
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Monogenic autoinflammatory syndromes (MAISs), are caused by pathogenic genetic variants in the innate immune system, leading to dysregulation and aberrant inflammasome activation spontaneously or with minimal triggering. The diagnosis and treatment of MAISs can be intricate, relying on an increased recognition of potential differential diagnoses. This review examines the clinical features of MAIS, with a special focus on uveitis. It also evaluates treatment options and assesses the effects of activating molecular and cytokine pathways.
Monogenic autoinflammatory syndromes (MAISs) arise from disorders of the innate immune system. Familial Mediterranean fever (FMF), tumour necrosis factor (TNF) receptor-associated periodic fever syndrome (TRAPS), mevalonate kinase deficiency (MKD), and cryopyrin-associated periodic syndrome (CAPS) are the most extensively studied monogenic autoinflammatory conditions.Citation1 The episodes of inflammation are caused by a dysregulated immune system, which leads to excessive production of pro-inflammatory cytokines, for example, interleukin-1β (IL-1β).
Inflammasomes are multiprotein complexes belonging to the family of pattern recognition receptors and are an integral part of the innate immune system.Citation2 These multiprotein complexes can sense pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). A well-characterised PAMP is lipopolysaccharide (LPS), located on the outer cell wall of gram-negative bacteria. DAMPs originate from host cells, encompassing tumour or dying cells, as well as substances that cells release in response to different types of stress.
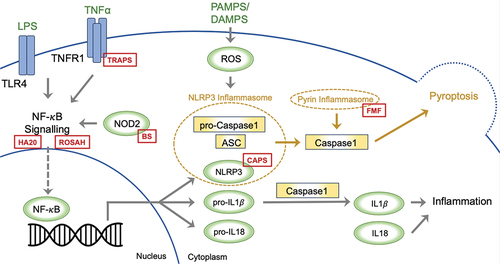
There are several inflammasomes, and in general, these complexes typically form around a cytoplasmic receptor belonging to the nucleotide-binding leucine-rich repeat-containing receptor (NLR) family (such as NLRP3; ), although other cytoplasmic receptors such as pyrin have been described. At present, a two-step framework is proposed to explain the commencement of NLRP3 inflammasome activation.Citation3 The first step involves priming of the system via the NF-κB signalling pathway, which promotes transcription of NLRP3, and the inactive pro-IL-1β and pro-IL18. The second step involves activation of the inflammasome upon recognition of a microbial, danger, or homeostasis pattern by the receptor. NLRP3 connects with the adaptor protein ASC as a response and recruits procaspase-1, which leads to the self-cleaving activation of caspase-1 (). The ensuing inflammatory response is highly dependent on caspase-1, which in turn results in cleavage of inactive pro-IL-1β and pro-IL-18 to the active IL-1β and IL-18. Caspase activation also leads to pyroptosis, a specific form of programmed cell death, that is executed through the cleavage of Gasdermin D. In illustrating the inflammasome activation, we highlight distinct locations where pathogenic variants lead to the development of MAISs. This figure highlights how particular genetic changes in the components of the inflammasome system can lead to the development of these autoinflammatory conditions.
Figure 1. A two-step model has been proposed to explain the initiation of NLRP3 inflammasome activation. The first step entails system priming through the NF-κB signalling pathway, promoting the transcription of NLRP3, pro-IL-1β, and pro-IL-18. In the second step, the inflammasome activates upon the recognition of PAMPS or DAMPS. NLRP3 associates with the adaptor protein ASC, prompting the recruitment of procaspase-1, which leads to caspase-1 self-cleaving activation. The ensuing inflammatory response significantly hinges on caspase-1, facilitating the conversion of inactive pro-IL-1β and pro-IL-18 into active IL-1β and IL-18. Caspase activation also triggers pyroptosis; a specific form of programmed cell death. The red boxes depict the sites within inflammasome activation, where pathogenic variants drive the development of monogenic autoinflammatory syndromes; ASC: Apoptosis-Associated Speck-Like Protein, BS: Blau Syndrome, CAPS: Cryopyrin-Associated Periodic Syndromes, DAMPs: Damage-Associated Molecular Patterns, FMF: Familial Mediterranean Fever, HA20: Haploinsufficiency A20, IL: Interleukin, LPS: Lipopolysaccharide. NLRP3: nucleotide-binding leucine-rich repeat-containing receptor 3, NOD2: Nucleotide-Binding Oligomerisation Domain-Containing Protein 2, NF-κB: Nuclear Factor Kappa B, PAMPs: Pathogen-Associated Molecular Patterns, ROSAH: Retinal dystrophy, Optic nerve oedema, Splenomegaly, Anhidrosis, and Headache Syndrome, TLR4: Toll-Like Receptor 4, TNFα: Tumour Necrosis Factor Alpha, TNFR1: Tumour Necrosis Factor Receptor 1, TRAPS: Tumour Necrosis Factor Receptor-Associated Periodic Syndrome.
