
ABSTRACT
Microglial activation is known to be an important event during innate immunity, but microglial inflammation is also thought to play a role in the etiology of neurodegenerative diseases. Recently, it was reported that autophagy could influence inflammation and activation of microglia. However, little is known about the regulation of autophagy during microglial activation. In this study, we demonstrated that mitochondrial fission-induced ROS can promote autophagy in microglia. Following LPS-induced autophagy, GFP-LC3 puncta were increased, and this was suppressed by inhibiting mitochondrial fission and mitochondrial ROS. Interestingly, inhibition of mitochondrial fission and mitochondrial ROS also resulted in decreased p62 expression, but Beclin1 and LC3B were unaffected. Taken together, these results indicate that ROS induction due to increased LPS-stimulated mitochondrial fission triggers p62 mediated autophagy in microglial cells. Our findings provide the first important clues towards understanding the correlation between mitochondrial ROS and autophagy.
Abbreviations: Drp1; Dynamin related protein 1, LPS; Lipopolysaccharide, ROS; Reactive Oxygen Species, GFP; Green Fluorescent Protein, CNS; Central Nervous System, AD; Alzheimer’s Disease, PD; Parkinson’s Disease, ALIS; Aggresome-like induced structures, iNOS; inducible nitric oxide synthase, Cox-2; Cyclooxygenase-2, MAPK; Mitogen-activated protein kinase; SODs; Superoxide dismutase, GPXs; Glutathione Peroxidase, Prxs; Peroxiredoxins
Graphical Abstract
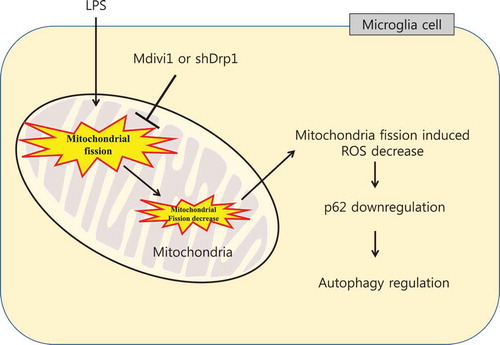
The mechanistic cascade between LPS-induced mitochondrial fission and autophagy in BV-2 microglial cells.
Microglia are immune cells that reside in the brain and play important roles in the central nervous system (CNS) to maintain homeostasis through an immune response [Citation1,Citation2]. Proper activation of microglia is beneficial for neuronal maintenance, but excessive activation of microglia can damage neurons. Therefore, a microglia-mediated inflammatory response is regarded as a common and early hallmark of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) [Citation3]. Therefore, the precise mechanisms that regulate microglial activation may be crucial for developing new neurodegenerative therapies.
Autophagy is the degradation and recycling system within cells, where defective organelles or pathogens are enveloped into double-membrane vesicles and fuse with lysosomes [Citation4]. In response to stress, the autophagy pathway plays a critical role in eliminating dysfunctional organelles or proteins. A growing number of studies suggest that autophagy is associated with controlling the activation and survival of immune cells, such as macrophages and microglia [Citation5,Citation6]. However, the exact mechanism by which autophagy acts during microglial activation is still unknown. Autophagy consists of two types: canonical autophagy, such as macro- and microautophagy, and chaperone-mediated autophagy. Microtubule-associated protein light chain 3 (LC3), Beclin1, and p62 (also known as SQSTM1) are critical for the regulation of canonical autophagy processes [Citation7,Citation8]. p62 is also required for non-canonical autophagy, and exhibits selective autophagy of aggresome-like induced structures (ALIS) [Citation9]. Previous studies have shown that p62 is increased in activated microglia and macrophages [Citation9,Citation10]. However, the precise p62-mediated autophagic regulatory mechanism that exists in microglial cells is not well elucidated.
