
ABSTRACT
Galactose oxidase and amine oxidase contain a cofactor which is generated by post-translational chemical modification to the corresponding amino acid side chains near the copper active center. Such cofactors provide proteins unusual catalytic ability that canonical amino acids cannot exert as well as their structural stability, and thereby are called as protein-derived cofactors. These cofactors and modifications are mostly derived from aromatic amino acid residues, especially Tyr, Trp, and His. Current information about unusual cofactors derived from two of those, heteroaromatic residues (Trp and His) is summarized, especially chemical properties and maturation process of the cross-links between cysteine and heteroaromatic amino acids (His–Cys and Trp–Cys cross-links).
Abbreviations: FMN: flavin mononucleotide; FAD: flavin adenine nucleotide; RNA: ribonucleic acid; PDC: protein-derived cofactor; GFP: green fluorescent protein; MIO: 3,5-dihydro-5-methylidene-4-imidazol-4-one; LTQ: lysyl tyrosylquinone; CTQ: cysteine tryptophylquinone; TTQ: tryptophan tryptophylquinone; E.coli: Escherichia coli; WT: wild type.
Grphical abstract
Protein-derived cofactors are mostly derived from aromatic amino acid residues, especially Tyr, Trp, and His, providing host proteins special functions.
Post-translational modification denotes the covalent and generally enzymatic modification of proteins. After ribosomal translation of mRNA into polypeptide chains, proteins may then frequently undergo a variety of the post-translational modification to form the mature protein product. They are pivotal for structure and several downstream functions because they involve the addition of the functional groups, for instance, glycosylation, ubiquitination, and isoprenylation [Citation1]. In some cases, cofactors also are covalently attached to proteins via post-translational modification. Among them, five types of covalent flavinylation have been identified up to the present [Citation2]. There are several modification manners, 6-S-cysteinyl-FMN in trimethylamine dehydrogenase [Citation3] and histamine dehydrogenase [Citation4,Citation5], 8-S-cysteinyl-FMN in monoamine oxidase B [Citation6], 8α-N1-histidyl FAD in cholesterol oxidase [Citation7], 8α-N3-histidyl FAD in vanillyl alcohol oxidase [Citation8], 8α-O-tyrosyl FAD in p-cresol methylhydroxylase [Citation9], and 8α-N1-histidyl-6-S-cysteinyl-FAD in glucooligosaccharide oxidase [Citation10]. Interestingly, these flavinylations have been shown to be an autocatalytic process [Citation11,Citation12]. Furthermore, there are enormous reports about the modification with smaller chemical groups such as phosphorylation, acetylation, formylation, methylation, and hydroxylation [Citation1].
Disulfide bond is the most common and widely prevalent of all known post-translational modifications, but it’s clearly different from those described above in terms of the formation of the cross-link between amino acid side chains [Citation13]. Over the last two decades, a large number of unusual post-translational modifications containing such cross-links have been discovered in ribosomally synthesized peptides thanks to mass spectrometric techniques combined with biochemical methods. Such peptides have recently been well overviewed and summarized by van der Donk et al. [Citation14]. Unusual post-translational modifications are not just confined to small peptide and, in the case of proteins, high-resolution X-ray crystal structures have allowed us to identify these modifications as comprehensively reviewed by many researchers [Citation15–Citation23]. To our best knowledge, more than 20 kinds of the post-translationally modified cross-links were newly discovered in the natural proteins so far.
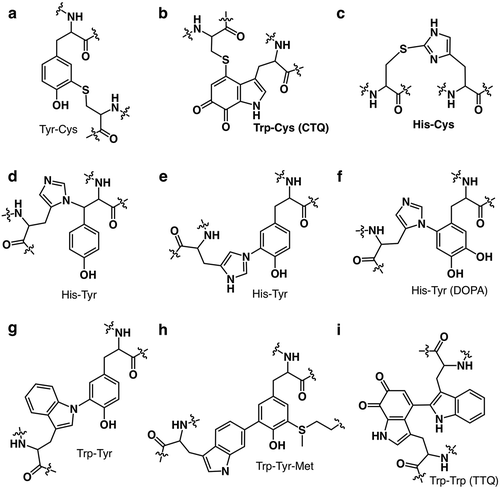
In order to obtain biological functionality, nature appears to attempt to exploit all sorts of chemical bonds, thioether, thioester, ether, ester, sulphenyl amide, sulfilimine, amide, amine, carbon–carbon, and their combinations as previously described in the literatures () [Citation15–Citation23]. In most cases, these cross-links provide the special functions to the protein as well as both intra and intermolecular structural stability. The examples include the fluorescence chromophore (4-(p-hydroxybenzylidene)-imidazolidin-5-one) of the green fluorescent protein (GFP) [Citation24], the colorimetric chromophore (bis-LTQ) of the ranasmurfin [Citation25], 3,5-dihydro-5-methylidene-4-imidazol-4-one (MIO) [Citation26], quinonoid redox cofactors and Tyr–Cys. The MIO, CTQ [Citation27,Citation28], TTQ [Citation29] and Tyr–Cys [Citation30] are directly involved in the catalytic cycles of histidine (phenylalanine) ammonia lyase, amine oxidases, amine dehydrogenase, and galactose oxidase as the redox prosthetic groups, respectively. Thus, they are referred to as protein-derived cofactors (PDC).
Figure 1. The structural representation of characteristic amino acids generated by the post-translational modifications from heteroaromatic amino acid side chains.