Variations in clinical presentation among patients with MAISs stem from both the specific pathogenic variant, as well as the distinctive cellular distribution of a given inflammasome and its associated substrates. The diagnosis of MAISs can be intricate, relying on an increased recognition of potential differentials. In this review, we delve into the clinical characteristics of MAISs, with a specific emphasis on uveitis, and explore the corresponding treatments for each condition. Our aim is to elucidate how these distinct molecular and cytokine pathways influence the selection of appropriate treatments for each condition.
Familial Mediterranean fever (FMF)
FMF is an autosomal recessive condition caused by pathogenic variants of the MEFV gene, encoding the pyrin protein.Citation6 FMF is predominantly seen in Mediterranean populations with highest prevalence in Arab, North African Jewish, Middle Eastern, Turkish, and Armenian ethnicities.Citation7 In Armenian and Israeli populations, the carrier rate varies from 1 in 4 to 1 in 8. However, the disease also occurs in other populations and is not exclusive to places with higher prevalence, displaying variation in genotype and phenotype ().Citation8
A pathogenic variant in the MEFV gene was the first to be implicated within the pathophysiology of FMF or any other autoinflammatory disease and is also relevant in Behçet’s disease and ankylosing spondylitis.Citation9 Pyrin is an innate immune system protein found predominantly in myeloid lineage cells, fibroblasts, and dendritic cells. Pyrin’s role as a pattern recognition receptor allows it to detect pathogen virulence activity and stimulate a pyrin inflammasome, which leads to an inflammatory response.Citation10 Normally, pyrin is regulated by a RhoA-dependent phosphorylation and subsequent interaction with the 14-3-3 protein. An increasing body of evidence suggests that MEFV pathogenic variants lead to changes in pyrin that block phosphorylation sites for kinases, resulting in a lowered threshold for activation of the pyrin inflammasome.Citation11 This leads to an increased secretion of IL-1b and IL-18, with the resulting pro-inflammatory effects.Citation12
The disease may initially present in children with recurrent attacks of fever alone but typically progresses to more classic features such as pleuritis, peritonitis, and arthritis within 3 years of onset.Citation13 Within the last decade, the potential clinical presentation has expanded and symptoms such as severe myalgia, protracted febrile myalgia syndrome, scrotal swelling, and cardiac involvement have been described in paediatric populations.Citation14 According to new diagnostic criteria, diagnosis requires the presence of more than two of five major criteria (fever, abdominal pain, chest pain, arthritis, and family history of FMF).Citation15 This new criterion has a higher sensitivity and specificity (86.5% and 93.6% in a Turkish population) than the previously used Tel Hashomer criteria.Citation16 However, the figures have not been replicated in populations with lower incidences of FMF.