Mitochondria are important organelles that produce energy for cells, which can be used to regulate cellular metabolism. Mitochondria undergo dynamic changes in form, through lengthening by fusion and shortening by fission [Citation11]. Mitochondrial dynamics contribute to the regulation of numerous cellular functions such as cell division, biogenesis, turnover and cell death [Citation12]. In addition, mitochondrial dynamics are associated with oxidative stress by controlling mitochondrial ROS generation [Citation13]. Therefore, an imbalance in mitochondrial dynamics can affect many sites of mitochondrial function. Recent studies have also demonstrated that mitochondria-derived reactive oxygen species (ROS) act as modulators in innate immunity through the regulation of MAPK activation and pro-inflammatory gene expression [Citation14,Citation15]. Furthermore, we previously demonstrated that mitochondrial dynamics are important for mediating microglial activation by regulating mitochondrial ROS, induced by lipopolysaccharide (LPS)-mediated excessive mitochondrial fission [Citation16,Citation17]. In addition, previous studies suggest that mitochondrial ROS contributes to modulation of the autophagy process [Citation18]. However, the relationship between mitochondrial fission and autophagy in LPS-induced activated microglial cells has not yet been fully elucidated.
In the present study, we investigated the mechanistic relationship between LPS-induced mitochondrial fission and autophagy in BV-2 microglial cells. Furthermore, we determined the effect of mitochondrial ROS resulting from LPS-induced mitochondrial fission on autophagy.
Materials and methods
Cell culture and treatment
BV-2 murine microglial cells were immortalized by infection with v-raf/c-myc recombination retroviruses [Citation19], which were kindly provided by Dr. Jau-Shyong Hong (National Institute of Environmental Health Sciences, NC, USA). BV-2 cells were cultured in DMEM (Welgene, Daegu, Korea) containing 10% FBS (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (Welgene). Cells were incubated at 37°C in a humidified 5% CO2 incubator (Sanyo, Osaka, Japan). BV-2 cells were pre-treated with Mdivi-1 (Sigma, St. Louis, MO, USA) and Mito-TEMPO (Sigma) for 1 h followed by stimulation with LPS from E. coli serotype O26:B6 (Sigma). The cells were treated with 100μM Chloroquine (CQ) (Sigma) for 4h before sampling.
Cloning and generation of lentivirus
A pEGFP-LC3 plasmid purchased from Addgene (Boston, MA, USA). The GFP-LC3 gene was amplified by PCR with LA Taq polymerase (Takara, Shiga, Japan), cloned into the pCR8/GW/TOPO vector (Invitrogen, Carlsbad, CA, USA), then inserted into the pLenti6.3/V5-DEST vector (Invitrogen) using LR clonase (Invitrogen). The Drp1 shRNA plasmid (TCR number: TRCN0000301169) was purchased from Sigma. Construction of the lentivirus was performed as previously described [Citation20].
Establishment of stable cell lines
shDrp1 and GFP-LC3 stable cell lines were established by infecting BV-2 cells with the lentivirus (MOI = 5) together with 8 μg/mL of polybrene, which increases the efficiency of lentivirus infection in mammalian cells. GFP-LC3 lentivirus-transduced BV-2 cells were cultured for 72 h, and then selected with 4 μg/mL blasticidin (Invitrogen) for several days. shDrp1 lentivirus-transduced BV-2 cells were selected using 8 μg/mL of puromycin (Sigma).
GFP-LC3 puncta formation assay
The number of GFP-LC3 puncta was determined using DE/DMI 3000B fluorescence microscopy (LEICA, Ernst-Leitz-Strasse, Wetzlar, Germany). The percentage of cells containing GFP-LC3 puncta was calculated by counting the number of the cells with GFP-LC3 fluorescence among total GFP-LC3 positive cells. A minimum of 100 cells from 4 randomly selected fields were scored using ImageJ software (NIH, Bethesda, MD, USA).
Western blotting
Protein lysates were extracted using PRO-PREP solution (Intron Biotechnology, Seongnam, Korea). Equal amounts of protein lysates were separated by electrophoresis on SDS-PAGE and transferred onto a nitrocellulose membrane (Pall Corporation, Port Washington, NY, USA). Membranes were blocked with skimmed-milk (BD Biosciences, Franklin Lakes, NJ, USA) and incubated overnight with primary antibodies against anti-Beclin1 (Abgent, CA, USA), anti-LC3B, anti-p62, anti-iNOS (Cell Signaling Biotechnology, Danvers, MA, USA), anti-Cox-2, anti-β-actin, anti-Drp1 (Santa Cruz Biotechnology Inc., TX, USA) at 4°C. Membranes were washed with TBST then incubated with horseradish peroxidase-conjugated secondary antibodies (Thermo Scientific, Waltham, MA, USA). After removal of excess antibodies by TBST washing, specific binding was detected using a chemiluminescence detection system (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions.
