
ABSTRACT
A phytase from Escherichia coli, AppA, has been the target of protein engineering to reduce the amount of undigested phosphates from livestock manure by making phosphorous from phytic acid available as a nutrient. To understand the contribution of each amino acid in the active site loop to the AppA activity, alanine and glycine scanning mutagenesis was undertaken. The results of phytase activity assay demonstrated loss of activity by mutations at charged residues within the conserved motif, supporting their importance in catalytic activity. In contrast, both conserved, non-polar residues and non-conserved residues tended to be tolerant to Ala and/or Gly mutations. Correlation analyses of chemical/structural characteristics of each mutation site against mutant activity revealed that the loop residues located closer to the substrate have greater contribution to the activity of AppA. These results may be useful in efficiently engineering AppA to improve its catalytic activity.
Abbreviations: AppA: pH 2.5 acid phosphatase; CSU: contacts of structural units; HAPs: histidine acid phosphatases; SASA: solvent accessible surface area; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SSM: site-saturation mutagenesis; WT: wild type
Graphical Abstract
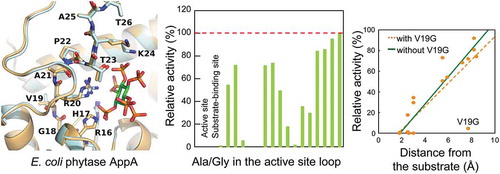
Alanine and glycine scanning mutagenesis revealed contribution of each amino acid in the active site loop to the activity of E. coli phytase AppA.
Phytase is the only category of enzymes that initializes the hydrolysis of phytic acid (inositol hexakisphosphate) () [Citation1–Citation6]. Phytic acid is a form of phosphorous storage in plant seeds [Citation7]. Many organisms, including plants, fungi, and bacteria, take advantage of phytases to extract phosphorous from phytic acids [Citation1]. However, since monogastric/agastric animals such as pigs, chickens, and fish cannot digest phytic acids in their feed, phytase is added to help them absorb phosphorous from phytic acids [Citation5,Citation8–Citation11]. Among phytases from various sources, Escherichia coli pH 2.5 acid phosphatase (AppA) has been intensely studied for its higher activity than another industrially utilized phytase from Aspergillus niger [Citation12,Citation13]. Since it was first identified as an acid phosphatase in 1979 [Citation14], crystallographic study and protein engineering research for a higher activity and thermostability have been performed due to the enthusiasm from livestock and fish feed industry [Citation15–Citation26]. In the case of livestock feed, although AppA is active at fairly high temperature (60℃), it has to withstand the 70–90℃ pelleting process heat while manufacturing livestock feed and remain highly active after the process [Citation6]. On the other hand, in the case of fish feed, phytases do not necessarily need to go through the pellet-heating treatment. Plant seed-derived ingredients in fish feed can be pre-treated with phytases, and phytic acid-bound phosphorous in the plant seeds is released as nutrient for fish [Citation11,Citation27,Citation28]. However, the efficiency of phosphorous release by phytases in fish feed or fish stomach is still not enough. To use AppA in livestock and fish feed in a cost-effective manner, the specific activity of AppA needs to be improved. Much effort has been invested to increase the specific activity of AppA using different strategies [Citation16,Citation17,Citation19–Citation22]. Several groups have designed AppA mutants based on its three-dimensional structure for enhanced activity [Citation23–Citation26]. For example, Wu et al. improved the specific activity of AppA by 18% through V89T mutation [Citation25]. However, significant improvements in activity have not been achieved to date.
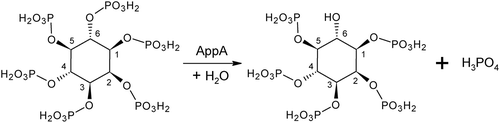
Figure 1. Hydrolytic reaction of phytic acid by AppA. The phosphate groups on a phytic acid (left) become free one by one, starting from the D-6 (L-4) position.
It has been proven from previous mutagenesis studies of enzymes that the mutations in active site loops cause drastic changes in the enzymatic activity [Citation29,Citation30]. This is because the amino acids in active site loops tend to be conserved over different species [Citation31–Citation33]. Thus, if conserved sites in the active site loop of an enzyme were mutated to other residues, the enzyme could either lose a large fraction of the activity [Citation33–Citation35] or increase the activity in a large increment [Citation30,Citation36–Citation39]. On the other hand, mutations in non-conserved residues in active site loops do not necessarily result in activity loss. In some cases, when a residue close to a conserved site having contact with substrate was mutated, an improvement in activity was observed [Citation38,Citation40]. For these reasons, if we carefully choose which site (either conserved or non-conserved) to mutate in active site loops, the activity of an enzyme can be improved. To do this careful selection, information on the role of each residue in the active site loop of an enzyme is required.
