
ABSTRACT
Bagremycin A and bagremycin B isolated from Streptomyces sp. Tü 4128 have activities against Gram-positive bacteria, fungi and also have a weak antitumor activity, which make them have great potential for development of novel antibiotics. Here, we report a draft genome 8,424,112 bp in length of S. sp. Tü 4128 by Illumina Hiseq2000, and identify the bagremycins biosynthetic gene cluster (BGC) by bioinformatics analysis. The putative bagremycins BGC includes 16 open reading frames (ORFs) with the functions of biosynthesis, resistance and regulation. Disruptions of relative genes and HPLC analysis of bagremycins production demonstrated that not all the genes within the BGC are responsible for the biosynthesis of bagremycins. In addition, the biosynthetic pathways of bagremycins are proposed for deeper inquiries into their intriguing biosynthetic mechanism.
Graphical Abstract
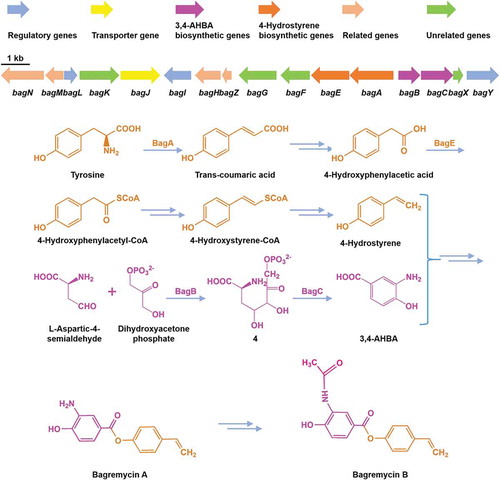
Clarification of gene cluster and pathway of bagremycins is beneficial to deeper inquiries into the intriguing biosynthetic mechanism and for novel antibiotics development.
Streptomyces spp. generally produce many natural products which have medical and industrial importance, such as antibiotics, antitumor agents, antifungal compounds, and vitamins. Over the decades, numerous natural products have been isolated and identified from various Actinobacteria strains, especially Streptomyces. However, it becomes more and more difficult to find new natural products in recent years. Moreover, due to the increasing incidence of drug-resistance, it is important to discover and identify novel antimicrobials by activating cryptic secondary biosynthetic gene clusters from known strains [Citation1–Citation3] or finding novel products from new strains [Citation4,Citation5].
Bagremycin A and bagremycin B isolated from Streptomyces sp. Tü 4128, are two novel antibiotics and show activities against Gram-positive bacteria and fungi as well as a weak antitumor activity against a human adenocarcinoma cell line [Citation6]. In particular, bagremycin A exhibits activity against fungal pathogen Candida albicans, whereas bagremycin B against necrotrophic pathogen Botrytis cinerea. Thus, bagremycins have great potentials to be applied in medicine and agriculture in the future by combinatorial biosynthesis and metabolic pathway engineering strategy. The structures of bagremycin A and bagremycin B were elucidated as 4-vinylphenyl-3-amino-4-hydroxybenzoate and 4-vinylphenyl-3-N-acetyl-4-hydroxy-benzoate respectively by X-ray and NMR spectra analysis. And, they were speculated to be derived from the condensation of para-coumaric acid and 3-amino-4-hydroxybenzoic acid (3, 4-AHBA) [Citation6].
In our previous study, we have cloned and characterized a novel tyrosine ammonia lyase-encoding gene bagA, which is involved in bagremycins biosynthesis in the S. sp. Tü 4128 strain. The intermediate para-coumaric acid could restore the bagremycin A production, but not the bagremycin B production in the bagA mtant [Citation7]. Subsequently, the other two genes bagB and bagC, which encode aldolase and 3-dehydroquinate synthase respectively, have also been cloned and identified to participate in bagremycins biosynthesis [Citation8]. In addition, a SARP family regulatory protein BagI has been proved to positively regulate bagremycins biosynthesis in the S. sp. Tü 4128 strain [Citation9]. However, the complete gene cluster and detailed biosynthetic mechanism of bagremycins are still unclear. In this study, we performed genome mining of the S. sp. Tü 4128 strain. And on this basis, we identified the bagremycins biosynthetic gene cluster (BGC) by bioinformatics analysis and genes disruption experiments, and speculated the bagremycins biosynthetic pathway.