With respect to ocular involvement in FMF, the first observation was made in 1959, when Michaelson et al. noted dotted lesions on fundoscopy in Bruch’s membrane identified as colloid bodies.Citation17 Since then, FMF has been linked to other ocular conditions such as acute posterior multifocal placoid pigment epitheliopathy and keratoconus where it may be a predisposing factor, especially if a patient is a carrier of a homozygous pathogenic variant.Citation18 The most common ocular finding in a meta-analysis was the gradual decline in choroidal thickness, documented in 101 patients (47.9%).Citation19 Retinal vasculitis was noted in 66 patients (31.2%), with rates of anterior uveitis of approximately 10%. A Turkish study in children with FMF found that during acute attacks, choroidal thickness was markedly increased compared to control groups as opposed to attack-free periods when there was no significant difference in thickness. This increase in choroidal thickness was also correlated with increased levels of inflammatory biomarkers, particularly C-reactive protein (CRP). On the one hand, the increased thickness was explained by the increase in inflammatory response and vascular permeability during acute attacks.Citation20 Another possibility is that it is related to other changes such as increased body temperature during acute attacks. Nevertheless, a study in 61 patients with anterior uveitis concluded an association with increased subfoveal choroidal thickness.Citation21 Of note, in a cohort of 32 patients with FMF, there was no difference detected in complement levels in FMF patients and healthy controls.Citation22
Tumour necrosis factor receptor-associated periodic fever syndrome (TRAPS)
TRAPS is an autosomal dominant MAIS linked to heterozygous variants of the TNFRSF1A gene responsible for generation of the Tumour Necrosis Factor Receptor 1 (TNFR1).Citation23 TRAPS is the second most common MAIS, with an estimated prevalence of one per million, reported to occur more frequently in Caucasians.Citation24 However, this may be due to ascertainment bias and unrepresentative of a true strong ethnic predominance as is true with FMF.Citation25
In TRAPS, pathogenic variants in TNFRSF1A alter the extracellular domain of TNFR1, impacting structure and interaction with its ligand, TNF.Citation26 Several molecular mechanisms have been proposed to explain the cellular disruptions involved in TRAPS, ultimately leading to pro-inflammatory cytokine production and recurrent fever. The pathogenic receptor can fail to undergo normal shedding from the cell surface, leading to a lack of soluble TNFR1 proteins that help dampen TNFR1 signalling.Citation27 A further potential mechanism is the accumulation of misfolded proteins within cells, which induces endoplasmic reticulum stress leading to an unfolded protein response, and the elevated production of reactive oxygen species in mitochondria inducing pro-inflammatory cytokine production.Citation23 Intracellular TNFR1 is also typically cleared through autophagy, a process that is also impaired in patients with TRAP further extending the activity of TNFR1.
Although clinical presentations can vary among different TRAPS phenotypes, a study involving a diverse group of 158 patients found that the median age for symptom onset was 4.3 years. Additionally, 9.1% of the patients experienced symptom onset after the age of 30.Citation25 The most common clinical features included fever, limb pain, abdominal pain, rash, and ocular involvement. Symptoms such as lymphadenopathy, periorbital oedema, and abdominal pain were more likely in children. One of the most serious complications of TRAPS is AA amyloidosis and occurred in 10% of patients at a median age of 43 years in the above study.Citation24 Over the past two decades, a series of diagnostic criteria have been developed to aid in the identification of TRAPS. These criteria focused on specific features including recurrent inflammatory symptoms, fever, abdominal symptoms, skin rashes, and lymphadenopathy in paediatric cases. In 2015, Gattorno et al. introduced an evidence-based classification system that aimed to integrate both genetic criteria (TNFRSF1A genotype) and specific clinical features to establish a diagnosisCitation1 with increased sensitivity and specificity. The Eurofever panel (https://www.printo.it/eurofever/registry) recommended that for cases where genetic testing is unavailable, an alternative criterion based on clinical variables, with ordinal scores for fever, migratory rash, periorbital oedema, myalgia, positive family history, absence of aphthous stomatitis, and absence of pharyngotonsillitis should be used.
A systematic review across the MAIS literature found that across 17 studies, 138 patients with TRAPS experienced ocular involvement.Citation19 The most common ocular manifestations were conjunctivitis (56.5%) and periorbital oedema (47.8%). Of note, two patients developed multifocal choroiditis (1.4%) and one patient developed bilateral panuveitis (0.7%). The case of panuveitis described a 7-year-old boy who presented with active bilateral panuveitis, with two choroidal lesions on the posterior pole of the right eye, and a macular rash associated with fever. They were subsequently diagnosed with TRAPS after genetic analysis found a pathogenic variant in the TNFRSF1A gene and treated with oral steroids and systemic immunosuppression.Citation28
Interestingly, the patient was healthy until 6 months prior to the current evaluation. At that time, he developed bilateral acute conjunctivitis, fever, and a widespread maculopapular rash.