Measurement of mitochondrial ROS levels and membrane potential
LPS induced mitochondrial ROS was assessed using MitoSOX (Invitrogen) and measurement of membrane potential was using JC-1 (Invitrogen). Trypsinized cells were incubated with 5μM MitoSOX and JC-1 at 37℃ for 15min, washed with PBS, and then analyzed with flow cytometer (BD Biosciences)
Statistical analysis
Data represent the mean and SD of three independent experiments (n = 3). Differences between experiments were tested for statistical significance using one-way and two-way ANOVA with GraphPad Prism software. A p-value of < 0.05 was deemed statistically significant and is indicated in the figures by an asterisk. P-values of < 0.01 and < 0.001 are indicated by two and three asterisks, respectively.
Results
LPS induces autophagy in BV-2 microglial cells
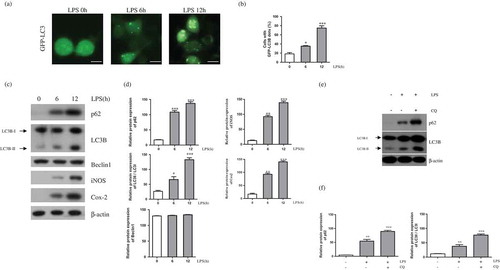
To determine whether LPS induces autophagy in BV-2 microglial cells, we generated GFP-LC3 stable expressing BV-2 cells. We treated GFP-LC3 expressing BV-2 cells with LPS for 0, 6, or 12 h. The number of GFP-LC3 puncta was significantly increased after LPS treatment for 6 h ()). It is well known that Beclin1 binds to LC3B-II, the converted form of LC3B-I, and interacts with p62 during autophagy initiation and autophagosome formation [Citation21]. We therefore measured levels of Beclin1, LC3B, and p62 protein expression following LPS treatment. Levels of iNOS and Cox-2 protein were used as markers of an LPS-induced inflammatory response, and both were increased in this system following LPS treatment ()). In addition, LC3B-II and p62 levels were significantly increased, whereas Beclin1 expression did not change. We also measured autophagy flux to confirming inducing effect of LPS on autophagic activity in BV-2 cells ()). Western blot analysis indicated that p62 and LC3B-II protein was increased after treatment of lysosome inhibitors chloroquine. These findings demonstrate that autophagy was induced in LPS-mediated activated microglial cells through increased LC3B-II and p62 expression.
Figure 1. LPS induces autophagy in BV-2 cells. (a) GFP-LC3 puncta were observed in GFP-LC3 expressing BV-2 cells treated with LPS (1μg/mL) for the indicated times (0, 6, and 12 h) using a fluorescence microscope. Scale bars represent 200 μm. (b) Graph showing number of GFP-LC3 puncta per cell. (c) Western blot analysis of p62, LC3B, Beclin1, iNOS, and Cox-2 in LPS treated BV-2 cells for 6 and 12 h. (d) Graphs represent quantification of western blot band intensity. (e) Western blot analysis to measure autophagic flux. LPS stimulated BV-2 cells were treated 4h before the cell harvest with chloroquine (100μM). (f) Graphs represent quantification of western blot band intensity. (b), (d) and (f) used one way ANOVA statistical analysis. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
LPS-induced autophagy is decreased by inhibiting mitochondrial fission
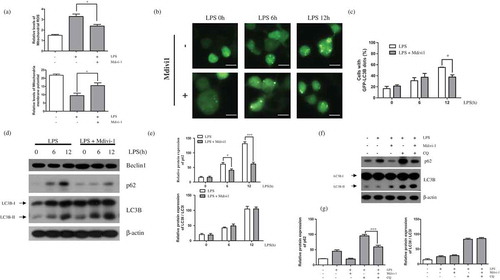
We then examined whether LPS-induced autophagy in BV-2 cells is involved in mitochondrial fission using Mdivi-1, an inhibitor of mitochondrial fission. We previously showed that Mdivi-1 successfully prevented LPS-induced mitochondrial fission in BV-2 cells [Citation16]. First, we confirmed capacity of Mdivi-1 that downregulates mitochondrial ROS and rescue effect of mitochondrial membrane potential through inhibition of mitochondrial fission in LPS stimulated condition ()). Next, our results from the present study showed that the LPS-induced increase in GFP-LC3 puncta formation was suppressed by Mdivi-1 treatment at 12 h ()). In addition, we confirmed the effect of mitochondrial fission inhibition on the LPS-induced increase in expression of LC3B-II and p62, p62 was decreased following Mdivi-1 treatment, but LC3B-II and Beclin1 were unchanged ()). We can also confirmed the effect of Mdivi-1 on autophagic flux ()). In contrast with LC3B, p62 didn’t accumulate after treatment of chloroquine with Mdivi-1, indicating that Mdivi-1 suppressed the basal level of autophagic flux through p62 in BV-2 cells.