Materials and methods
Bacterial strains and culture conditions
The bacterial strains and plasmids used in this study are listed in . S. sp. Tü 4128 is deposited as all Tü-strains in the culture collection of the Department of Microbiology/Biotechnology, University of Tübingen. The head of the Department is Prof. Dr. Wolfgang Wohlleben ([email protected]). Escherichia coli JM83 was used for preparation of the recombinant plasmids and DNA manipulation. E. coli ET12567/pUZ8002 [Citation10,Citation11] was used as a donor host for conjugal transfer into S. sp. Tü 4128. S. sp. Tü 4128 was used for genome mining and gene functional researches.
Table 1. Strains and plasmids used in this study.
E. coli strains were grown in liquid or on solid Luria-Bertani (LB) media at 37°C. S. sp. Tü 4128 and its mutants were cultured as previously [Citation7]. Briefly, they were grown in liquid YEME medium (10 g/L yeast extract, 5 g/L polypeptone, 10 g/L glucose, 3 g/L maltose, 5 mM MgCl2·2H2O, 340 g/L sucrose) or on solid SMA medium (20 g/L mannitol, 20 g/L soybean meal, 20 g/L agar) at 28ºC. ISP4 medium (10 g/L soluble starch, 1 g/L K2HPO4, 5 g/L MgSO4·7H2O, 1 g/L NaCl, 2 g/L (NH4)2SO4, 2 g/L CaCO3, 15 g/L agar, 0.001 g/L FeSO4·7H2O, 0.001 g/L MnCl2·4H2O, 0.001 g/L ZnSO47H2O, 0.02 mol/L MgCl2) containing 80 mM MgCl2 was used for conjugal transfer. Fermentation medium (10 g/L glucose, 10 g/L glycerin, 10 g/L soluble starch, 2.5 g/L corn steep liquor, 5 g/L polypeptone, 2 g/L yeast extract, 1 g/L NaCl, 3 g/L CaCO3, pH 7.2) was used for bagremycin production assays. 50 μg/mL kanamycin, 50 μg/mL apramycin, 30 μg/mL chloramphenicol and/or 25 μg/mL nalidixic acid were added into the media when necessary.
Genome sequencing, mining and sequence analysis
Genomic DNA of S. sp. Tü 4128 was extracted according to standard protocols [Citation12]. A genome sequencing was performed by BIG (Shenzhen, China) using Illumina Hiseq2000. Reads were assembled by SOAPdenovo 1.05, and the draft genome was performed prediction and functional annotation of open reading frames (ORFs) by Rapid Annotation using Subsystem Technology (RAST) [Citation13]. Furthermore, secondary metabolite gene clusters were analyzed using antiSMASH [Citation14].
Construction of gene disruption mutants
pECU2 was used to inactivate the genes as previously described [Citation8]. Briefly, two homologous arms of the relative genes to be disrupted were ligated into pECU2 to generate disruption plasmids pTDRN, pTDRM, pTDRL, pTDRK, pTDRG, pTDRF, pTDRE, pTDRX and pTDRY. E. coli ET12567/pUZ8002 containing individual disruption plasmid and S. sp. Tü 4128 were mixed and spread onto ISP4 agar plates, incubated for 16–18 h at 37ºC and then overlaid with 1 mL sterile ddH2O supplemented with kanamycin and nalidixic acid. The plates were continuously incubated at 28°C for further 3 to 4 days. Kanamycin resistant but apramycin sensitive exconjugants were screened for double crossover events by PCR and transcriptional analysis. Total DNA isolation, plasmid DNA preparations, RNA extraction and reverse transcription PCR were performed as previously described [Citation15]. Thus, we constructed various disruption mutants, TDRN (ΔbagN), TDRM (ΔbagM), TDRL (ΔbagL), TDRK (ΔbagK), TDRG (ΔbagG), TDRF (ΔbagF), TDRE (ΔbagE), TDRX (ΔbagX) and TDRY (ΔbagY). Primers for construction of bag mutants are listed in Table S1.