Mevalonate kinase deficiency (MKD)
MKD is a recessively inherited pleiomorphic condition encompassing a range of diseases, (hyperimmunoglobulinaemia D [HIDS], periodic fever syndrome, and MVA [mevalonic aciduria]), with mild to severe complications.Citation29 This group of phenotypes is caused by pathogenic variants in the mevalonate kinase encoding gene with varying degrees of severity according to varying levels of normal enzyme activity. MKD is one of the rarest MAISs with around 300 patients reported worldwide including 30 patients with MVA. Across the literature, a higher concentration of patients within European areas have been reported particularly in Western Europe and the Netherlands, where there is a particular incidence of HIDS.Citation30 In comparison to 80 cases reported in The Netherlands, only 20 HIDS cases have been reported in the US. Whilst the data is sparse and studies are limited, an estimate for the prevalence of MKD has been estimated as 1.3 per million in European countries for patients younger than 19 years of age.Citation31
Loss-of-function variants in the mevalonate kinase gene, which is responsible for generating the mevalonate kinase enzyme, have been linked to the aetiology of MKD. The mevalonate kinase enzyme follows HMG-CoA reductase (the target for statins) in the mevalonate pathway and converts mevalonic acid to 5-phosphomevalonic acid. The mevalonate pathway produces cholesterol while also generating nonsterol isoprene compounds. The reduced function of the enzyme leads to mevalonic acid accumulation and deficiency of downstream compounds. While the precise pathogenesis of MKD remains unclear due to a lack of representative models, several studies have convincing evidence suggesting that the pro-inflammatory state is secondary to reduced isoprene compounds.Citation29 The use of statins to block the mevalonate pathway demonstrated that isoprenoid deficiency contributed to inflammasome activation and cytokine production. Additionally, approaches to increase cellular isoprenoid reduce inflammation in an experimental model.
The HIDS phenotype of MKD has been characterised as a milder manifestation presenting before 1 year of age, with recurrent bouts of fever, adenopathy, rash, and abdominal and joint pain, the latter potentially secondary to cholestasis and arthritis. The disease episodes are potentially cyclical and triggered by stress or vaccinations.Citation29 A more severe form of MKD is MVA, also emerging in infancy with features such as frontal bossing, developmental delay, myopathies, and central nervous system involvement (e.g. psychomotor issues, ataxia, and seizures). HIDS and MVA are two extremes of a continuous spectrum of MKD disease. A diagnosis of MKD is typically confirmed by observing elevated IgD levels during flare-ups, reduced mevalonate kinase activity during symptom-free periods, and the presence of mevalonate in the urine. The Eurofever/PRINTO clinical classification criteria defined the following features to establish a diagnosis at least three of six criteria: age at onset <1 year, gastrointestinal symptoms, painful lymph nodes, aphthous stomatitis, triggers, and maculopapular rash.Citation5 A recent study into the sensitivity and specificity of Eurofever MKD diagnostic criteria in a cohort of 119 patients found that the new criteria had a sensitivity and specificity of 87.5% and 60%.Citation32 However, when this included genetic and clinical variables, it had a higher sensitivity and specificity of 85% and 100%, respectively.
The classical description of ophthalmological findings of MVA, the severe form of MKV, includes blue sclerae, uveitis, central cataracts, optic atrophy, and retinitis pigmentosa. In a meta-analysis of patients with MAISs, which included 32 cases with MKD, it was found that uveitis was more common in MKD than in any other MAIS (90.6%).Citation19 Anterior uveitis (71.9%) was more common than intermediate uveitis (21.9%). A single case of early-onset bilateral granulomatous panuveitis with subsequent development of secondary glaucoma and total cataracts has been reported.Citation33
Cryopyrin-associated periodic syndrome (CAPS)
CAPS is a MAIS encompassing a range of diverse, autosomal dominant phenotypes caused by IL1ß-mediated systemic inflammation. The group includes familial cold autoinflammatory syndrome type 1 (FCAS1), Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disorder (NOMID). These conditions exist on a spectrum of severity with FCAS1 the least severe and NOMID the most aggressive.Citation34 In contrast to FMF, CAPS does not exhibit significant ethnic predominance and while FCAS and MWS may be associated with familial inheritance patterns, NOMID is caused by random pathogenic variants.Citation35
The uniting cause of all CAPS phenotypes is a pathogenic gain-of-function variant affecting the NLRP3 gene.Citation36 NLRP3 codes for the NLRP3 (also called cryopyrin) protein, which normally functions as an intracellular pattern recognition receptor, are able to recognise PAMPS and DAMPS. The ensuing activated NLRP3 inflammasome comprises NLRP3, the adaptor protein ASC, and pro-caspase-1, which in turn activates the inflammatory cytokine, IL-1β. The pathogenic gain-of-function variant results in a loss of an autoinhibitory step in NLRP3 activation causing excessive generation of the active inflammasome without the presence of PAMPS and DAMPS stimuli.Citation37