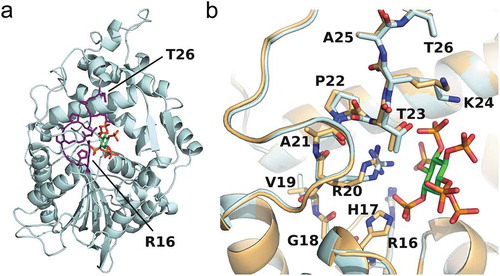
The structure of AppA consists of two domains: an α-helical domain and an α/β domain, between which exists the cavity for the substrate (phytic acid) to be catalyzed [Citation15] ()). The active site loop of AppA (residues 16–26; residue numbering excluding the N-terminal 22-residue signal sequence) belongs to its α-helical domain and lies in front of the substrate-binding pocket ()). According to molecular dynamics simulations of AppA, this loop is suggested to play an important role in expressing catalytic activity [Citation41]. The active site loop of AppA contains a conserved motif of RHGXRXP (residues 16–22), where X denotes any amino acid. Among the residues in this motif, the functions of R16, H17, and R20 have been determined from the mutagenesis study by Ostanin et al. [Citation42]. Based on the complete loss in AppA activity of R16A and H17N mutants, the authors concluded that R16 and H17 are the substrate-binding site and active site, respectively. On the other hand, R20A mutant showed a very low activity (less than 0.4%) compared to wild-type AppA, suggesting its role in binding to the substrate. The interactions between R20 and the substrate were subsequently confirmed from the crystal structure [Citation15]. Yet, the roles of eight other residues in AppA active site loop remain unknown.
Figure 2. Structure of E. coli phytase AppA.(a) Overall structure of AppA. The active site loop is shown in purple, and the substrate (phytic acid) is shown in green. (b) The enlarged view of AppA around its active site loop (sticks) and the substrate (green sticks). The AppA structures in the substrate-bound form (light cyan) and in the free form (gold) are overlaid. Blue, red, and orange denote the nitrogen, oxygen, and phosphorous atoms, respectively. The figures were drawn using the PyMOL Molecular Graphics System, Schrodinger, LLC.
Alanine or glycine scanning mutagenesis is a method widely used to elucidate the contribution of an original residue to the activity of an enzyme [Citation43–Citation46]. Alanine or glycine is used because it represents an average of 20 naturally occurring amino acids. Bromberg and Rost (2008) reported that the average value of mutational effects upon mutating all residues in a protein to all the non-native residues correlated best with the mutational effect of alanine substitution [Citation46]. The second-best correlation was observed for glycine substitution [Citation46]. These results indicate that alanine or glycine should be used in mutagenesis studies to find out the function of the original residue. Furthermore, alanine scanning mutagenesis can elucidate the effects to the enzymatic activity from the side chain of a mutation site [Citation43,Citation46], while glycine scanning mutagenesis can examine the contribution of the main chain structure of the mutation site to the activity by increasing the mobility of the main chain [Citation44,Citation45].
Based on these considerations, here we carried out alanine and glycine scanning mutagenesis on AppA, to examine the contribution of each residue in the active site loop to its activity. E. coli phytase AppA was our choice of phytase for the present mutational study, because it is already in industrial use. We found that Ala and/or Gly mutations at conserved residues with charged sidechains eliminated the activity. However, both conserved, non-polar residues and non-conserved residues tended to be tolerant to Ala and/or Gly mutations. Correlation analyses between mutant activity and AppA properties at each mutation site suggest that the loop residues located closer to the substrate have greater contribution to the activity of AppA. A strategy to improve AppA activity based on the present results is discussed.
Materials and methods
Plasmids and strains
The appA gene was amplified from E. coli BL21(DE3) genome extracted using nexttec 1-step DNA Isolation Kit for Bacteria (nexttec GmbH, Hilgertshausen, Germany). Using the purified genome, appA was amplified by PCR using a forward primer appA_F and reverse primer appA_AS (see Supplementary Table S1) and was inserted into the vector plasmid pETDuet-1 (Merck Millipore, Darmstadt, Germany) at the NdeI and AvrII restriction sites, yielding the plasmid pETDuet-AppA. His6-tag was inserted at the C-terminus of AppA protein for purification and named pETDuet-AppA+His.
The addition of His6-tag to pETDuet-AppA plasmid was carried out as follows. First, primers for His6-tag introduction were prepared by amplifying two short primers containing complimentary linker and His6-tag sequences (AppA+His_F, 5ʹ-cat acc ggc gtg cag ttt ggg tag cag cgg tca tca tca tca tca tca t-3ʹ, AppA+His_R, 5ʹ-gca gcc tag gtt agt cag tta atg atg atg atg atg atg acc gct gct acc-3ʹ) by PCR [98℃ for 10 sec, 1 cycle; 98℃ for 10 sec, 68℃ for 5 sec, 72℃ for 30 sec, 30 cycles] using KOD-Plus-Neo DNA polymerase (Toyobo, Osaka, Japan). The PCR product was purified using FastGene Gel/PCR Extraction Kit (Nippon Genetics, Tokyo, Japan) and used as primers to introduce His6-tag in the subsequent PCR [94℃ for 2 min, 1 cycle; 98℃ for 10 sec, 68℃ for 4 min, 30 cycles] using the protocols of the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Methylated template plasmids were digested by DpnI restriction enzyme (Takara Bio, Shiga, Japan). The reaction solution was then used to transform E. coli JM109 competent cells (Nippon Gene, Tokyo, Japan) with pETDuet-AppA+His plasmid. The plasmid was extracted from the cultivated cells using FastGene Plasmid Mini Kit (Nippon Genetics, Tokyo, Japan). Finally, E. coli BL21(DE3)pLysS competent cells (BioDynamics Laboratory, Tokyo, Japan) were transformed with pETDuet-AppA+His plasmid for protein expression. This plasmid was utilized to construct Ala and Gly mutants by the protocols of the QuikChange site-directed mutagenesis kit using the primers listed in Supplementary Table S1. All primers used in this study were synthesized by Eurofins Genomics (Tokyo, Japan).