Construction of the bagE complementation and overexpression strains
A 1.3-kb fragment covering the coding region of bagE was amplified by PCR using the primer pair from the genomic DNA of S. sp. Tü 4128. Then, the fragment was digested with NdeI/XbaI restriction enzymes and cloned into the plasmid pIB139 [Citation16], resulting plasmid pBE139, where bagE is controlled by the strong constitutive promoter ermE*p. After propagation into E. coli ET12567/pUZ8002, pBE139 plasmid was transformed into bagE disruption mutant TDRE and wild-type strain S. sp. Tü 4128 by conjugal transfer to generate bagE complementation strain TCPE and overexpression strain TOEE, respectively. In addition, the plasmid pIB139 was also transformed into bagE disruption mutant TDRE and wild-type strain S. sp. Tü 4128, resulting in control strains TCP139 and TOE139.
Production and analysis of secondary metabolites
The S. sp. Tü 4128 wild-type and mutant strains were cultivated in 500 mL flasks with 80 mL fermentation medium at 28 °C for 15 days in a shaking incubator (190 rpm). The supernatants were collected by centrifugation at 4 °C (8000 rpm for 15 min) to measure bagremycins production. Extract and HPLC analysis of bagremycins were carried out as previously described [Citation6,Citation7]. The obtained supernatants were adjusted to pH 5 by HCl, and the flocculent precipitates were removed by centrifugation. Then, the supernatants were extracted with equal volumes of ethyl acetate three times. The organic phases were evaporated, and the residues were dissolved in 0.8 mL methanol and analysed by HPLC using a Eclipse XDB-C18 (4.6 × 250 mm) column, eluting with the mobile phase acetonitrile/0.1% phosphoric acid (2:3, v/v) at a flow rate of 1 mL/min and employing UV detection at 280 nm.
Nucleotide sequence accession number
The nucleotide sequences of the bagremycins biosynthetic gene cluster from S. sp. Tü 4128 has been submitted to the GenBank database under accession number MH697666.
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession QTSY00000000. The version described in this paper is version QTSY01000000.
Results
Sequencing and genome mining of S. sp. Tü 4128
In our previous study, we have cloned and sequenced the bagremycin biosynthetic genes bagA, bagB and bagC by high-efficiency thermal asymmetric interlaced (HE-TAIL) PCR and genomic walking kit [Citation7,Citation8]. However, when we tried to carry on cloning the flanking fragments, difficulties and problems came, that maybe due to the high G + C content, complicated secondary structure, or repeated sequence, and we could not obtain the flanking sequence. Thus, we chose genome sequencing by Illumina sequencing technology, and obtained a draft genome of S. sp. Tü 4128.
As a result, 84 scaffolds were composed from 717 contigs using Velvet assembler. The N50 of the scaffolds was 0.5 Mb and the largest one was 1.0 Mb. Combination of scaffolds resulted in a estimated draft genome sequence consisted of 8,424,112 bp, with a GC content of 72.1%. Genome analysis predicted 7,412 ORFs with an average length of 941 bp.
Sequence analysis by anti SMASH predicted that 25 biosynthetic gene clusters for secondary metabolism are exist in the genome of S. sp. Tü 4128, including polyketides (PKs, pks1-pks3), non-ribosomal peptides (NRPs, nrps1-nrps6), terpenes (terp1-terp6), bacteriocins (bcin1-bcin2), siderophores (sid1-sid2) and others (but, ect, lant, mel, nrps-mel and oth) (Table S2). Some of the clusters are highly homologous to the known ones, while the others are uncharacterized, indicating the potential to find novel bioactive products.