Due to the rare occurrence and diverse clinical presentation, diagnosing CAPS is challenging, leading to a significant delay between the onset of symptoms and the establishment of a definitive diagnosis.Citation36 Symptoms of CAPS may present in acute attacks or because of organ damage due to chronic inflammation. Acute attacks are often triggered by a range of external factors including cold, stress, infections, trauma, or sleep deprivation. The clinical presentation of CAPS was first noted when reports of MWS in 1962 described the triad of urticaria, deafness, and amyloidosis.Citation38 Subsequently, initial reports of NOMID in 1975 described a Still’s disease-like rash, deforming arthropathy, intellectual disability, and uveitis. A common symptom in all CAPS phenotypes is neutrophilic, non-symmetrical urticaria lasting 24 h. The rash is often non-painful but is burning and sensitive to touch. Whilst MWS generally presents in early childhood with a rash, arthralgia, myalgia, and fever during attacks, NOMID is more likely in early infancy with neurological symptoms common including chronic meningitis, increased intracranial pressure, developmental delay, seizures, and sensorineural hearing loss. The new Eurofever/PRINTO classification criteria in 2019 developed require the presence of a confirmatory NLRP3 genotype and at least one of the major features, namely: urticarial rash, red eye (conjunctivitis, episcleritis, uveitis), or sensorineural hearing loss. In the absence of the relevant NLRP3 genotype at least two of the features are required.Citation5
A range of ocular manifestations have been described in CAPS. A study into a pool of 1353 patients with MAISs with ocular involvement found that conjunctivitis and papillitis were found significantly more often in CAPS compared to other MAISs.Citation19 In this meta-analysis, 680 patients with CAPS were identified. The average age of onset for ocular involvement was 12.5 years, compared to a mean onset age of 5.5 years for CAPS itself. The most frequent ocular symptom was conjunctivitis (62.4%), followed by uveitis (28.4%). The breakdown of uveitis cases included 130 with anterior uveitis (19.1%), five with posterior uveitis (1%), two with intermediate uveitis and panuveitis (0.3%), and one with bilateral papillitis and uveitis (0.2%).
Blau syndrome (BS)
Blau-Jabs syndrome, commonly referred to as Blau Syndrome (BS), is a rare autosomal dominant MAIS. BS is caused by pathogenic variants in the pattern recognition receptor Nucleotide-Binding Oligomerisation Domain-Containing Protein 2 (NOD2). The syndrome is characterised by the triad of arthritis, uveitis, and skin rash.Citation39 BS/Early Onset Sarcoidosis (EOS) and systemic sarcoidosis are chronic granulomatous conditions displaying the common histologic feature of noncaseating granulomas, which can impact similar organ systems. Nonetheless, patients exhibit variations with respect to age of onset, genetic factors, and prevailing clinical characteristics. The incidence of BS remains uncertain. The yearly incidence of combined granulomatous disorders (encompassing BS, Early Onset Sarcoidosis, and Sarcoidosis) before the age of 18 has been documented at a range of 0.06 to 1.02 cases per 100,000 individuals.Citation40 Individuals with BS typically manifest symptoms before reaching the age of 5, in contrast to children with “adult-type” sarcoidosis, who present during adolescence. BS predominantly affects children of Asian, Caucasian, or Hispanic heritage, while “adult-type” sarcoidosis emerging during childhood more commonly affects individuals of African American descent (80%).Citation41 BS follows an autosomal dominant inheritance pattern, while EOS arises from a pathogenic variant that occurs sporadically in the same gene. The prevailing consensus indicates that the underlying pathogenesis is a NOD2 gain-of-function variant, resulting in the activation of proteins that would remain inactive under normal circumstances. NOD2 pathogenic variants cause increased generation of NOD2 protein, NF-κB activation, and proinflammatory cytokine production.
The clinical picture for BS is typified by a triad of symmetric arthritis, granulomatous dermatitis, and recurrent uveitis with onset below 4 years of age.Citation42 A rash is often the first sign with papulonodular rashes and subcutaneous nodules being common. The arthritis resembles rheumatoid arthritis and can cause finger deformations and wrist stiffness. When making a diagnosis of BS, a choice of diagnostic procedures can be used to identify BS features. These include skin biopsy to identify noncaseating granulomas, and X-rays to identify carpal dysplasia, camptodactyly, abnormal ulna, and second metacarpal bone shape. Definitive confirmation of the disease is achieved through the genetic identification of pathogenic variants in NOD2.