Cell cultivation
Auto-induction medium was used for cultivation of E. coli BL21(DE3)pLysS competent cells transformed with plasmids constructed above [Citation47]. Cultivation was initiated by inoculating colonies into 96-well plates with non-inducing ZYM-505 medium (100 μl per well). After the overnight cultivation at 37℃, 10 μl from each well was transferred to a new well in 96-well plate with 150 μl inducing ZYM-5052 medium. Each strain was cultivated using 3 wells for one sample. Cells were harvested in 1.5 ml tubes after 20 h of cultivation, washed twice with Tris-HCl buffer (50 mM Tris-HCl, pH 7.4), and kept at -80℃ until use.
Phytase assay
Phytase assay was carried out based on the molybdenum blue method [Citation48]. Cells were first thawed on ice, and 200 μl Tris-HCl buffer (pH 7.4) was added to each sample to suspend the cells. Following the addition of 100 μl lysozyme solution (10 mg/ml in Tris-HCl buffer, pH 7.4), cells were lysed by incubating at 37℃ for 1 h. Supernatant of cell lysates was obtained by centrifuging the lysed cells (4℃, 20,600 × g, 30 min).
The amount of the soluble form of AppA contained in the supernatant was quantified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gels. Ten microliters of supernatant or pellet sample was loaded to each lane of a gel. Coomassie Brilliant Blue-stained gel images were acquired using a Gel Doc EZ imager (Bio-Rad, Hercules, CA, USA). Quantification of AppA band was performed using the ImageLab software (Bio-Rad). To normalize possible differences in the loading amount of the supernatant samples, band intensity of E. coli protein in each lane (shown with black arrows in Supplementary Figure S1) was analyzed using the ImageLab software and was divided by that in the lane for wild-type AppA. The resultant number was termed as a “correction factor.” The amount of soluble AppA contained in the supernatant was normalized by dividing the band intensity of AppA protein by the corresponding correction factor (Supplementary Figure S2(b)). Even after this correction, the relative amounts of soluble AppA protein did not change beyond the standard errors of five-times measurement.
Phytase reaction was started by mixing 10 μl diluted supernatant of cell lysates (200 times dilution) and 70 μl substrate solution (1 mM sodium phytate in 250 mM sodium acetate buffer, pH 4.5) in 96-well plates, and incubated at 60℃ for 15 min. The enzyme reaction was quenched by adding 70 μl trichloroacetic acid to the reaction solution. Following the addition of 70 μl color developing solution (0.54% ammonium molybdate, 2.16% ascorbic acid in 0.34 M sulfuric acid), the 96-well plate was placed under 50℃ for 15 min for phosphate-molybdenum complex formation. An absorbance at 820 nm was measured (Synergy HTX multi-mode reader, BioTek, Vermont, USA) and used to calculate the apparent activity for each sample using the KH2PO4 standard curve (Supplementary Figure S2(a)). Finally, the activity was standardized by the amount of the soluble form of AppA contained in lysate supernatant (). The activity measurements were carried out five times, and means ± standard errors are shown. For each experiment, three samples of the wild type were used (total of fifteen samples for wild type).
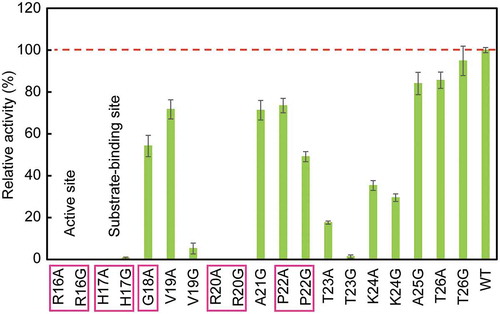
Figure 3. Relative activity of Ala and Gly mutants compared with wild-type AppA. Mutants of conserved residues are boxed, and those of active site and substrate-binding site are designated. The broken line shows the activity of wild type (100%). WT denotes wild type. The activity measurements were carried out five times, and means ± standard errors are shown.