Identification of bagremycins biosynthetic gene cluster
To further identify the bagremycins biosynthetic gene cluster, we scanned the known sequences of bagA/bagB/bagC genes from genome of S. sp. Tü 4128, and found the target on scaffold 69, which consists of 505 kb and contains 434 ORFs. Sequence analysis demonstrated that 13 genes besides bagA/bagB/bagC are speculated to be involved in bagremycins biosynthesis, the combination of which was named bagremycins biosynthetic gene cluster (bag BGC) (). In addition, amino acids of 7 ORFs and 5 ORFs, which are located on the two sides of the flanking region of bag BGC respectively, are homologous to the clustered sequences from various Streptomyces, indicating that they may be participated in primary metabolism and do not belong to the bag BGC.
Figure 1. Genetic organization of bagremycins biosynthetic gene cluster (BGC). The proposed BGC contains 11 putative biosynthetic and regulatory genes, 1 resistance gene and 4 unrelated genes.
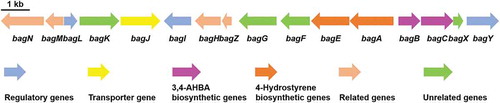
The bag BGC spans a DNA region of 17,321 bp and contains 16 ORFs, which is almost identical to the ferroverdin biosynthetic gene cluster (fer BGC, Genbank accession no. MG708299.1 and MG564782.1) (Table S3). Structural analysis revealed that ferroverdin seemed be compounded by three bagremycins as precursors [Citation17,Citation18]. However, the sequence of fer BGC was directly submitted to NCBI database and there was virtually no reporting on the research of its biosynthetic genes and pathway. Within the bag GBC, bag A, encoding a tyrosine ammonia lyase, has been proved to be participated in biosynthesis of the intermediate trans-coumaric acid [Citation7]; bag B and bagC, encoding a aldolase and a 3-dehydroquinate synthase respectively, have been proved to be participated in biosynthesis of the intermediate 3,4-AHBA [Citation8]; bagJ, a transporter encoding gene, is responsible for pumping out the synthetic bagremycins from intracellular [Citation19]; and bagI, a SARP-family transcriptional activator encoding gene, is responsible for positively regulating the biosynthesis of bagremycins [Citation9]. Beyond these genes, the others are still unidentified through experiments. We firstly predicted the function of the coding products by protein sequence alignments (), and found another two regulators are existed in the bag BGC, a MarR-family transcriptional regulator encoded by bagL and a LuxR-family transcriptional regulator encoded by bagY. In addition, the bag BGC also contains two decarboxylase encoding genes bagN and bagM, two putative FAD-dependent oxygenase encoding genes bagK and bagG, a O-aminophenol oxidase encoding gene bagH, one copper chaperon encoding gene bagZ, a putative phospho-2-dehydro-3-deoxyheptonate aldolase encoding gene bagF, a phenylacetate-coenzyme A ligase bag E, and a hypothetical protein encoding gene bagX.
Table 2. Proposed functions of ORFs within the bagremycins biosynthetic gene cluster.
Verification of the bagremycins biosynthetic genes by gene disruption
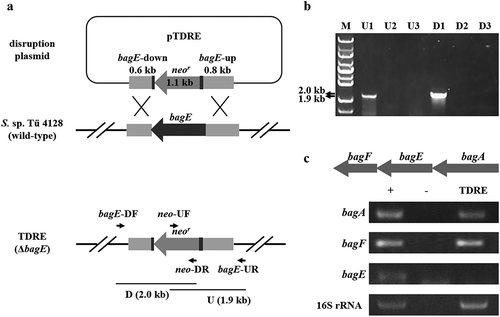
Furthermore, to figure out whether these genes within bag BGC are relative to the biosynthesis of bagremycins or not, we performed individual gene disruption by homologous recombination. Taking the disruption of bagE as an example (), the internal region of bagE was replaced by a neomycin resistance cassette to construct a bagE mutant TDRE. After resistant screening, the conjugants were identified by PCR. Only when double crossover happened, 1.9 kb U and 2.0 kb D fragments could be amplified by using the genome DNA as template (,)). The obtained mutant was finally confirmed by semi-quantitative reverse transcription and polymerase chain reaction (sqRT-PCR), and the result showed that transcription of bagE was completely hindered in the mutant TDRE, while the transcriptions of the upstream and downstream genes bagA and bagF were not affected, indicating the bagE gene was successfully disrupted ()). The mutants of other genes were also obtained by this method and identified by PCR (Figure S1).