Figure 2. Effect of LPS-induced mitochondrial fission on autophagy. (a) BV-2 cells were treated with LPS (1μg/mL) for 12h in the absence or presence of Mdivi-1 (25μM). The cells were incubated with MitoSOX and JC-1 for 15min at 37℃ and mitochondrial ROS and membrane potentials were analyzed by flow cytometry. (b) GFP-LC3B puncta were observed in GFP-LC3 expressing BV-2 cells treated with LPS for indicated times, in the presence or absence of Mdivi-1 using a fluorescence microscope. Scale bars represent 200 μm. (c) Graph indicates numbers of GFP-LC3 puncta per cell. (d) p62, LC3B, and Beclin1 expression in LPS-treated BV-2 cells with or without Mdivi-1 pre-treatment was determined using western blotting. (e) Graphs represent quantification of western blot band intensity. (f) Western blot analysis to measure authphagic flux. LPS stimulated BV-2 cells were treated 4h before the cell harvest with chloroquine (100μM) in the absence or presence of Mdivi-1. (g) Graphs represent quantification of western blot band intensity. (a), (g) used one way ANOVA and (c), (e) used two way ANOVA statistical analysis. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
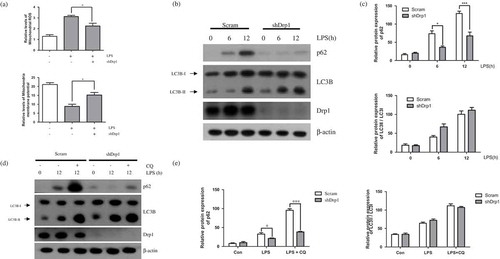
Next, we sought to determine whether LPS-induced mitochondrial fission affects p62-related autophagy using shRNA-mediated knockdown of Drp1 (shDrp1), a regulatory protein of mitochondrial fission [Citation22]. Like as Mdivi-1, we measured capacity of shDrp1 on mitochondrial ROS and membrane potential ()). Drp1 downregulated BV-2 cells indicated that suppression of mitochondrial ROS and alleviation of mitochondrial membrane potential in LPS stimulated conditions. Our data showed that the increased expression of p62 by LPS stimulation was reversed in shDrp1-expressing BV-2 cells, but that LC3B was not affected by shDrp1 treatment ()). Furthermore, accumulation of p62 also decreased after treatment of chloroquine but not LC3B ()). This data also support that downregulation of LPS-induced mitochondrial fission regulates autophagic flux through p62. These findings indicate that suppression of mitochondrial fission can regulate LPS-induced p62-related autophagy in BV-2 microglial cells.