In a meta-analysis of 238 patients with BS, the proportion of cases with uveitis was the highest of any other MAISs (95.4%).Citation19 Of these cases 109 presented with granulomatous anterior uveitis (48%), 6 with intermediate uveitis (2.6%), 27 with posterior uveitis (11.9%), and 99 with panuveitis (43.6%). Ocular symptoms typically appeared in both eyes and often followed a chronic course of inflammation. These symptoms usually manifested later than joint and skin issues. Uveitis was consistently accompanied by either joint or skin symptoms, with no cases of isolated ocular disease reported.
Unfortunately, uveitis tends to be severe when present, with up to 30% of patients with BS developing moderate-to-severe visual impairment.Citation43 In contrast to juvenile idiopathic arthritis (JIA)-related uveitis, which is more commonly non-granulomatous, BS typically presents as a bilateral panuveitis accompanied by peripheral multifocal choroidal scars.Citation43–45
Retinal dystrophy, optic nerve oedema, splenomegaly, anhidrosis, and headache (ROSAH)
Williams et al. identified a pathogenic variant, in the Alpha-protein kinase 1 (ALPK1) gene in 2019, associated with a condition marked by a range of symptoms including retinal dystrophy, optic nerve oedema, splenomegaly, anhidrosis, and headaches, which they termed ROSAH syndrome.Citation46 The activation of the kinase domain in ALPK1 leads to the phosphorylation of the TRAF-interacting protein with a forkhead-associated domain, known as TIFA. This phosphorylation sets off a cascade that results in the formation of a TIFA–TRAF6 complex, ultimately leading to the activation of NF-κB signalling pathways. This process is crucial for innate immunity responses.Citation47 The pathogenic variants identified in ROSAH appear to have gain-of-function with increased innate immune activation and enhanced NF-κB signalling.Citation48
ROSAH is a newly described autosomal dominant MAIS, with few reports on identifying features and successful treatments. In the largest cohort of 27 patients, nearly all patients exhibited at least one inflammatory feature that included recurrent fever or abdominal pain, malaise, and headaches. The fever episodes were reported to predominantly last 24 h and self-resolve. Arthralgia was noted in 76% of participants, with cases of deforming erosive arthritis in 33%.
Ocular features included bilateral optic nerve swelling, which was nearly always present, and associated with uveitis during acute episodes, as well as progressive visual field loss. The mean age of presenting with visual symptoms was 15 years of age in this cohort.Citation49 At early stages, the retina can appear normal; however, with more advanced disease, there has been evidence of progressive nummular retinal pigmentation, vascular attenuation, and retinal pigment epithelium atrophy.
A20 haploinsufficiency (HA20)
A20 haploinsufficiency (HA20) is a rare autosomal dominant autoinflammatory disease caused by a heterozygous loss-of-function pathogenic variant in TNF-Induced Protein 3 (TNFAIP3), which encodes for the NF-
B regulatory protein A20 or TNAP3.Citation50 TNFAIP3 has a role in deubiquitinase activity and inhibits pro-inflammatory mediators such as NF-
B kinase subunit gamma and receptor-interacting protein kinase 1.Citation51 HA20 was first described in six unrelated families with systemic inflammation in childhood, manifesting with symptoms similar to Behçet’s syndrome, with pathogenic variants causing defective deubiquitinase activity of A20, increased NF-
B signalling and phosphorylation of the c-Jun N-terminal kinase and p38 mitogen-activated protein kinases.Citation50 In these families, five were described to have recurring Behçet’s disease and one patient from a Turkish cohort study carried pathogenic variants in TNFAIP3. Clinical features of Behçet’s syndrome and HA20 can be differentiated,Citation50 and this distinction is key to treatment decisions, as for instance, response to colchicine in HA20 is less effective than in Behçet’s syndrome.Citation52