Figure 2. Construction of bagE disruption mutant TDRE. (a) Schematic representation of bagE disruption. The internal region of bagE was replaced by neomycin resistance gene (neor). (b) Identification of bagE mutant TDRE by PCR. Line M indicated the DNA molecular weight marker. Line U and D indicated PCR products of the fragments containing the bagE-upstream and bagE-downstream regions, respectively. 1, 2 and 3 represent three conjugants selected by resistant screening and are used for PCR identification. c Transcriptional analysis of bagE and its flanking genes in the wild-type strain S. sp. Tü 4128 (+) and the bagE mutant TDRE. 16S rRNA was used for internal control. – indicated the negative control.
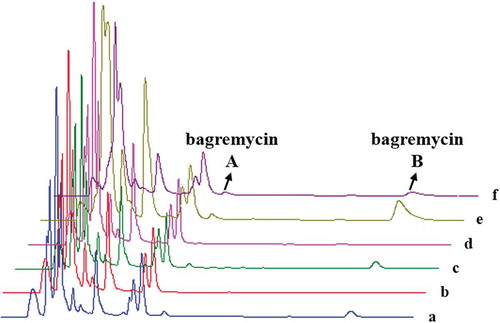
Subsequently, we analyzed the bagremycins production of these mutants by HPLC. As results (), bagremycin A and bagremycin B productions were not affected by disruption of the bagL, bagK, bagG, bag F, bagX and bagY genes, indicating these genes are not participated in bagremycins biosynthesis. Moreover, disruption of the bagN, bagM, bagZ and bagH genes partly inhibited bagremycins productions, suggesting that the four genes seem to be related to the biosynthesis of bagremycins, but not the essential genes. Importantly, disruption of bagE gene completely inhibited bagremycins production, showing that bagE is a critical gene participated in the biosynthesis of bagremycins.
Table 3. Analysis for bagremycins productions of relative genes disruption mutants.
Furthermore, to confirm the phenotype of the bagE mutant TDRE was exclusively related to the absence of bagE, a bagE complementation strain TCPE was constructed and the bagremycins productions were analyzed. The complementation of bagE restored the bagremycins production (). Thus, we concluded that the protein encoded by bagE was involved in the biosynthetic pathway of bagremycins. Moreover, in order to analyze the in vivo effect of bagE overexpression in wild-type strain S. sp. Tü 4128, we also constructed a bagE overexpression strain TOEE. HPLC analysis revealed that overexpression of bagE significantly increased bagremycin B production (). Sequence alignment revealed that BagE is a phenylacetate-CoA ligase (PCL), and conserved in the homologues from other strains, including Streptomyces and non-Streptomyces (Figure S2).
Figure 3. HPLC chromatograms of organic extracts of wild-type strain S. sp. Tü 4128 (a), bagE mutant TDRE (b), bagE complementation strain TCPE (c), bagE complementation control (d), bagE overexpression strain TOEE (e), bagE overexpression control (f).
Discussion
Bagremycins are two novel trans-coumaric acid derived antibiotics, which were firstly found in the strain S. sp. Tü 4128 isolated from the soil of Java [Citation6]. Because of their good activities against Gram-positive bacteria, fungi as well as human adenocarcinoma cells, bagremycins have great potential to be applied in medicine and agriculture in the future by combinatorial biosynthesis and metabolic pathway engineering strategy.