Results
Activity assay of AppA and its Ala/Gly mutants
To figure out the contribution of each amino acid in active site loop (residues 16–26) to AppA activity, the 11 residues were mutated to Ala and/or Gly by site-directed mutagenesis. Two residues that were initially Ala (A21 and A25) were replaced only by Gly, and one residue originally Gly (G18) was substituted with Ala. In eight other residues, both Ala and Gly took their positions. Thus, 19 mutants were prepared. After transforming E. coli BL21(DE3)pLysS with constructed plasmids, wild-type and mutant AppAs were successfully expressed by cultivating the cells. Using the cell lysate supernatants, phytase assay was carried out under the AppA optimal conditions (60℃ and pH 4.5) with sodium phytate as substrate. The 820 nm absorbance of molybdenum blue complex formed by molybdenum and PO43- liberated from substrate hydrolysis was measured, and phytase activity was calculated (Supplementary Figure S2(a)). As a negative control, the supernatant sample of an E. coli strain harboring pETDuet-1 (empty vector plasmid) was used in the AppA activity assay. No AppA activity was detected in the negative control, showing that there was no detectable background of AppA activity derived from the wild-type AppA from the genome of host E. coli cells. When comparing enzyme activities of different mutants, protein expression levels of each mutant can heavily influence the activity values. Hence, to normalize each mutant activity with its AppA protein expression level, the activities were divided by the AppA band intensity from SDS-PAGE analysis. Comparing the band intensities of wild type and mutants, there was no significant difference in the amount of soluble AppA protein (Supplementary Figure S2(b)). Lastly, each mutant activity was expressed as a relative value against wild-type activity (). This method enabled us to evaluate activity of 19 mutants and wild type of AppA in a timely manner.
As a result of phytase assay (), eight mutants (R16A, R16G, H17A, H17G, V19G, R20A, R20G, and T23G) lost more than 90% of the activity compared to the wild type. Besides, comparing the Ala and Gly mutants at the same position, we found that V19G showed a greater activity loss than V19A by more than 50%.
Correlation analysis
To investigate the mutational effect on AppA activity, we examined correlations between the relative activity of a mutant and structural/chemical properties of each mutation site, including solvent accessible surface area (SASA), B-factor, number of interactions with substrate and other structural units in AppA, distance from the substrate, and degree of conservation for each residue ( and Supplementary Figure S3). SASA values were predicted by GetArea server [Citation49] using crystal structures of AppA with substrate (PDB ID: 1DKQ) and without substrate (PDB ID: 1DKN) to examine differences induced by conformational changes upon substrate binding ()). There is a little conformational change in the main chain of active site loop by substrate binding; however, the side chains of V19, R20, P22, T23, and K24 turn towards the substrate and move closer towards the substrate-binding pocket ()). B-factor values for the Cα atoms of residues 16–26 were taken from the same PDB entries. The correlation analysis showed that regardless of the existence of substrate, there were no concrete correlations between SASA/B-factor of each mutation site and mutant activity (Supplementary Figure S3(a-d)). Nevertheless, when the substrate was present, a weak positive correlation between the B-factor and relative activity was confirmed (Suppleme-ntary Figure S3(c)). This suggests that the residues whose motions become less mobile upon substrate binding are important for function. Conversely, residues flexible when bound to substrate may not contribute much for function and thus can be candidates for further mutagenesis to improve the AppA activity.
Figure 4. Correlation analysis. Correlation plots of relative activity of a mutant with (a) the number of contacts of each mutation site with substrate and other structural units in AppA, and (b) the distance from substrate to each mutation site. In both panels, a red continuous line shows the regression line, and the corresponding correlation coefficient, r, and the p-value are shown. In (b), a blue continuous line shows the regression line obtained using the data without V19G.
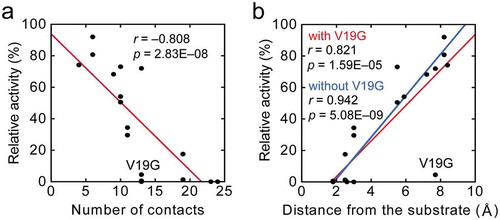
The number of interactions between each amino acid in the active site loop and the substrate/other parts of AppA was counted with the help of “ligand-protein contacts” and “contacts of structural units” (CSU) softwares [Citation50] using the crystal structure of the AppA-substrate complex (PDB ID: 1DKQ). The number of contacts varied from 4 to 24, with the two Arg from the conserved motif (R16 and R20) having more contacts (24 and 23, respectively) in contrast to others ()). The correlation analysis yielded a strong negative correlation between mutant activity and the number of interactions of each mutation site with substrate and other structural units in AppA, which exhibited an overall trend of greater activity loss with the increase in interactions ()).
The distance from the substrate to each mutation site was obtained using the PyMOL software (Molecular Graphics System, Schrodinger, LLC) by measuring the distance between any of the six phosphate groups on the substrate (phytic acid) and the side chain of the original residue at a mutation site. The analysis incorporating all the mutants produced a significantly high correlation coefficient ()). Despite this tendency, the V19G mutant was completely off the linearity. When the value for V19G was removed, the correlation was strengthened further ()).
Sequence conservation among HAPs
Among the 11 residues in the AppA active site loop, five (R16, H17, G18, R20, and P22) are conserved within the AppA superfamily (histidine acid phosphatases (HAPs) superfamily) as active site motif (RHGXRXP). Besides, a previous site-directed mutagenesis study of AppA showed that R16 and H17 are responsible for the substrate binding and catalysis, respectively [Citation42]. Among conserved residues, ones with charged side chains (R and H) showed complete (or nearly complete) loss of activity, while ones without charge (G and P) retained more than 50% activity ().