Figure 3. Effect of Drp1 downregulation on LPS-induced autophagy. (a) BV-2 cells were treated with LPS (1μg/mL) for 12h in the absence or presence of Drp1. The cells were incubated with MitoSOX and JC-1 for 15min at 37℃ and mitochondrial ROS and membrane potentials were analyzed by flow cytometry. (b) Western blotting for p62 and LC3B was performed in LPS treated BV-2 cells expressing either scrambled shRNA (scram) or shDrp1 expressing BV2 cells. (c) Graphs represent quantification of western blot band intensity. (d) Western blot analysis to measure authphagic flux. LPS stimulated BV-2 cells were treated 4h before the cell harvest with chloroquine (100μM) in the absence or presence of Drp1. (e) Graphs represent quantification of western blot band intensity. (a) was used one way ANOVA and (c), (e) was used two way ANOVA statistical analysis. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Mitochondrial ROS trigger LPS-induced autophagy
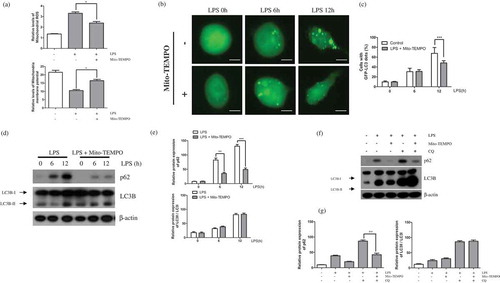
Mitochondrial fission induces mitochondrial ROS [Citation23]. Therefore, we examined the relationship between mitochondrial ROS produced by LPS-mediated mitochondrial fission and the induction of autophagy. For this, we treated BV2 microglial cells with Mito-TEMPO, a mitochondria-targeted ROS scavenger. We previously determined the concentration of Mito-TEMPO required to inhibit LPS-mediated mitochondrial ROS generation [Citation17]. As we expected, treatment of Mito-TEMPO in LPS stimulated BV-2 cells downregulates mitochondrial ROS and alleviates membrane potential ()). Here, we showed that the accumulation of LC3-GFP by LPS was reversed by pre-treatment of LC3-GFP expressing BV-2 cells with Mito-TEMPO ()). Additionally, the enhanced expression of p62 in LPS-induced activated BV-2 cells was suppressed by Mito-TEMPO treatment ()). Downregulation of mitochondrial ROS through Mito-TEMPO treatment also suppress accumulation of p62 but not LC3B in treatment of chloroquine with Mito-TEMPO ()). These results indicate that LPS-induced mitochondrial fission is key trigger of p62-mediated non-canonical autophagy through an increase in mitochondrial ROS.
Figure 4. Mitochondrial ROS affects LPS-induced autophagy. (a) BV-2 cells were treated with LPS (1μg/mL) for 12h in the absence or presence of Mito-TEMPO (200μM). The cells were incubated with MitoSOX and JC-1 for 15min at 37℃ and mitochondrial ROS and membrane potentials were analyzed by flow cytometry. (b) The effect of mitochondrial ROS on GFP-LC3 puncta formation was addressed using GFP-LC3-expressing BV-2 cells treated with LPS for indicated times in the presence or absence of Mito-TEMPO using a fluorescence microscope. Scale bars represent 200 μm. (c) Graph represents number of GFP-puncta per cell. (d) Western blot analysis of p62, LC3B, and Beclin1 expression in LPS-treated BV-2 cells in the presence or absence of Mito-TEMPO. (e) Graphs represent quantification of western blot band intensity. (f) Western blot analysis to measure authphagic flux. LPS stimulated BV-2 cells were treated 4h before the cell harvest with chloroquine (100μM) in the absence or presence of Mito-TEMPO. (g) Graphs represent quantification of western blot band intensity. (a), (g) was used one way ANOVA and (c), (e) was used two way ANOVA statistical analysis. Data are presented as mean ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
Discussion
Mitochondrial ROS plays a critical role in cellular homeostasis as a secondary messenger and is involved in numerous cellular signaling pathways [Citation24,Citation25]. However, excessive ROS production due to increased mitochondrial fission can disrupt cellular function and trigger cell death [Citation26,Citation27]. We previously suggested that mitochondrial ROS induced by LPS-stimulated mitochondrial fission is involved in pro-inflammatory responses in microglial cells [Citation16,Citation28]. Recently, many studies have suggested that mitochondrial ROS may regulate the autophagy process [Citation18,Citation29]. However, the precise relationship between mitochondrial ROS and autophagy in activated microglial cells has not been elucidated. In the present study, we investigated the correlation between mitochondrial fission and the autophagic process, and the regulatory pathway of mitochondrial fission-induced mitochondrial ROS and autophagy. Our results suggest that modulation of mitochondrial fission-induced ROS is important for controlling autophagy in microglia.