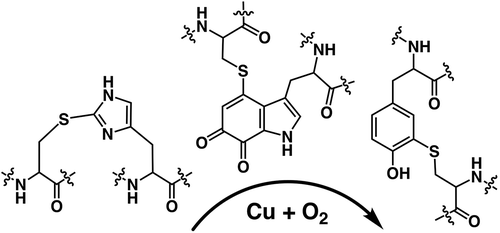
Since Tyr–Cys ()) has been identified in several kinds of proteins to date, thus one may consider that the Tyr–Cys bond is not unusual anymore. A covalent bond is formed autocatalytically between the Cε atom of tyrosine and the Sγ atom of nearby cysteine in copper-dependent galactose oxidase [Citation30] and glyoxal oxidase [Citation31], eukaryotic iron-dependent cysteine dioxygenases [Citation32], heme-containing sulfite reductase from Mycobacterium tuberculosis [Citation33], and cytochrome c nitrite reductase from Thioalkalivibrio nitratireducens [Citation34]. The covalently bound tyrosine and cysteine are located in the active sites of these enzymes and are important for their functions. In the case of galactose oxidase, formation of the Tyr radical is necessary for substrate oxidation. This radical is stabilized both by the Tyr–Cys bond itself and by the π-π stacking interaction with a nearby Trp residue [Citation35]. In the case of human cysteine dioxygenase, the Tyr–Cys cofactor enables the Tyr hydroxyl group to stabilize the oxygen radical formed during catalysis, thus preventing the formation of damaging hydroxyl radicals [Citation36]. No structure/function relationship for the Tyr–Cys bond in M. tuberculosis nitrite reductase has been suggested by explaining the reduced activity of the mutant forms of this enzyme without the Tyr–Cys bond [Citation33].
As exemplified by Tyr–Cys bond, PDCs are very hot topics in biochemistry, particular, enzymology field. Additionally, the existing of these cross-links makes it more difficult to make annotation of the unknown function proteins and genome mining approach to search out the new functional proteins. As described above, PDCs are mostly derived from aromatic amino acid residues, especially Tyr, Trp, and His ()[Citation21]. The modification and diversity of Tyr in biology have strongly attracted attention, yet those of the other two, heteroaromatic residues (Trp and His) have not been so well-known. In this review, current information of such unusual cofactors that the author’s group obtained so far is summarized, especially chemical properties and maturation process of the cross-links between cysteine and heteroaromatic amino acids [His–Cys ()) and Trp–Cys(CTQ, )) cross-link].
His–Cys in copper enzymes
Ergothioneine was first discovered in the sclerotia of the ergot fungus in 1909 as a thiourea derivative of the betaine of histidine and contains a sulfur atom bonded to the 2-position of the imidazole ring. Despite of enormous research for 100 years, the functions of ergothioneine in vivo still remain obscure, but it has been proposed that the sulfur atom is incorporated from cysteine via the oxidative sulfurization of the imidazole moiety by biosynthetic genes (EgtB) product [Citation37]. Similar chemical moiety to ergothioneine has been discovered in the copper enzymes in the proximity of copper-binding site. Namely, His-Cys cross-link was identified in the Neurospora crassa tyrosinase on the course of amino acid sequence analysis [Citation38]. Then, X-ray crystal structural analysis has identified His-Cys cross-link in Octopus hemocyanin [Citation39], plant (Ipomoea batatas) catechol oxidase [Citation40], and mushroom tyrosinase (Agaricus bisporus) [Citation41]. Interestingly, thioether linkage is present in one of the copper-ligating histidines, which is covalently bound by nearby cysteine. It was reported that the Ala mutation of this cysteine residue led to the inactivation of tyrosinase [Citation42]. One has proposed that its formation is triggered by the nucleophilic attack to Cε of histidine by cysteine thiol [Citation43]. However, its function and the formation mechanism still remain unknown.
The author and collaborators have recently reported that the apo-form of recombinant pro-tyrosinase (inactive precursor) from Aspergillus oryzae can be over-expressed in E. coli by using the full-length of gene (melB) [Citation44,Citation45]. The copper-bound pro-tyrosinase has no catalytic activity, but the trypsin-treatment transforms the holo-pro-form into the active form of the enzyme, in which the shielding domain (Glu458–Ala616) is cleaved off [Citation46]. The reported crystal structure of mushroom tyrosinase seems to have a similar state of this C-terminal absent form of melB [Citation41]. The X-ray structure of melB pro-tyrosinase is the first example of the crystal structure of a pro-form eukaryotic tyrosinase with an auxiliary domain [Citation47]. The recombinant protein from the E. coli expression system was crystallized by the conventional way, hanging drop method. The final structure of the apo-form (copper-depleted form) was refined, displaying an ellipsoid shape with dimensions of 95 Å × 65 Å × 45 Å ()). A number of very tight interactions between monomers through extensive hydrophobic and charge–charge interactions have been observed supporting the homo-dimeric state at physiological condition described in our previous study [Citation46]. The monomers attached with each other in a side-to-side fashion along a two-fold axis; thus, the substrate-binding cavities face opposite ways so as not to interfere with each other ()) [Citation47].