The degree of conservation for each position of the active site loop was calculated by dividing the number of AppA homologs with the same amino acid as AppA at a certain position by the total number of amino acid sequences considered. Here, amino acid sequences of 62 enzymes homologous to AppA were obtained from the NCBI Conserved Domain Database under the protein category of “HP_HAP_like” (phosphatases with His residue in the active site) that AppA belongs to (Supplementary Figure S4). For simplicity, only one enzyme from one species was chosen when calculating the degree of conservation. The second phytase from E. coli, AppA2, was included as an exception to contrast with AppA. As shown in Supplementary Figure S3(e), the degree of conservation at each mutation site correlates weakly with the mutant activity. This weak correlation may be because some mutations at conserved residues did not result in reduction of the activity and even mutations at some non-conserved residues significantly reduced the activity.
Although no evident correlation between the degree of conservation and the activity was found, certain sequence variations arose from the multiple sequence alignment ( and Supplementary Figure S4). The alignment revealed that T23, which comes just after the conserved motif RHGXRXP, is partially conserved. On the other hand, amino acids at the positions A21, K24, A25, and T26 varied among species. Besides, the equivalent position of V19 had a strong conservation tendency to have either Ala or negatively charged residue (Glu or Asp) (found in 19, 14, and 13 homologs, respectively). Among the 62 AppA homologs, Val at residue 19 was rather rare (5 out of 62 homologs). Furthermore, it is noteworthy that the position of A21, one of the variable residues in the middle of the conserved motif, is occupied by bulky hydrophobic residues (Tyr and Phe) in more than half of the phosphatases examined.
Figure 5. Sequence logo showing the degree of conservation of active site loop residues based on the multiple sequence alignment of 63 HAPs (Supplementary Figure S4). The horizontal axis shows the residue number in AppA, and the vertical axis shows the probability of a residue in a position. The larger the logo of a residue, the more likely the residue is placed in a certain position in the multiple alignment. The figure was created by WebLogo application [Citation52].
![Figure 5. Sequence logo showing the degree of conservation of active site loop residues based on the multiple sequence alignment of 63 HAPs (Supplementary Figure S4). The horizontal axis shows the residue number in AppA, and the vertical axis shows the probability of a residue in a position. The larger the logo of a residue, the more likely the residue is placed in a certain position in the multiple alignment. The figure was created by WebLogo application [Citation52].](/cms/asset/34313a80-0fc2-4251-b57e-b82e28fae485/tbbb_a_1571897_f0005_oc.jpg)
Discussion
Mutations at conserved residues in the active site loop
We performed Ala and Gly substitutions on AppA active site loop region aiming to analyze the contribution of each residue to AppA activity. We found that charged conserved sites resulted in significant decreases when mutated to Ala and/or Gly (). The reason why charged conserved residues (R16, H17, and R20) forfeited their activities is straightforward. R16 and R20 both have multiple interactions with the scissile 3-phosphate of substrate and are both positioned at a very close proximity in the crystal structure ()). Without positively charged side chains, AppA would not be able to hold any negatively charged phosphate groups on the substrate. H17, on the other hand, is in charge of being phosphorylated during the enzymatic reaction. Hence, its positive charge also cannot be replaced by aliphatic residues.
Although G18 and P22 are both conserved sites, Ala and/or Gly mutations did not cause much effect on their activities (). This may be because G18 and P22 have no interaction with substrate and fewer contacts with other structural units in AppA ()). However, when we compared the AppA expression levels of G18A, P22A, and P22G (Supplementary Figure S2(b)), P22A and P22G had lower expression levels of AppA compared to wild type, while G18A had almost the same AppA expression level as wild type. This might suggest the conservation of P22 for keeping the loop structure and maintaining the stability of a protein.
Mutations at non-conserved residues in the active site loop
When non-conserved residues were mutated, AppA did not experience severe activity loss overall (). Especially, A25 and T26 were barely affected by mutations. In a mutagenesis study by Wu et al., A25F mutant of AppA showed 79% activity compared with wild type [Citation25]. This result almost coincides with ours on A25G. On the contrary, mutations to T23 and K24 substantially decreased their activity, suggesting the importance of these residues in AppA catalytic activity. Moreover, T23 mutants lost more activity than K24 mutants (), possibly because of its more contacts with substrate in a closer proximity ()). T23 has one water-mediated interaction with substrate [Citation15], which could stabilize AppA structure by hydration. Also, while K24 is not conserved, T23 is partially conserved () even though it is outside the conserved active site motif. Altogether, T23 can be considered as both a structurally and catalytically vital residue in AppA. Hence, mutations on T23 and K24 could induce large effects on AppA activity, depending on with which amino acid to be replaced. In a work done by Zhang et al., a K24E mutant had approximately 40% activity at 60℃ [Citation51]. The replacement of K24 with a smaller residue may not be suitable as it loosens the packing of the binding pocket.