In our previous study, we have proved that bagA, bagB and bagC genes were involved in the biosynthesis bagremycins [Citation7,Citation8]. In this paper, for the first time, we successfully identified the bag BGC by genome mining and screened the relative genes to bagremycins production. Interestingly, we found that not all genes within the BGC were participated in the biosynthesis of bagremycins. Moreover, we also demonstrated that a PCL encoding gene bagE is crucial for the biosynthesis of bagremycins. It has been reported that PCL family proteins could activate the acyl or aryl acids to acyl-AMP or aryl-AMP respectively using ATP, after activation, the carboxy group is transferred to the thiol group of CoA, forming a thioester and releasing AMP [Citation20,Citation21]. All members of this family show a highly conserved AMP-binding domain, however, their substrate specificity may be different [Citation20,Citation22], such as some PCLs could also catalyze the reaction with 4-hydroxyphenylacetic acid as a substrate with a lower activity. Sequence alignment of BagE with other known PCL family proteins showed a high similarity with the conserved residues constituting motifs 94SSGSTGNPT102. Thus, we proposed that BagE might take part in the formation of 4-Hydrostyrene. The function of bagE participated in the biosynthesis of bagremycins need to be further verified.
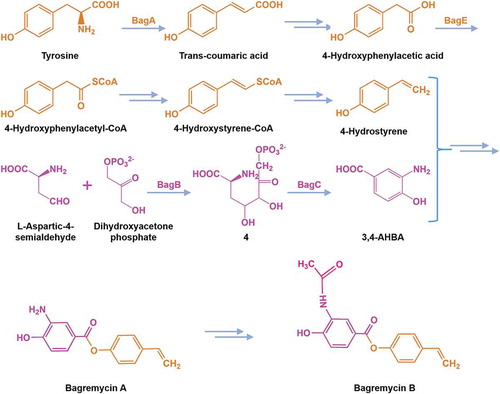
From the study, we further predicted the bagremycins biosynthetic pathway (). BagA converts L-Tyrosine to produce Trans-coumaric acid, which further generates 4-hydroxyphenylacetic acid after decarboxylation reaction, then 4-hydroxyphenylacetic acid converts into 4-hydroxyphenylacetyl-CoA through thioesterification of BagE, the activated 4-hydroxyphenylacetyl-CoA is converted into 4-hydroxystyrene-CoA through reduction reaction, subsequently converting into 4-hydroxystyrene. On the other hand, L-aspartic-4-semialdehyde and dihydroxyacetone phosphate are catalyzed by BagB to result in the intermediate 4, which is converted to 3,4-AHBA by BagC. Finally, 4-hydroxystyrene and 3,4-AHBA are converted into bagremycin A, then synthesizing bagremycin B ().
Figure 4. Proposed biosynthetic pathway of bagremycins.
Author Contributions
Ye J, Wu HZ and Zhang HZ designed the experiments; Zhu YX and Ye J carried out the experiments; Zhu YX, Hou BB, Ye J and Zhang HZ analyzed the data; Hou BB and Wu HZ wrote the manuscript. All of the authors assisted with critical reading of the manuscript.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplementary_Material.docx
Download MS Word (4.4 MB)Acknowledgments
We are particularly thankful to Prof. Hans-Peter Fiedler for providing S. sp. Tü 4128 strain.
Disclosure statement
The authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kalan L, Gessner A, Thaker MN, et al. A cryptic polyene biosynthetic gene cluster in Streptomyces calvus is expressed upon complementation with a functional bldA gene. Chem Biol. 2013;20:1214–1224.
- Xu J, Zhang J, Zhuo J, et al. Activation and mechanism of a cryptic oviedomycin gene cluster via the disruption of a global regulatory gene, adpA, in Streptomyces ansochromogenes. J Biol Chem. 2017;292:19708–19720.