Figure 2. The superimposed crystal structure of melB pro-tyrosinase with other tyrosinases. (a) whole structure of apo (white, PDB code:3W6Q) and holo form (green, PDB code:3W6W) (b) Copper-binding domains of mushroom (blue, PDB code: 2y9w), plant catechol oxidase (orange, PDB code: 1BT3), and the functional unit g of octopus hemocyanin (pink, PDB code: 1JS8). The protein main chain is displayed as ribbon and key cysteine residues are highlighted as sticks and green sphere indicates copper.
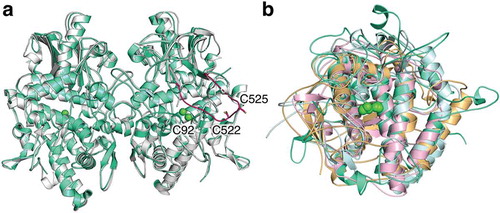
Each monomer has two structural distinct domains: the N-terminal and C-terminal sides are designated as copper-binding domain (Ser1–Lys457) and shielding domain (Glu458–Ala616), respectively. The structure of copper-binding domain is similar with that of mushroom tyrosinase reported previously (PDB code: 2y9w) [Citation41] and those of the other dinuclear copper proteins such as plant catechol oxidase (PDB code: 1BT3) [Citation40] and the functional unit g of octopus hemocyanin (PDB code: 1JS8, )) [Citation39].
In these copper proteins, the highly conserved six histidine residues as the copper ligands are present in a four-helix bundle of the copper-binding domain as a core structure and they sit in the active site of melB tyrosinase as well observed [three histidine imidazoles for each copper ion, CuA (His67, His94, and His103, melB numbering) and CuB (His328, His332 and His 371, melB numbering)]. In this structure, as expected, no bound copper was observed and intriguingly the Cys92, which forms a thioether linkage to one of these histidines (His94), does not bind covalently with His94 via a thioether linkage. It should be noted that two conformers of Cys92 were observed, demonstrating that this cysteine residue flexibly flips () and )) [Citation47].
Figure 3. The crystal structure of melB pro-tyrosinase copper-binding site. (a) apo form (b) holo form.
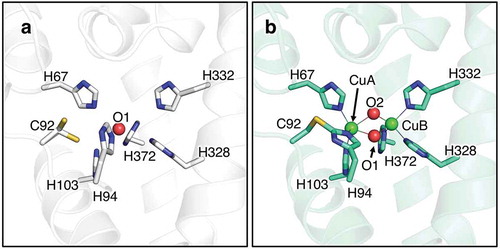
The high-resolution crystal structure of holo-form (copper bound from) of melB pro-tyrosinase was also determined at a resolution of 1.39 Å. The subunit structure is almost identical to that of apo-form, indicating that copper binding does not affect the tyrosinase structure ()). In stark contrast, its copper-binding site seems to be clearly different from that of apo-form ()). All of the conserved six histidines ligated to two coppers, CuA and CuB and the thioether linkage between Cys92 and His94 was seen. Furthermore, the distance between CuA and CuB is 3.5 Å, suggesting that this is met-form (dinuclear copper (II) state). These results strongly support that the formation of His-Cys cross-link occurs in the process of the copper incorporation [Citation47].
Generation of His–Cys cross-link involving with copper incorporation
Through in vitro reconstitution of copper-binding site, the author and collaborators revealed that His-Cys cross-link formation proceeds autocatalytically in the pro-tyrosinase without the aid of any other external catalysts [Citation45]. Namely, the apo-form of pro-tyrosinase exhibited that of met-form (dinuclear copper (II) state) after the treatment of CuSO4. Besides this, formation of His–Cys cross-linkage was examined by free thiol (cysteine) quantification experiment. Before the treatment of CuSO4, the thiol number of wild type (WT) and C92A mutant was determined to be 6 mol/subunit and 5 mol/subunit, respectively. However, both values changed to 3 mol/subunit after the treatment of CuSO4. The values were contrary to normal expectations, but consistent with the hypothesis that one disulfide bond produced via the oxidation catalyzed by CuSO4 in addition to His-Cys cross-link formation. On the other hand, holo-C92A mutant showed the spectral features due to the (μ-η2:η2-peroxo)dicopper(II) species, which is the oxygen-bound form in side-on fashion and one state during the catalytic cycle in matured tyrosinase [Citation48]. This presumably suggests that Cu(II) ions are reduced by two cysteines to give deoxy-form (dinuclear copper (I) state, , d2→e). This species can react with molecular dioxygen producing the (μ-η2:η2-peroxo)dicopper(II) species. Furthermore, comparison of the behavior in the CuSO4 treatment under aerobic conditions between WT and C92A mutant has suggested that the (μ-η2:η2-peroxo)dicopper(II) species is involved as a key reactive intermediate for the His–Cys formation [Citation45]. In the case of the formation of Tyr–Cys cross-link, it has been demonstrated that such kind of active oxygen-copper species triggers its initial step. Tyr–Cys formation requires at least a two-electron oxidation of the Cu(II)-containing precursor enzyme, where was found to be autocatalytic, requiring only copper and O2. The possible mechanisms for the Tyr–Cys coupling reaction were proposed so far, which is an initial electron transfer from Tyr to Cu ion followed by either a radical coupling reaction or nucleophilic attack of thiolate [Citation49]. Furthermore, the reaction proceeded much more efficiently when Cu(I) was used [Citation50]. In this case, the activation of dioxygen is a key step to produce thiyl radical for the active species.
Figure 4. The proposed mechanism of copper incorporation into fungal pro-tyrosinase.