The most notable difference between the effects of Ala and Gly mutations at the same position was observed for V19A and V19G mutants, which had a huge gap in activity loss caused by mutation (; 29% and 95% activity loss, respectively). By alanine scanning mutagenesis, we can investigate the contribution of the side chain of a residue, while glycine scanning gives us insights on the main chain contribution to the enzyme activity. Thus, the fact that the V19A mutation did not reduce much of the AppA activity suggests that the side chain of the position V19 does not greatly affect the catalytic activity. In fact, we can observe V19 facing the opposite side of the substrate from ), thus supporting the insight that the side chain of V19 is unlikely to affect the AppA catalytic ability. This can also be confirmed from the result of HAPs amino acid sequence alignment ( and Supplementary Figure S4). Unlike V19 in AppA, Asp and Glu are the two most abundant residues other than Ala at the position equivalent to V19. This means that V19 position can be occupied by bulky, charged amino acids. In contrast, the significant reduction in the AppA activity of V19G indicates that the increased flexibility of the main chain caused by the mutation is not favorable for the AppA enzymatic reaction. Since the residue before V19 on the amino acid sequence is G18, V19G mutation makes it two consecutive Gly, which drastically increases the flexibility of the loop, affecting the mobility of the whole AppA active site loop. Thus, although the V19 site was distant from the substrate, the effect of Gly substitution extended to the whole loop.
This notion is also supported by detailed investigation of crystal structures of AppA-substrate complex (PDB ID: 1DKQ) and A. fumigatus phytase-substrate complex (PDB ID: 1SKB) using the CSU software. Since A. fumigatus phytase originally has Ala in the equivalent position of V19 in AppA, which is A61, it can be a good model of the V19A mutant of AppA. We found that the Cγ atom of V19 forms hydrophobic interactions with R20 in AppA, while the Cβ atom of A61 forms hydrophobic interactions with R62 in A. fumigatus phytase. These results suggest that the presence of hydrophobic interactions of V19 with R20, which is essential for substrate binding, could stabilize the R20-substrate interactions and retain more than 70% activity in the V19A mutant. In contrast, removal of the hydrophobic interactions by the V19G mutation could destabilize the interactions between R20 and substrate, leading to drastic reduction in the activity. As a result, the V19G went off from the correlation line as shown in ).
HAPs sequence alignment based on specific activity
The difference in the activity of HAP phytases from different species should be ascribed to the differences in non-conserved amino acids, since HAPs share the conserved active site motif (RHGXRXP). Thus, we performed an alignment of amino acid sequences of phytases from various organisms with known phytase specific activity (Supplementary Figure S5) [Citation4]. The amino acid sequences were sorted in descending order based on the specific activity values, and we especially focused on the residues around the active site loop (residues 16–26 in AppA). Comparing the specific activity of different phytases, Yersinia intermedia phytase was found to possess a 11% higher phytase activity than E. coli phytase AppA [Citation13,Citation20]. However, considering the experimental errors of AppA activity assay, the specific activity of AppA is not significantly lower than that of Y. intermedia phytase. When we compared the amino acid sequences of AppA and Y. intermedia phytase, there were two different residues in the active site loop of both phytases, namely A21S and A25Q substitutions (residue numbers correspond to AppA sequence). Introduction of such mutations in AppA might increase its specific activity. In fact, the present studies showed that both A21 and A25 were tolerant to Gly mutation, suggesting that site-saturation mutagenesis (SSM) at these positions may improve AppA activity.
When we compared the non-conserved sites of V19, T23, K24, A25, and T26, it was observed that phytases with lower specific activity tend to have Ala or Gly in the equivalent positions (e.g. V19 is replaced by Ala in eight phytases with specific activity less than 40% of AppA activity). These observations are consistent with our experimental results.
Other differences in residues outside of the active site loop can be detected among the groups of higher activity and lower activity, such as at residue 12. While the top three phytases with the highest specific activity (those from Y. intermedia, E. coli, and Y. kristensenii) have Val at residue 12, four phytases with lower specific activity have Gln at the same position (those from A. ficuum, A. niger, A. oryzae, and Kodamaea ohmeri BG3). Such information may also be useful in improving AppA activity.
Future perspectives
In this study, to search for mutation sites that can possibly improve AppA activity, we substituted each site of AppA active site loop with Ala and/or Gly. Since Ala or Gly represents an average of 20 naturally occurring amino acids [Citation46], we can expect that the activity of Ala/Gly mutants shows the average value of mutational effects when the mutation site is substituted with all other possible amino acids. Thus, if the remaining AppA activity after Ala/Gly substitutions was relatively high, one or some of possible mutants at this site would likely to display a higher activity than wild-type AppA. Hence, if we carry out SSM on the residues that are tolerant to Ala/Gly substitutions, we would be able to obtain AppA mutants with improved AppA activity. The structure-sequence-function relationship of AppA active site loop elucidated in this study would facilitate the engineering of AppA and other phytases with similar structure to elevate their catalytic activity.
The present results showed that some mutations at non-conserved residues in the active site loop, including V19, A21, A25, and T26, retained more than 50% of AppA activity relative to wild type. By carrying out SSM at those four sites, we may be able to obtain an AppA mutant with a better activity. SSM is a common method used to search for an amino acid most suitable for a certain enzymatic function (activity, substrate specificity, etc.) by creating and evaluating the activity of a library of mutants where one residue is replaced by all the non-native amino acids [Citation29]. On the other hand, previous study has reported that the V89T mutation outside of the active site loop improved the AppA activity [Citation25]. Combination of mutations at the active site loop and that outside of the loop may further improve the activity of AppA.
