d.pdf
Download PDF (52.9 KB)Table_S1_c.pdf
Download PDF (33 KB)Table_S1_b.pdf
Download PDF (38.8 KB)Table_S1_a.pdf
Download PDF (16.9 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Arikit S, Xia R, Kakrana A, et al. An atlas of soybean small RNAs identifies phased siRNAs from hundreds of coding genes. Plant Cell. 2014 Dec;26(12):4584–4601.
- Liu W, Yu W, Hou L, et al. Analysis of miRNAs and their targets during adventitious shoot organogenesis of Acacia crassicarpa. PLoS One. 2014 Apr;9(4):e93438.
- Axtell MJ, Bowman JL. Evolution of plant microRNAs and their targets. Trends Plant Sci. 2008 Jul;13(7):343–349.
- Ding Y, Ma Y, Liu N, et al. microRNAs involved in auxin signalling modulate male sterility under high-temperature stress in cotton (Gossypium hirsutum). Plant J. 2017 Sep;91(6):977–994.
- Fan G, Wang L, Dong Y, et al. Genome of Paulownia (Paulownia fortunei) illuminates the related transcripts, miRNA and proteins for salt resistance. Sci Rep. 2017 Apr 28;7(1):1285.
- Sosa-Valencia G, Romero-Perez PS, Palomar VM, et al. Insights into the function of the phasiRNA-triggering miR1514 in response to stress in legumes. Plant Signal Behav. 2017 Mar;12(3):e1284724.
- Wang H, Jiao X, Kong X, et al. A signaling cascade from miR444 to RDR1 in rice antiviral RNA silencing pathway. Plant Physiol. 2016 Apr;170(4):2365–2377.
- Li JM, Zhou YR, Sun ZT, et al. Identification and profiling of conserved and novel microRNAs in Laodelphax striatellus in response to rice black-streaked dwarf virus (RBSDV) infection. Genom Data. 2015 Mar;3:63–69.
- Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005 May;6(5):376–385.
- Mallory AC, Bouche N. MicroRNA-directed regulation: to cleave or not to cleave. Trends Plant Sci. 2008 Jul;13(7):359–367.
- Dukowic-Schulze S, Sundararajan A, Ramaraj T, et al. Novel meiotic miRNAs and indications for a role of phasiRNAs in meiosis. Front Plant Sci. 2016 Jun;7:762.
- Allen E, Xie Z, Gustafson AM, et al. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005 Apr;121(2):207–221.
- Howell MD, Fahlgren N, Chapman EJ, et al. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell. 2007 Mar;19(3):926–942.
- Vaucheret H. MicroRNA-dependent trans-acting siRNA production. Sci STKE. 2005 Sep;2005(300):pe43.
- Gupta M, Bhaskar PB, Sriram S, et al. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2017 May;36(5):637–652.
- Nonogaki H. MicroRNA gene regulation cascades during early stages of plant development. Plant Cell Physiol. 2010 Nov;51(11):1840–1846.
- Flores T, Karpova O, Su X, et al. Silencing of GmFAD3 gene by siRNA leads to low alpha-linolenic acids (18:3) of fad3-mutant phenotype in soybean [Glycine max (Merr.)]. Transgenic Res. 2008 Oct;17(5):839–850.
- Li X, Xie X, Li J, et al. Conservation and diversification of the miR166 family in soybean and potential roles of newly identified miR166s. BMC Plant Biol. 2017 Feb;17(1):32.
- Li W, Wang T, Zhang Y, et al. Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J Exp Bot. 2017 Jul;68(16):4727–4729.
- Tyczewska A, Gracz J, Kuczynski J, et al. Deciphering the soybean molecular stress response via high-throughput approaches. Acta Biochim Pol. 2016 Nov;63(4):631–643.
- Bustos-Sanmamed P, Bazin J, Hartmann C, et al. Small RNA pathways and diversity in model legumes: lessons from genomics. Front Plant Sci. 2013 Jul;4:236.
- Yu D, Ma X, Zuo Z, et al. Bioinformatics resources for deciphering the biogenesis and action pathways of plant small RNAs. Rice (N Y). 2017 Dec;10(1):38.
- Ma X, Tang Z, Qin J, et al. The use of high-throughput sequencing methods for plant microRNA research. RNA Biol. 2015 Jul;12(7):709–719.
- Meng Y, Shao C, Wang H, et al. The regulatory activities of plant microRNAs: a more dynamic perspective. Plant Physiol. 2011 Dec;157(4):1583–1595.
- Yu D, Meng Y, Zuo Z, et al. NATpipe: an integrative pipeline for systematical discovery of natural antisense transcripts (NATs) and phase-distributed nat-siRNAs from de novo assembled transcriptomes. Sci Rep. 2016 Feb;9(6):21666.
- Shamimuzzaman M, Vodkin L. Identification of soybean seed developmental stage-specific and tissue-specific miRNA targets by degradome sequencing. BMC Genomics. 2012 Jul;16(13):310.
- Jones SI, Vodkin LO. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS One. 2013 Mar;8(3):e59270.
- Song QX, Liu YF, Hu XY, et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011 Jan;10(11):5.
- Yu L, Shao C, Ye X, et al. miRNA Digger: a comprehensive pipeline for genome-wide novel miRNA mining. Sci Rep. 2016 Jan;6:18901.