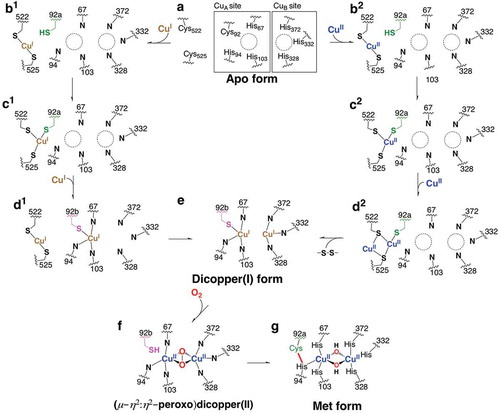
In the crystal structure of melB tyrosinase, the two cysteine residues can be found corresponding to the disulfide bond that was described in the previous paragraph. The shielding domain bears an unusual long loop protruding from the juxtaposed two-β strands near copper-binding site and bending with a small angle in the middle of loop (), magenta) [Citation47]. On its turnover, the conserved CXXC motif (Cys522 and Cys525) was found, which is an essential motif in copper chaperones [Citation51,Citation52]. Three mutants (C552A, C525A, and C522A/C525A) showed the lower copper-uptaking ability than that of WT in vivo. This has demonstrated that this CXXC motif is closely related to the incorporation of copper ions into the active site [Citation47]. After this finding, Rompel et al. also identified this motif in the crystal structure of mushroom pro-tyrosinase and designated this unusual flexible part as a “copper grappler” implying the role to shuttle copper ions to the active site of pro-tyrosinase [Citation53].
The Cys92 sits spatially between the CXXC motif and copper-binding site. One conformer faces to the CXXC motif, while another one points to CuA binding site ()). Taking these findings together, we have proposed a mechanism of the formation of His-Cys cross-link in the process of copper incorporation as shown in . In both the cases of Cu(I) (b1→d1) and Cu(II) (b2→d2), the first copper ion is “grabbed” by the CXXC motif with Cys522 and Cys525 and transferred close to Cys92 (a→b→c). Another copper ion is carried in a similar manner. Whereas this process directly produces a dicopper(I) form in the case of Cu(I) (d1→e), the reduction takes place in the case of Cu(II) to give the same species (d2→e). As a via point, Cys92 may rotate from the CXXC side to the dicopper binding side enhancing the copper(I) transportation. From the dicopper(I) form thus generated, the (μ-η2:η2-peroxido)dicopper(II) species is produced through the reaction with dioxygen (e→f). We have proposed that this (μ-η2:η2-peroxido)dicopper(II) species accounts for triggering the cross-link formation between His94 and Cys92 on the basis of these detailed spectroscopic and mutational studies described above [Citation45]. There are several possibilities regarding mechanistic details of the oxidative coupling reaction between His94 and Cys92. Hydrogen atom abstraction from the thiol group of Cys92 by the peroxide species generates a thiyl radical intermediate, which then attacks to the ε carbon of the imidazole group of His94 to induce the C–S bond formation. Then, the generated imidazolyl radical intermediate containing the C–S bond releases another hydrogen atom to the oxyl radical of the generated Cu(II)(O•)(OH)Cu(II) species to complete the posttranslational modification (f→g). On the other hand, the hydroxylation of Cys92 in the precursor protein, which may be well-known metal-induced oxygenation of thiolates, is possibly another potential mechanism. This involves the initial oxygenation of Cys92 to a sulfenic acid, which could act as an electrophile toward His94. In analogy with the biosynthesis of ergothioneine [Citation37], the hydroxylation of thiol of Cys92 is assumed to occur via a direct nucleophilic attack to peroxide species to give a cysteine sulfenic acid as a possible alternative way. Then, imidazole attacks to a sulfur atom of sulfenic acid invoking the dissociation of hydroxyl group from the sulfur atom. Similar way was proposed in Tys–Cys and sulfenic acid may act as an electrophile in His–Cys formation.
It is noteworthy that such Cys mutation resulted in the complete loss or significant decrease in the catalytic activity [Citation42,Citation45]. Thus, His-Cys cross-link play significant roles in the catalytic properties, where thioether substituent seems to perturbate the donor effect of imidazole moiety of copper-ligating histidine. Furthermore, the cross-link may also increase catalytic activity by controlling deleterious interactions of the thiolate or thiol of the uncross-linked Cys with Cu ion or substrates in the case of cysteine dioxygenases [Citation54], although uncross-linked Cys92 is responsible for copper incorporation process.
Cys-Trp cross-link in cysteine tryptophylquinone (CTQ)
The discovery of PQQ-containing enzymes [Citation55] has been followed by the incredible finding of a protein tyrosine-derived cofactor, TOPA quinone (TPQ) in an eukaryotic copper amine oxidase [Citation55]. Then, a tryptophan tryptophyl quinone cofactor (TTQ) has been found in methylamine dehydrogenase [Citation29], and lysine tyrosyl quinone (LTQ) has been seen in lysyl oxidase as well [Citation56], where a tyrosine-based quinone is cross-linked to lysine. Given these findings of the several variants derived from tryptophan and tyrosine structure, unusual quinone cofactors have turned out to be rich in terms of molecular structures. Quinohemoprotein amine dehydrogenase (QHNDH) is expressed in Paracoccus denitrificans [Citation27] and Pseudomonas putida [Citation28] when adding biological amines in the media as the sole source of carbon, nitrogen, and energy. QHNDHs are soluble enzymes that can catalyze the oxidative deamination of amines to formaldehydes and ammonia. QHNDH has a αβγ heterotrimeric structure and, surprisingly, the smallest γ subunit has a peculiar quinone cofactor, in which a tryptophyl quinone is cross-linked to a cysteine to form cysteine tryptophylquinone (CTQ).