Along with structural and chemical properties of thermophilic and psychrophilic enzymes from preceding work, the present results may be useful for designing a phytase highly active within a preferred temperature range. Thermostable phytases have been designed for the applications to animal feed industry as phytase degrades phytic acids in the feed and helps animals take up phosphates as their nutrition. Likewise, cold-active phytases would benefit fish by extracting phosphorous from phytic acids under the cold aquatic environment. In either case, highly active phytases at certain temperature ranges would prevent undigested phytic acids from polluting the environment and being wasted as a limited phosphorous source.
Author Contribution
MW and MA conceived and designed the research. MW and YH carried out the experiments and analyzed the data. MW and MA wrote the manuscript. All authors read and approved the final manuscript.
Supplemental_Material.pdf
Download PDF (4.4 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Konietzny U, Greiner R. Molecular and catalytic properties of phytate-degrading enzymes (phytases). Int J Food Sci Technol. 2002;37(7):791–812.
- Haefner S, Knietsch A, Scholten E, et al. Biotechnological production and applications of phytases. Appl Microbiol Biotechnol. 2005;68(5):588–597.
- Rao DE, Rao KV, Reddy TP, et al. Molecular characterization, physicochemical properties, known and potential applications of phytases: an overview. Crit Rev Biotechnol. 2009;29(2):182–198.
- Yao MZ, Zhang YH, Lu WL, et al. Phytases: crystal structures, protein engineering and potential biotechnological applications. J Appl Microbiol. 2012;112(1):1–14.
- Lei XG, Weaver JD, Mullaney E, et al. Phytase, a new life for an “old” enzyme. Annu Rev Anim Biosci. 2013;1:283–309.
- Rebello S, Jose L, Sindhu R, et al. Molecular advancements in the development of thermostable phytases. Appl Microbiol Biotechnol. 2017;101(7):2677–2689.
- Reddy NR, Sathe SK, Salunkhe DK. Phytates in legumes and cereals. Adv Food Res. 1982;28:1–92.
- Jackson LS, Li MH, Robinson EH. Use of microbial phytase in channel catfish Ictalurus punctatus diets to improve utilization of phytate phosphorus. J World Aquacult Soc. 1996;27(3):309–313.
- Lei XG, Stahl CH. Nutritional benefits of phytase and dietary determinants of its efficacy. J Appl Anim Res. 2000;17(1):97–112.
- Greiner R, Konietzny U. Phytase for food application. Food Technol Biotech. 2006;44(2):125–140.
- Kumar V, Sinha AK, Makkar HP, et al. Phytate and phytase in fish nutrition. J Anim Physiol Anim Nutr (Berl). 2012;96(3):335–364.
- Wyss M, Brugger R, Kronenberger A, et al. Biochemical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): catalytic properties. Appl Environ Microbiol. 1999;65(2):367–373.
- Huang H, Luo H, Yang P, et al. A novel phytase with preferable characteristics from Yersinia intermedia. Biochem Biophys Res Commun. 2006;350(4):884–889.
- Tetu C, Dassa E, Boquet PL. Unusual pattern of nucleoside polyphosphate hydrolysis by the acid phosphatase (optimum pH = 2.5) of Escherichia coli. Biochem Biophys Res Commun. 1979;87(1):314–322.
- Lim D, Golovan S, Forsberg CW, et al. Crystal structures of Escherichia coli phytase and its complex with phytate. Nat Struct Biol. 2000;7(2):108–113.
- Rodriguez E, Wood ZA, Karplus PA, et al. Site-directed mutagenesis improves catalytic efficiency and thermostability of Escherichia coli pH 2.5 acid phosphatase/phytase expressed in Pichia pastoris. Arch Biochem Biophys. 2000;382(1):105–112.
- Garrett JB, Kretz KA, O’Donoghue E, et al. Enhancing the thermal tolerance and gastric performance of a microbial phytase for use as a phosphate-mobilizing monogastric-feed supplement. Appl Environ Microbiol. 2004;70(5):3041–3046.
- Cao L, Wang W, Yang C, et al. Application of microbial phytase in fish feed. Enzyme Microb Technol. 2007;40(4):497–507.
- Zhu W, Qiao D, Huang M, et al. Modifying thermostability of appA from Escherichia coli. Curr Microbiol. 2010;61(4):267–273.
- Fu D, Li Z, Huang H, et al. Catalytic efficiency of HAP phytases is determined by a key residue in close proximity to the active site. Appl Microbiol Biotechnol. 2011;90(4):1295–1302.
- Yao MZ, Wang X, Wang W, et al. Improving the thermostability of Escherichia coli phytase, appA, by enhancement of glycosylation. Biotechnol Lett. 2013;35(10):1669–1676.
- Wang X, Yao MZ, Yang B, et al. Enzymology and thermal stability of phytase appA mutants. RSC Adv. 2015;5(54):43863–43872.