- Shao C, Chen M, Meng Y. A reversed framework for the identification of microRNA-target pairs in plants. Brief Bioinform. 2013 May;14(3):293–301.
- Choi SI, Maeng YS, Kim KS, et al. Autophagy is induced by raptor degradation via the ubiquitin/proteasome system in granular corneal dystrophy type 2. Biochem Biophys Res Commun. 2014 Aug;450(4):1505–1511.
- Fukunaga R, Han BW, Hung JH, et al. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell. 2012 Oct;151(3):533–546.
- Deng W, Wang Y, Liu Z, et al. HemI: a toolkit for illustrating heatmaps. PLoS One. 2014 Nov;9(11):e111988.
- Zhang Y. miRU: an automated plant miRNA target prediction server. Nucleic Acids Res. 2005 Jul;33(Web Server issue):W701–W704.
- Addo-Quaye C, Eshoo TW, Bartel DP, et al. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008 May;18(10):758–762.
- German MA, Luo S, Schroth G, et al. Construction of parallel analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc. 2009 Feb;4(3):356–362.
- Llave C, Xie Z, Kasschau KD, et al. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002 Sep;297(5589):2053–2056.
- Ma X, Shao C, Wang H, et al. Construction of small RNA-mediated gene regulatory networks in the roots of rice (Oryza sativa). BMC Genomics. 2013 Jul;14:510.
- Yu L, Meng Y, Shao C, et al. Are ta-siRNAs only originated from the cleavage site of miRNA on its target RNAs and phased in 21-nt increments? Gene. 2015 Sep;569(1):127–135.
- Zabala G, Campos E, Varala KK, et al. Divergent patterns of endogenous small RNA populations from seed and vegetative tissues of Glycine max. BMC Plant Biol. 2012 Oct;12:177.
- Bazin J, Bustos-Sanmamed P, Hartmann C, et al. Complexity of miRNA-dependent regulation in root symbiosis. Philos Trans R Soc Lond B Biol Sci. 2012 Jun;367(1595):1570–1579.
- Li H, Deng Y, Wu T, et al. Misexpression of miR482, miR1512, and miR1515 increases soybean nodulation. Plant Physiol. 2010 Aug;153(4):1759–1770.
- Shikata M, Koyama T, Mitsuda N, et al. Arabidopsis SBP-box genes SPL10, SPL11 and SPL2 control morphological change in association with shoot maturation in the reproductive phase. Plant Cell Physiol. 2009 Dec;50(12):2133–2145.
- Shao F, Lu Q, Wilson IW, et al. Genome-wide identification and characterization of the SPL gene family in Ziziphus jujuba. Gene. 2017 Sep;627:315–321.
- Padmanabhan MS, Ma S, Burch-Smith TM, et al. Novel positive regulatory role for the SPL6 transcription factor in the N TIR-NB-LRR receptor-mediated plant innate immunity. PLoS Pathog. 2013 Mar;9(3):e1003235.
- Laxmi A. DUF581 is plant specific FCS-like zinc finger involved in protein-protein interaction. PLoS One. 2014 Jul;9(6):e99074.
- He Y, Tang W, Swain JD, et al. Networking senescence-regulating pathways by using Arabidopsis enhancer trap lines. Plant Physiol. 2001 Jun;126(2):707–716.
- He Y, Gan S. A novel zinc-finger protein with a proline-rich domain mediates ABA-regulated seed dormancy in Arabidopsis. Plant Mol Biol. 2004 Jan;54(1):1–9.
- Arabidopsis Interactome MC. Evidence for network evolution in an Arabidopsis interactome map. Science. 2011 Jul;333(6042):601–607.
- Chen HM, Chen LT, Patel K, et al. 22-Nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc Natl Acad Sci U S A. 2010 Aug;107(34):15269–15274.
- Li X, Bao H, Wang Z, et al. Biogenesis and function of multivesicular bodies in plant immunity. Front Plant Sci. 2018;9:979.
- Shahriari M, Hulskamp M, Schellmann S. Seeds of Arabidopsis plants expressing dominant-negative AtSKD1 under control of the GL2 promoter show a transparent testa phenotype and a mucilage defect. Plant Signal Behav. 2010 Oct;5(10):1308–1310.
- Pel HJ, Rozenfeld S, Bolotin-Fukuhara M. The nuclear Kluyveromyces lactis MRF1 gene encodes a mitochondrial class I peptide chain release factor that is important for cell viability. Curr Genet. 1996 Jun;30(1):19–28.
- Zhang J, Zhang S, Li S, et al. A genome-wide survey of microRNA truncation and 3ʹ nucleotide addition events in larch (Larix leptolepis). Planta. 2013 Apr;237(4):1047–1056.
- Ebhardt HA, Tsang HH, Dai DC, et al. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009 May;37(8):2461–2470.
- Lee WC, Lu SH, Lu MH, et al. Asymmetric bulges and mismatches determine 20-nt microRNA formation in plants. RNA Biol. 2015 Sep;12(9):1054–1066.
- Taylor RS, Tarver JE, Foroozani A, et al. MicroRNA annotation of plant genomes - Do it right or not at all. Bioessays. 2017 Feb;39(2):1600113.
- Chen HM, Li YH, Wu SH. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A. 2007 Feb;104(9):3318–3323.
- Vazquez F, Hohn T. Biogenesis and biological activity of secondary siRNAs in plants. Scientifica (Cairo). 2013 Feb;2013:783253.