The author and collaborators have attempted to characterize the physicochemical features of CTQ. QHNDH has two hemes c in the largest β subunit. Thereby, it is difficult to directly observe the UV-vis spectra of CTQ due to the spectral overlapping of CTQ with the very intense absorption band of hemes. Thereby, the author and collaborators isolated the monomeric γ subunit from the complex structure of QHNDH by size-exclusion chromatography after denaturation of QHNDH upon urea addition. During the redox titration with several electron-transfer mediators, the fully oxidized and reduced forms of the γ subunit exhibited a characteristic absorption peak at 380 nm and a shoulder band around 315 nm, respectively. The author determined the two-electron redox potential of CTQ in the isolated γ subunit to be 65 mV (vs. SHE) at pH 7.0 by the redox titration. Based on the pH dependence of the redox potential, the proton-coupled electron transfer of CTQ changes its mode. Namely, the two-proton two-electron transfer takes place at pH less than 8.6, whereas one-proton two-electron transfer at pH more than 8.6. This value (8.6) corresponds to the pKa value (Ka: acid-dissociation constant), which can be assigned to one of the phenolic OH groups of the quinol form [Citation57].
The redox reaction of CTQ in the isolated γ subunit proceeds via two-electron transfer fashion, thus a semiquinone radical was not observed at all. However, the signal due to semiquinone form of CTQ was observed by ESR spectroscopy of the intact QHNDH partial reduced with dithionite [Citation58]. Naturally, the redox properties in the native protein environment are different from isolated one and are necessary for the elucidation of the catalytic mechanism of enzymes. Recently, some enzymes containing CTQ have been discovered without any other cofactors such as heme molecules [Citation59,Citation60]. These enzymes are a member of a family of CTQ-containing oxidases, so-called LodA-like protein and the members of this class that have been functionally characterized to date are amino acid oxidases. In the crystal structures, lysine ε-oxidase (LodA) from Marinomonas mediterranea (PDB code: 3WEU) [Citation59] and glycine oxidase (GoxA) from Pseudomonas luteoviolacea (PDB code: 6BYW) [Citation61] have none of the cofactors or metals, but the protein-derived CTQ cofactor as the sole cofactor. Thus, in GoxA, the physicochemical properties of the CTQ have been characterized directly in spectroscopic techniques [Citation62]. Based on our results, these recent findings will provide the new insight into the catalytic mechanism of the CTQ in the near future.
Inactive form of QHNDH
The formation of a covalent Trp-Cys cross-link between the indole ring and the sulfur atom of Cys is involved in the biosynthesis of CTQ, which occurs simultaneously insertion of two oxygen atoms into the indole ring of Trp. The formation mechanism of CTQ is still a mystery and its pathway in QHNDH seems to be distinct from that in LodA-like protein [Citation63–Citation66]. In the case of QHNDH, the biosynthetic pathway of CTQ is proposed as follows. First, transcription of the qhpADCBEF operon as well as qhpG is activated by the binding of QhpR with amines. After the expression of proteins, triple intrapeptidyl thioether cross-links are formed within the intact γ subunit (QhpC) by the radical SAM enzyme QhpD [Citation63]. The QhpG which is a flavoenzyme may catalyze the hydroxylation of Trp (CTQ precursor) in the cross-linked γ subunit to form pre-CTQ, mono-hydroxylated Trp [Citation64]. The leader sequence of the γ subunit is cleaved off by the subtilisin-like protease QhpE [Citation65]. In the periplasm, the α subunit (QhpA) incorporated two hemes c in the periplasm, which may assist the formation of the Trp-Cys cross-link [Citation66].
In stark contrast, the pathway of CTQ biogenesis in LodA appears to be much simpler than that in QHNDH. The lod operon harbors only two genes, lodA and lodB. The lodA encodes a CTQ-containing enzyme precursor, LodA, and the lodB was predicted to encode a flavoenzyme. Active LodA (matured enzyme) was obtained only when lodA was expressed with lodB at the same time. In a recombinant Escherichia coli expression system without lodB gene, lodA produces an inactive precursor protein with a monohydroxlated Trp residue [Citation67]. This was also observed for the precursor of QHNDH that was expressed in the absence of QhpD [Citation64]. Similarly, co-expression of goxA and goxB which is analogous to lodA and lodB, respectively, also produced a GoxA with glycine oxidase activity [Citation60,Citation61]. Thus, the mono-hydroxylation of the precursor Trp residue is a key step to share with both systems. However, the step is catalyzed by flavoenzyme, QhpG, in QHNDH, whereas takes place prior to the post-translational modifications catalyzed by the flavoprotein, LodB and GoxB, in LodA-like protein [Citation67]. In the latter case, it is possible for free copper to catalyze the initial hydroxylation of the unmodified precursor by the binding to an Asp residue near a Trp residue that forms CTQ.