Due to the rare occurrence of HA20, both its annual incidence and the details of its clinical symptoms, disease severity, systemic complications, and treatment approaches remain unclear. Further cases of HA20 have been reported in the literature, particularly in Japanese families, since the first description. From a Japanese family with three cases spanning three generations, two demonstrated an identical novel TNFAIP3 pathogenic variant.Citation53 One of the family members had arthralgia, proximal limb muscle pain, recurrent aphthous stomatitis, aphthous ulcer of the palpebral conjunctiva and haemorrhoids. Another family member had polyarthritis and anterior uveitis starting in infancy and ultimately passed away at the age of 65 due to gastrointestinal bleeding. Another Japanese family investigated for presumed Behçet’s disease, with six patients over four generations presenting with frequent oral ulcers, genital ulcers, and erythema nodosum-like lesions but no ocular lesions, similarly identified a common heterozygous missense pathogenic variant in A20/TNFAIP3, with all carrying a specific heterozygous C234Y variant in the ovarian tumour domain.Citation54 There is one case report of a germline heterozygous variant in TNFAIP3 causing A20 haploinsufficiency in a 7-month-old Japanese boy, with an unusual presentation of autoimmune lymphoproliferative syndrome, a condition characterised by chronic lymphoproliferation and autoimmunity.Citation55 Expanding the clinical spectra of heterozygous loss-of-function pathogenic variants in TNFAIP3 is a case report of a 14-year-old British boy, presenting at age 10 with insulin-dependent diabetes, cytopaenias, hepatitis, enteropathy, and interstitial lung disease, who was responsive to haematopoietic stem cell transplantation.Citation56 Some case reports have also described patients presenting with a lupus-like phenotype,Citation57 inflammatory bowel disease,Citation58 and haemophagocytic lymphohistiocytosis.Citation59
Although no unifying diagnostic criteria exist, efforts have been made to improve the description of HA20, such as in a case series of 16 patients derived from seven families, in whom a genetic diagnosis of HA20 was established.Citation50 In this case series, the age of disease presentation was highly variable, ranging from the first week of life to 29 years of age. Authors described the frequency and severity of clinical phenotype as variable, with early onset recurrent oral, genital, and/or gastrointestinal ulcers described as characteristic features of HA20. Other clinical manifestations reported include musculoskeletal symptoms, gastrointestinal complaints, cutaneous lesions, episodic fever, and recurrent infections. Of the ocular findings in this case series, severe and treatment-refractory uveitis in two sisters and retinal vasculitis with chorioretinal scarring and macular fibrosis and anterior uveitis in another young girl were described. In between flares, acute phase reactants were often within normal limits in most cases; however, there were some instances of increased levels of CRP and ESR pre-treatment, and the presence of autoantibodies was variable. In a meta-analysis of 89 patients, the median age of onset was 6 years old, with the main manifestations being recurrent oral ulcers (70%), recurrent fever (42%), gastrointestinal ulcers (40%), skin lesions (38%), genital ulcers (36%), and musculoskeletal disorders (34%).Citation60
Classification
Currently, there exists no clear consensus regarding the categorisation of MAIs. The 2022 updated phenotypic classification provided by the International Union of Immunological Societies expert committee on Inborn Errors of Immunity (IEI) is tailored towards clinicians at the bedside, focusing on the clinical attributes and laboratory features of distinct IEI conditions, including a section on auto-inflammatory disorders.Citation61 Alternative classification methods have focused on a taxonomy dictated by molecular pathways involved,Citation62 or the dermatological features that aid in differentiating the various conditions.Citation63
For patients with MAISs, a useful classification strategy relies on identifying the dysregulated primary cytokine. This approach holds potential therapeutic value by allowing targeted intervention (). For this review, we will focus on this strategy further to emphasise potential therapeutic targets and treatments. Determining the predominant cytokine or pathway involved in the disease could pave the way for the creation of focused therapeutic approaches for MAISs.Citation64
Table 1. Monogenic autoinflammatory syndromes clinical features.
Table 2. Affected pathways and suggested treatments. Biologics are increasingly used upfront alongside non-specific targets such as steroids.
Treatment
The evidence base for the treatment of monogenic autoinflammatory uveitides is primarily based on expert consensus and existing treatment regimens used to treat monogenic autoinflammatory diseases. Recent years have seen advancements in our understanding of the genetic underpinnings and pathogenesis of these conditions. Specifically, the role of the inflammasome in causing dysregulated production of IL-1ß has been clarified, particularly in the most commonly discussed autoinflammatory diseases.