- Fei B, Xu H, Cao Y, et al. A multi-factors rational design strategy for enhancing the thermostability of Escherichia coli AppA phytase. J Ind Microbiol Biotechnol. 2013;40(5):457–464.
- Fei B, Xu H, Zhang F, et al. Relationship between Escherichia coli AppA phytase’s thermostability and salt bridges. J Biosci Bioeng. 2013;115(6):623–627.
- Wu TH, Chen CC, Cheng YS, et al. Improving specific activity and thermostability of Escherichia coli phytase by structure-based rational design. J Biotechnol. 2014;175:1–6.
- Wang X, Du J, Zhang ZY, et al. A rational design to enhance the resistance of Escherichia coli phytase appA to trypsin. Appl Microbiol Biotechnol. 2018;102(22):9647–9656.
- Storebakken T, Shearer KD, Roem AJ. Availability of protein, phosphorus and other elements in fish meal, soy-protein concentrate and phytase-treated soy-protein-concentrate-based diets to Atlantic salmon, Salmo salar. Aquaculture. 1998;161(1–4):365–379.
- Vielma J, Ruohonen K, Peisker M. Dephytinization of two soy proteins increases phosphorus and protein utilization by rainbow trout, Oncorhynchus mykiss. Aquaculture. 2002;204(1–2):145–156.
- Tripathi A, Varadarajan R. Residue specific contributions to stability and activity inferred from saturation mutagenesis and deep sequencing. Curr Opin Struct Biol. 2014;24:63–71.
- Morley KL, Kazlauskas RJ. Improving enzyme properties: when are closer mutations better? Trends Biotechnol. 2005;23(5):231–237.
- Worth CL, Gong S, Blundell TL. Structural and functional constraints in the evolution of protein families. Nat Rev Mol Cell Biol. 2009;10(10):709–720.
- Gong S, Worth CL, Bickerton GR, et al. Structural and functional restraints in the evolution of protein families and superfamilies. Biochem Soc Trans. 2009;37(Pt 4):727–733.
- Echave J, Spielman SJ, Wilke CO. Causes of evolutionary rate variation among protein sites. Nat Rev Genet. 2016;17(2):109–121.
- McLaughlin RN Jr., Poelwijk FJ, Raman A, et al. The spatial architecture of protein function and adaptation. Nature. 2012;491(7422):138–142.
- Abriata LA, Palzkill T, Dal Peraro M. How structural and physicochemical determinants shape sequence constraints in a functional enzyme. PLoS One. 2015;10(2):e0118684.
- Petrosino JF, Palzkill T. Systematic mutagenesis of the active site omega loop of TEM-1 β-lactamase. J Bacteriol. 1996;178(7):1821–1828.
- McCarthy JK, Uzelac A, Davis DF, et al. Improved catalytic efficiency and active site modification of 1,4-β-D-glucan glucohydrolase A from Thermotoga neapolitana by directed evolution. J Biol Chem. 2004;279(12):11495–11502.
- Iwakura M, Maki K, Takahashi H, et al. Evolutional design of a hyperactive cysteine- and methionine-free mutant of Escherichia coli dihydrofolate reductase. J Biol Chem. 2006;281(19):13234–13246.
- Nayeem A, Chiang SJ, Liu SW, et al. Engineering enzymes for improved catalytic efficiency: a computational study of site mutagenesis in epothilone-B hydroxylase. Protein Eng Des Sel. 2009;22(4):257–266.
- Tinberg CE, Khare SD, Dou J, et al. Computational design of ligand-binding proteins with high affinity and selectivity. Nature. 2013;501(7466):212–216.
- Shivange AV, Schwaneberg U, Roccatano D. Conformational dynamics of active site loop in Escherichia coli phytase. Biopolymers. 2010;93(11):994–1002.
- Ostanin K, Harms EH, Stevis PE, et al. Overexpression, site-directed mutagenesis, and mechanism of Escherichia coli acid phosphatase. J Biol Chem. 1992;267(32):22830–22836.
- Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244(4908):1081–1085.
- Jin T, Peng L, Mirshahi T, et al. The βγ subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol Cell. 2002;10(3):469–481.
- Valbuena JJ, Vera R, Garcia J, et al. Plasmodium falciparum normocyte binding protein (PfNBP-1) peptides bind specifically to human erythrocytes. Peptides. 2003;24(7):1007–1014.
- Bromberg Y, Comprehensive RB. in silico mutagenesis highlights functionally important residues in proteins. Bioinformatics. 2008;24(16):i207–i212.
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–234.
- Shivange AV, Serwe A, Dennig A, et al. Directed evolution of a highly active Yersinia mollaretii phytase. Appl Microbiol Biotechnol. 2012;95(2):405–418.
- Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19(3):319–333.
- Sobolev V, Sorokine A, Prilusky J, et al. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15(4):327–332.
- Zhang J, Liu Y, Gao SP, et al. Site-directed mutagenesis and thermal stability analysis of phytase from Escherichia coli. Biosci Biotech Res Comm. 2016;9(3):357–365.
- Crooks GE, Hon G, Chandonia JM, et al. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190.