On the other hand, P. denitrificans expresses three soluble hemoproteins with QHNDH induced by butylamine. One of those, designated as “heme 2”, exhibited two bands of molecular mass 59.5 kDa and 36.5 kDa on SDS-PAGE [Citation58]. This band profile was almost identical with that of QHNDH, but heme 2 showed no QHNDH activity. Thus, the author and collaborators attempted to characterize this hemoprotein. The protein was slowly activated upon the addition of substrates, butylamine, and phenethylamine, and far more swiftly activated upon reduction with dithionite and dithiothreitol, presumably indicating that heme 2 is inactive form of QHNDH (silent form) [Citation68]. The heme 2 was inert to 4-nitrophenylhydrazine and its γ subunit showed a UV-vis spectrum with a peak at 390 nm, which is quite different from that of the native enzyme and completely identical to that of hydroxylamine-treated one, strongly demonstrating that silent form contains a 6-oxime form of CTQ as a chromophore (). Furthermore, this has been confirmed by mass spectroscopy, where the molecular mass was larger than that of the native enzyme by 15.1 (nitrogen plus hydrogen atom) [Citation68].
Figure 5. The proposed molecular structure of CTQ-6-oxime in QHNDH.
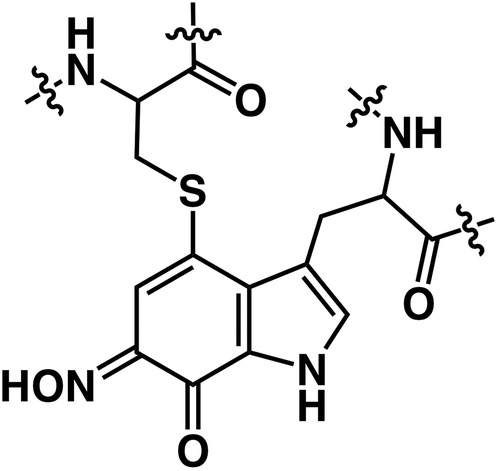
There are two possibilities how the silent form produces. QHNDH dissociates ammonium cation from biological amines during the catalytic cycle. Thus, given ammonia is further metabolized by ammonia monooxygenase-like enzyme to produce hydroxylamine molecule. One is the following; this reactive species may attach to the fully matured CTQ in QHNDH to suppress its activity. The another is that silent form is generated via nucleophilic attack by hydroxylamine after the activation of mono-hydroxylated Trp in the course of CTQ formation. The further information will reveal both the mechanisms of CTQ formation as well as the role of silent form of QHNDH.
Conclusion
The author reviewed recent progress in research on a new class of post-translational modification, protein-derived cofactors containing the covalent cross-link between amino acid residues in proteins. The biosynthetic pathway of His-Cys and Trp-Cys cross-link has been examined in the analogy of Tyr-Cys cross-link. These investigations have demonstrated that their mechanism shares the occurrence of metal-active oxygen species as the key step in the post-translational chemical modifications. The characterization of the chemical properties of these two cross-links has revealed that this modification controls the host enzyme activities. Further accumulation of knowledge will not only be driving force to discover new post-translational cross-linkings but also can provide challenges to create the unprecedent protein-derived cofactors in artificial proteins or natural proteins.
Acknowledgments
This review article partly includes the summary of the work which received the “JSBBA Award for Young Scientists”, and the author sincerely thanks the members of the selection committee. Our group’s studies presented in this review were mainly carried out at Kyoto University and Osaka University. The author sincerely thanks all the students and staff who helped with this study. The author especially thanks Prof. Kenji Kano (Kyoto University) who proposed the topic of this study and provided abundant guidance and encouragement. The author would also like to thank Prof. Shinobu Itoh (Osaka University) who gave me significant information and encouragement. This study was also supported by many collaborators who belong to various research institutes, and the author thanks them sincerely for their kind support.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chemie Int Ed. 2005;44:7342–7372.
- Heuts DPHM, Scrutton NS, McIntire WS, et al. What’s in a covalent bond? On the role and formation of covalently bound flavin cofactors. Febs J. 2009;276:3405–3427.
- Steenkamp DJ, McIntire W, Kenney WC. Structure of the covalently bound coenzyme of trimethylamine dehydrogenase. Evidence for a 6-substituted flavin. J Biol Chem. 1978;253:2818–2824.
- Fujieda N, Satoh A, Tsuse N, et al. 6-S-cysteinyl flavin mononucleotide-containing histamine dehydrogenase from Nocardioides simplex: molecular cloning, sequencing, overexpression, and characterization of redox centers of enzyme. Biochemistry. 2004;43:10800–10808.
- Fujieda N, Tsuse N, Satoh A, et al. Production of completely flavinylated histamine dehydrogenase, unique covalently bound flavin, and iron-sulfur cluster-containing enzyme of nocardioides simplex in Escherichia coli, and its properties. Biosci Biotechnol Biochem. 2005;69:2459–2462.
- Zhou BP, Lewis DA, Kwan SW, et al. Flavinylation of monoamine oxidase B. J Biol Chem. 1995;270:23653–23660.
- Coulombe R, Yue KQ, Ghisla S, et al. Oxygen access to the active site of cholesterol oxidase through a narrow channel is gated by an Arg-Glu pair. J Biol Chem. 2001;276:30435–30441.
- Fraaijet MW, Van Den Heuvel RHH, Van Berkel WJH, et al. Covalent flavinylation is essential for efficient redox catalysis in vanillyl-alcohol oxidase. J Biol Chem. 1999;274:35514–35520.
- Scott Mathews F, Chen ZW, Bellamy HD, et al. Three-dimensional structure of p-cresol methylhydroxylase (flavocytochrome c) from Pseudomonas putida at 3.0-Å resolution. Biochemistry. 1991;30:238–247.