- Challis GL. Exploitation of the Streptomyces coelicolor A3(2) genome sequence for discovery of new natural products and biosynthetic pathways. J Ind Microbiol Biotechnol. 2014;41:219–232.
- Cioni JP, Doroghazi JR, Ju KS, et al. Cyanohydrin phosphonate natural product from Streptomyces regensis. J Nat Prod. 2014;77:243–249.
- Challinor VL, Bode HB. Bioactive natural products from novel microbial sources. Ann N Y Acad Sci. 2015;1354:82–97.
- Bertasso M, Holzenkämpfer M, Zeeck A, et al. Bagremycin A and B, novel antibiotics from Streptomyces sp. Tü 4128. J Antibiot (Tokyo). 2001;54:730–736.
- Zhu Y, Liao S, Ye J, et al. Cloning and characterization of a novel tyrosine ammonia lyase-encoding gene involved in bagremycins biosynthesis in Streptomyces sp. Biotechnol Lett. 2012;34:269–274.
- Zhu Y, Xu D, Liao S, et al. Cloning and characterization of bagB and bagC, two co-transcribed genes involved in bagremycin biosynthesis in Streptomyces sp. Tü 4128. Ann Microbiol. 2013;63:167–172.
- Liu F, Xu D, Zhang Y, et al. Identification of BagI as a positive transcriptional regulator of bagremycin biosynthesis in engineered Streptomyces sp. Tü 4128. Microbiol Res. 2015;173:18–24.
- Macneil DJ, Klapko LM. Transformation of Streptomyces avermitilis by plasmid DNA. J Ind Microbiol. 1987;2:209–218.
- Huang H, Grove A. The transcriptional regulator TamR from Streptomyces coelicolor controls a key step in central metabolism during oxidative stress. Mol Microbiol. 2013;87:1151–1161.
- Kieser T, Bibb MJ, Buttner MJ, et al. Practical Streptomyces genetics. Norwich, United Kingdom: The John Innes Foundation; 2000.
- Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
- Weber T, Blin K, Duddela S, et al. AntiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:237–243.
- Hou B, Lin Y, Wu H, et al. The novel transcriptional regulator LmbU promotes lincomycin biosynthesis through regulating expression of its target genes in Streptomyces lincolnensis. J Bacteriol. 2018;200:e00447–17.
- Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, et al. Increasing the efficiency of heterologous promoters in actinomycetes. J Mol Microbiol Biotechnol. 2002;4:417–426.
- Tabata N, Tomoda H, Omura S. Ferroverdins, inhibitors of cholesteryl ester transfer protein produced by Streptomyces sp. WK-5344. II. Structure elucidation. J Antibiot. 1999;52:1108–1113.
- Maciejewska M, Pessi IS, Arguelles-Arias A, et al. Streptomyces lunaelactis sp. nov., a novel ferroverdin A-producing Streptomyces species isolated from a moonmilk speleothem. Antonie Van Leeuwenhoek. 2015;107:519–531.
- Zhang Y, Wu H, Ju C, et al. Identification of bagJ as a resistant gene for novel antibiotic bagremycins in Streptomyces sp.Tü 4128. J East China Univ Sci Technol. 2017;2:184–192.
- Lamas-Maceiras M, Vaca I, Rodríquez E, et al. Amplification and disruption of the phenylacetyl-CoA ligase gene of Penicillium chrysogenum encoding an aryl-capping enzyme that supplies phenylacetic acid to the isopenicillin N-acyltransferase. Biochem J. 2006;395:147–155.
- Erb TJ, Ismail W, Fuchs G. Phenylacetate metabolism in thermophiles: characterization of phenylacetate-CoA ligase, the initial enzyme of the hybrid pathway in Thermus thermophilus. Curr Microbiol. 2008;57:27–32.
- EI-Said Mohamed M. Biochemical and molecular characterization of phenylacetate-coenzyme A ligase, an enzyme catalyzing the first step in aerobic metabolism of phenylacetic acid in Azoarcus evansii. J Bacteriol. 2000;182:286–294.