We observed that LPS-mediated activation of BV-2 microglia cells induced formation of autophagy. LPS is the most fascinating stimulus and induces autophagy in microglial cells [Citation30,Citation31], therefore we selected LPS to induce autophagy in activated microglia cells in our experiments. Induction of autophagy was confirmed by the presence of GFP-LC3 puncta and expression of Beclin1, LC3B, and p62. Consistent with earlier studies [Citation32], we found that LC3B and p62 expression levels and the number of GFP-LC3 puncta were significantly increased 12 h after LPS stimulation. These results indicate that LPS-induced inflammation causes an elevation in autophagy in microglial cells.
The mitochondrial fission related factor, Drp1 plays important role in controlling autophagy [Citation33]. Therefore, we determined the relationship between autophagy and mitochondrial dynamics in microglial cells. To do this, we observed changes in autophagy induction in LPS-induced activated microglial cells following inhibition of mitochondrial fission via Mdivi-1 or Drp1 knockdown. Our data showed that Mdivi-1 pre-treatment inhibited LPS-induced autophagy, and that the increased response to LPS was also decreased following shDrp1 expression. From these results, we suggest that modulation of LPS-mediated mitochondrial fission may affect autophagy in microglial cells.
p62 is well known for targeting particular cellular contents for canonical autophagy, but it also plays a role in non-canonical autophagy [Citation34]. Furthermore, p62 expression is increased in macrophages and microglia under autophagy-induced conditions, and is decreased following downregulation of ROS [Citation10,Citation35]. Therefore, we examined the correlation between mitochondrial ROS and p62-dependent non-canonical autophagy. Inhibition of the LPS-induced increase in mitochondrial ROS using Mito-TEMPO resulted in a decrease in both autophagy formation and p62 expression, whereas the canonical autophagy related factors, LC3B and Beclin1, were unchanged. These results suggest that mitochondrial ROS in microglial cells may have a role in regulating p62-dependent non-canonical autophagy.
LPS-induced autophagy in macrophages is regulated by the p38 MAPK pathway and induction of p62 expression is also regulated by p38 MAPK [Citation36–Citation38]. Furthermore, growing reports suggest that mitochondrial fission and mitochondrial ROS play an important role in modulating immunoreactions by regulating various signaling pathways, such MAPK [Citation16,Citation17,Citation39]. Our results indicate that LPS-induced autophagy is regulated by MAPK activation, which is controlled by mitochondrial fission-induced mitochondrial ROS. However, further investigations will be required to uncover the precise mechanism underlying p62-dependent non-canonical autophagy induction in microglia.
Mitochondrial ROS plays a pro-inflammatory role [Citation40], whereas autophagy has the characteristics of an anti-inflammatory response in activated innate immune cells [Citation41,Citation42]. In particular, inhibition of LPS-induced mitochondrial ROS by suppression of LPS-mitochondrial fission (Mdivi-1 or shDrp1) or by direct inhibition (Mito-TEMPO) downregulated p62-dependent autophagy, which is regarded as an anti-inflammatory response. Based on these findings, we suggest that autophagy may occur as a defense mechanism during excessive activation of LPS-induced microglia cells, and that mitochondrial ROS induced by mitochondrial fission is a key regulator of autophagy, where it functions to negatively regulate the excessive inflammatory response. However, more research will be required to determine the relationship between mitochondrial ROS and autophagy, and in particular, the role of intercellular antioxidant enzymes such as SODs, GPXs, and Prxs in the regulation of mitochondrial derived ROS.