- Huang CH, Lai WL, Lee MH, et al. Crystal structure of glucooligosaccharide oxidase from Acremonium strictum: a novel flavinylation of 6-S-cysteinyl, 8α-N1-histidyl FAD. J Biol Chem. 2005;280:38831–38838.
- Mewies M, McIntire WS, Scrutton NS. Covalent attachment of flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) to enzymes: the current state of affairs. Protein Sci. 1998;7:7–21.
- Jin J, Mazon H, van den Heuvel RH, et al. Covalent flavinylation of vanillyl-alcohol oxidase is an autocatalytic process. Febs J. 2008;275:5191–5200.
- Sevier CS, Kaiser CA. Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. 2002;3:836–847.
- Arnison PG, Bibb MJ, Bierbaum G, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160.
- Okeley NM, Van Der Donk WA. Novel cofactors via post-translational modifications of enzyme active sites. Chem Biol. 2000;7:R159–R171.
- Halcrow MA. Chemically modified amino acids in copper proteins that bind or activate dioxygen. Angew Chem. 2001;40:346–349.
- Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292.
- Davidson VL. Generation of protein-derived redox cofactors by posttranslational modification. Mol Biosyst. 2011;7:29–37.
- Yukl ET, Wilmot CM. Cofactor biosynthesis through protein post-translational modification. Curr Opin Chem Biol. 2012;16:54–59.
- Davidson VL, Wilmot CM. Posttranslational biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone. Annu Rev Biochem. 2013;82:531–550.
- Ravikiran B, Mahalakshmi R. Unusual post-translational protein modifications: the benefits of sophistication. RSC Adv. 2014;4:33958–33974.
- Jones LH, Narayanan A, Hett EC. Understanding and applying tyrosine biochemical diversity. Mol Biosyst. 2014;10:952–969.
- Klinman JP, Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2014;114:4343–4365.
- Ormö M, Cubitt AB, Kallio K, et al. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395.
- Oke M, Ching R, Carter L, et al. Unusual chromophore and cross-links in ranasmurfin: a blue protein from the foam nests of a tropical frog. Angew Chem Int Educ. 2008;47:7853–7856.
- Schwede TF, Rétey J, Schulz GE. Crystal structure of histidine ammonia-lyase revealing a novel polypeptide modification as the catalytic electrophile. Biochemistry. 1999;38:5355–5361.
- Datta S, Mori Y, Takagi K, et al. Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc Natl Acad Sci. 2001;98:14268–14273.
- Satoh A, Kim JK, Miyahara I, et al. Crystal structure of quinohemoprotein amine dehydrogenase from Pseudomonas putida. Identification of a novel quinone cofactor encaged by multiple thioether cross-bridges. J Biol Chem. 2002;277:2830–2834.
- McIntire W, Wemmer D, Chistoserdov A, et al. A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science. 1991;252:817–824.
- Ito N, Phillips SEV, Stevens C, et al. Novel thioether bond revealed by a 1.7 A crystal structure of galactose oxidase. Nature. 1991;350:87–90.
- Whittaker MM, Kersten PJ, Cullen D, et al. Identification of catalytic residues in glyoxal oxidase by targeted mutagenesis. J Biol Chem. 1999;274:36226–36232.
- McCoy JG, Bailey LJ, Bitto E, et al. Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci U S A. 2006;103:3084–3089.
- Schnell R, Sandalova T, Hellman U, et al. Siroheme- and [Fe4-S4]-dependent NirA from Mycobacterium tuberculosis is a sulfite reductase with a covalent Cys-Tyr bond in the active site. J Biol Chem. 2005;280:27319–27328.
- Polyakov KM, Boyko KM, Tikhonova TV, et al. High-resolution structural analysis of a novel octaheme cytochrome c nitrite reductase from the haloalkaliphilic bacterium Thioalkalivibrio nitratireducens. J Mol Biol. 2009;389:846–862.
- Whittaker JW. Free radical catalysis by galactose oxidase. Chem Rev. 2003;103:2347–2363.
- Ye S, Wu X, Wei L, et al. An insight into the mechanism of human cysteine dioxygenase. Key roles of the thioether-bonded tyrosine-cysteine cofactor. J Biol Chem. 2007;282:3391–3402.
- Seebeck FP. In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J Am Chem Soc. 2010;132:6632–6633.
- Lerch K, Il E. Amino acid sequence of tyrosinase from Neurospora crassa. Proc Natl Acad Sci. 1978;75:3635–3639.
- Cuff ME, Miller KI, van Holde KE, et al. Crystal structure of a functional unit from Octopus hemocyanin. J Mol Biol. 1998;278:855–870.
- Klabunde T, Eicken C, Sacchettini JC, et al. Crystal structure of a plant catechol oxidase containing a dicopper center. Nat Struct Biol. 1998;5:1084–1090.
- Ismaya WT, Rozeboom HJ, Weijn A, et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50:5477–5486.
- Nakamura M, Nakajima T, Ohba Y, et al. Identification of copper ligands in Aspergillus oryzae tyrosinase by site-directed mutagenesis. Biochem J. 2000;350(Pt 2):537–545.
- Virador VM, Reyes Grajeda JP, Blanco-Labra A, et al. Cloning, sequencing, purification, and crystal structure of Grenache (Vitis vinifera) polyphenol oxidase. J Agric Food Chem. 2010;58:1189–1201.
- Obata H, Ishida H, Hata Y, et al. Cloning of a novel tyrosinase-encoding gene (Melb) from Aspergillus oryzae and its overexpression in solid-state culture (Rice Koji). J Biosci Bioeng. 2004;97:400–405.
- Fujieda N, Ikeda T, Murata M, et al. Post-translational his-cys cross-linkage formation in tyrosinase induced by copper(II)-peroxo species. J Am Chem Soc. 2011;133:1180–1183.
- Fujieda N, Murata M, Yabuta S, et al. Multifunctions of MelB, a fungal tyrosinase from Aspergillus oryzae. Chembiochem. 2012;13:193–201.
- Fujieda N, Yabuta S, Ikeda T, et al. Crystal structures of copper-depleted and copper-bound fungal pro-tyrosinase: insights into endogenous cysteine-dependent copper incorporation. J Biol Chem. 2013;288:22128–22140.
- Solomon EI, Heppner DE, Johnston EM, et al. Copper active sites in biology. Chem Rev. 2014;114:3659–3853.
- Firbank SJ, Rogers MS, Wilmot CM, et al. Crystal structure of the precursor of galactose oxidase: an unusual self-processing enzyme. Proc Natl Acad Sci. 2001;98:12932–12937.
- Whittaker MM, Whittaker JW. Cu(I)-dependent biogenesis of the galactose oxidase redox cofactor. J Biol Chem. 2003;278:22090–22101.
- O’Halloran TV, Culotta VC. Metallochaperones, an intracellular shuttle service for metal ions. J Biol Chem. 2000;275:25057–25060.
- Prohaska JR, Gybina AA. Intracellular copper transport in mammals. J Nutr. 2004;134:1003–1006.
- Mauracher SG, Molitor C, Al-Oweini R, et al. Latent and active abPPO4 mushroom tyrosinase cocrystallized with hexatungstotellurate(VI) in a single crystal. Acta Crystallogr Sect D Biol Crystallogr. 2014;70:2301–2315.
- Davies CG, Fellner M, Tchesnokov EP, et al. The Cys-Tyr cross-link of cysteine dioxygenase changes the optimal pH of the reaction without a structural change. Biochemistry. 2014;53:7961–7968.
- Janes S, Mu D, Wemmer D, et al. A new redox cofactor in eukaryotic enzymes: 6-hydroxydopa at the active site of bovine serum amine oxidase. Science. 1990;248:981–987.
- Wang SX, Mure M, Medzihradszky KF, et al. A crosslinked cofactor in lysyl oxidase: redox function for amino acid side chains. Science. 1996;273:1078–1084.
- Fujieda N, Mori M, Kano K, et al. Spectroelectrochemical evaluation of redox potentials of cysteine tryptophylquinone and two hemes c in quinohemoprotein amine dehydrogenase from Paracoccus denitrificans. Biochemistry. 2002;41:13736–13743.
- Takagi K, Torimura M, Kawaguchi K, et al. Biochemical and electrochemical characterization of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans. Biochemistry. 1999;38:6935–6942.
- Okazaki S, Nakano S, Matsui D, et al. X-ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in L-lysine epsilon-oxidase from Marinomonas mediterranea. J Biochem. 2013;154:233–236.
- Sehanobish E, Williamson HR, Davidson VL. Roles of conserved residues of the glycine oxidase GoxA in controlling activity, cooperativity, subunit composition, and cysteine tryptophylquinone biosynthesis. J Biol Chem. 2016;291:23199–23207.
- Andreo-Vidal A, Mamounis KJ, Sehanobish E, et al. Structure and enzymatic properties of an unusual cysteine tryptophylquinone-dependent glycine oxidase from Pseudoalteromonas luteoviolacea. Biochemistry. 2018;57:1155–1165.
- Ma Z, Davidson VL. The redox properties of a cysteine tryptophylquinone-dependent glycine oxidase are distinct from those of tryptophylquinone-dependent dehydrogenases. Biochemistry. 2019;58:2243–2249.
- Nakai T, Ito H, Kobayashi K, et al. The radical S-Adenosyl-L-methionine enzyme QhpD catalyzes sequential formation of intra-protein sulfur-to-methylene carbon thioether bonds. J Biol Chem. 2015;290:11144–11166.
- Ono K, Okajima T, Tani M, et al. Involvement of a putative [Fe-S]-cluster-binding protein in the biogenesis of quinohemoprotein amine dehydrogenase. J Biol Chem. 2006;281:13672–13684.
- Nakai T, Ono K, Kuroda S, et al. An unusual subtilisin-like serine protease is essential for biogenesis of quinohemoprotein amine dehydrogenase. J Biol Chem. 2012;287:6530–6538.
- Nakai T, Deguchi T, Frébort I, et al. Identification of genes essential for the biogenesis of quinohemoprotein amine dehydrogenase. Biochemistry. 2014;53:895–907.
- Chacón-Verdú MD, Campillo-Brocal JC, Lucas-Elío P, et al. Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-epsilon-oxidase and glycine oxidase from Marinomonas mediterranea. Biochim Biophys Acta - Proteins Proteomics. 2015;1854:1123–1131.
- Fujieda N, Mori M, Ikeda T, et al. The silent form of quinohemoprotein amine dehydrogenase from Paracoccus denitrificans. Biosci Biotechnol Biochem. 2009;73:524–529.