Dysregulation of microglial inflammation plays a role in the pathology of neurodegenerative diseases [Citation43]. There is increasing evidence to suggest that regulation of autophagy is important in neurodegenerative diseases [Citation44]. Findings from the current study provide the first evidence to suggest the existence of a regulatory mechanism between p62-dependent autophagy and mitochondria fission-induced mitochondrial ROS. Furthermore, our findings suggest that suppression of mitochondrial fission-induced ROS could provide a useful strategy to investigate the regulation of autophagy in microglial cells, which in turn would be important for alleviating the effects of neurodegenerative diseases.
Author Contribution
Unbin Chae, Han Seop Kim, Hyun-Shik Lee, Sang-Rae Lee and Dong-Seok Lee designed the study. Unbin Chae and Han Seop Kim performed the experiments. Unbin Chae, Sang-Rae Lee and Dong-Seok Lee wrote the paper.
Disclosure statement
The authors have no conflicting interests.
Additional information
Funding
References
- Greter M, Merad M. Regulation of microglia development and homeostasis. Glia. 2013;61:121–127.
- Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263.
- Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737.
- Yang Z, Zhang N, Shen H, et al. Microglial activation with reduction in autophagy limits white matter lesions and improves cognitive defects during cerebral hypoperfusion. Curr Neurovasc Res. 2014;11:223–229.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
- Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol. 2010;12:207–209.
- Fujita K, Maeda D, Xiao Q, et al. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427–1432.
- Francois A, Terro F, Janet T, et al. Involvement of interleukin-1beta in the autophagic process of microglia: relevance to Alzheimer’s disease. J Neuroinflammation. 2013;10:151.
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013;5.
- Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251.
- Bhatt MP, Lim YC, Kim YM, et al. C-peptide activates AMPKalpha and prevents ROS-mediated mitochondrial fission and endothelial apoptosis in diabetes. Diabetes. 2013;62:3851–3862.
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402.
- Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011;208:417–420.
- Park J, Choi H, Min JS, et al. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J Neurochem. 2013;127:221–232.
- Park J, Min JS, Kim B, et al. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci Lett. 2015;584:191–196.
- Li ZY, Yang Y, Ming M, et al. Mitochondrial ROS generation for regulation of autophagic pathways in cancer. Biochem Biophys Res Commun. 2011;414:5–8.
- Blasi E, Barluzzi R, Bocchini V, et al. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237.
- Kim TS, Choi HS, Ryu BY, et al. Real-time in vivo bioluminescence imaging of lentiviral vector-mediated gene transfer in mouse testis. Theriogenology. 2010;73:129–138.
- Kang R, Zeh HJ, Lotze MT, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580.
- Ferrier V. Mitochondrial fission in life and death. Nat Cell Biol. 2001;3:E269.
- Wu S, Zhou F, Zhang Z, et al. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. Febs J. 2011;278:941–954.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990.
- Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141.
- Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer. 2014;14:709–721.
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749–762.
- Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–417.
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–540.
- Yuan H, Perry CN, Huang C, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–479.
- Chen Z, Jalabi W, Shpargel KB, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–11715.
- Han HE, Kim TK, Son HJ, et al. Activation of autophagy pathway suppresses the expression of iNOS, IL6 and cell death of LPS-stimulated microglia cells. Biomol Ther (Seoul). 2013;21:21–28.
- Kageyama Y, Hoshijima M, Seo K, et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. 2014;33:2798–2813.
- Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631.
- Pursiheimo JP, Rantanen K, Heikkinen PT, et al. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28:334–344.
- Fukata M, Chen A, Vamadevan AS, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881.
- Matsuzawa T, Fujiwara E, Washi Y. Autophagy activation by interferon-gamma via the p38 mitogen-activated protein kinase signalling pathway is involved in macrophage bactericidal activity. Immunology. 2014;141:61–69.
- Rubio N, Verrax J, Dewaele M, et al. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radical Bio Med. 2014;67:292–303.
- Kasahara E, Sekiyama A, Hori M, et al. Mitochondrial density contributes to the immune response of macrophages to lipopolysaccharide via the MAPK pathway. FEBS Lett. 2011;585:2263–2268.
- Emre Y, Hurtaud C, Nubel T, et al. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J. 2007;402:271–278.
- Jo EK, Shin DM, Choi AM. Autophagy: cellular defense to excessive inflammation. Microbes Infect. 2012;14:119–125.
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268.
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997.