Conventional therapies
Colchicine
Colchicine is an alkaloid extracted from Lily family plants Colchicum autumnale and Gloriosa superba. The use of colchicine in the treatment of gout has been recognised for centuries, but its use has also expanded to other conditions including Behçet’s disease, pericarditis, and cutaneous vasculitides.Citation69 Several clinical trials have demonstrated the effectiveness of colchicine in treating Familial Mediterranean Fever (FMF), including its ability to reduce amyloidosis. These findings have led the European Alliance of Associations for Rheumatology (EULAR) to recommend colchicine as a first-line therapy for both adult and paediatric FMF patients.Citation70
Colchicine has several anti-inflammatory effects. Primarily it functions via inhibition of leukocyte chemotaxis by an interaction with tubulin, leading to microtubule dysfunction. By binding non-polymerised tubulin, colchicine causes movement inhibition of intracellular granules. Other effects of colchicine include effects on TNF signalling by both reducing production by macrophages and effects of TNF receptors.Citation69 It also inhibits phospholipase A2 activity, phagocytosis, and the release of lysosomal enzymes. Colchicine has also shown an ability to suppress the activation of caspase 1 leading to the inability to convert pro-IL-1 to active IL-1.Citation71
Colchicine has a narrow therapeutic range due to a half-life following oral ingestion of between 7 and 9 h. It is metabolised in the liver and excreted in the biliary, intestinal, and renal systems. Its use in pregnancy and nursing patients is considered relatively safe, if hepatic and renal function is intact.Citation72
NSAIDs
Non-steroidal anti-inflammatory drugs (NSAIDs) exert their therapeutic effects via inhibition of cyclooxygenases, altering arachidonic acid in prostaglandins, and via thromboxanes. They have been used as symptomatic treatment of the monogenic autoinflammatory diseases either in isolation or as an additional therapy to other drug regimens.Citation65 Although the Eurofever registry has documented complete response in a minority of patients treated with NSAIDs alone, they do appear to provide symptomatic benefit in 70–80% of patients.
Corticosteroids
Corticosteroid therapies, such as prednisolone, have pleiotropic effects in control of inflammation via suppression of multiple pro-inflammatory pathways, direct effects of leukocyte migration, and inhibition of fibroblast function.Citation73 This leads to disease control both acutely (less pain and oedema) and long term (reduced cell fibrosis associated changes). They exert their effect via binding to a steroid response element intracellularly leading to activation of a transcription factor and consequent gene expression effects: up-regulated anti-inflammatory proteins, and suppression of the pro-inflammatory NF-B pathway.
In FMF that is not sufficiently controlled with colchicine alone, the use of glucocorticoids can help further reduce disease activity and this aligns with data from the Eurofever registry.Citation65 Acute control of disease can be further enhanced with the use of intravenous methylprednisolone during attacks of fever, abdominal pain, or pleuritic pain.Citation74
Case reports of ocular involvement in patients with FMF described positive response to the addition of acute steroid therapy to manage uveitis flare-ups.Citation7 In instances of anterior uveitis, positive outcomes were observed with the use of steroid drops. Alternatively, for intermediate or posterior uveitis cases, systemic steroids such as oral prednisolone or intravenous methylprednisolone were found to be effective.
In patients with TRAPS, the use of oral prednisolone can help relieve attacks with a 91% effectiveness in controlling inflammatory attacks.Citation65 However, these treatments do not appear to alter the development of amyloidosis in these patients, nor do they reduce the frequency of attacks. Therefore, their use should ideally be limited to managing acute flare-ups.
Patients with TRAPS should be considered for biologic therapy to help minimise the ongoing steroid dose. During flare-ups, patients with MKD also respond positively to acute steroid therapy. However, an even smaller proportion achieve complete remission solely through steroid treatment compared to patients with TRAPS (9% vs 41%). In patients with CAPS, steroid therapy is also used with benefit in 80% of patients to provide additional relief during attacks; however, their use does not resolve the underlying inflammation or the frequency of attacks. In patients with CAPS, steroids should be ideally avoided as primary maintenance therapy.Citation75
Patients with BS are commonly treated for ocular inflammation including uveitis and other systemic involvement with steroid therapy. A multicenter case series of 50 patients with BS worldwide identified 75% of eyes of patients treated with topical steroid drops, with 26 of 38 patients with ocular involvement treated with systemic corticosteroids.Citation44 However, 76% of patients included in the case series continued to experience persistent uveitis activity despite the use of steroid therapies, with or without alternative immunosuppressants, reflecting the difficulty in controlling the disease entirely in this patient population.
Thalidomide
Thalidomide has been used to treat dermatological diseases such as lupus and mucosal ulcers, in addition to its more common usage in multiple myeloma.Citation76 Its anti-inflammatory properties arise from a mixture of inhibition of TNF-, interferon- (IFN-) synthesis, leukocyte chemotaxis, and angiogenesis. There have been sporadic case reports of its use in the treatment of the autoinflammatory monogenic including case series of colchicine-resistant FMF (crFMF) and MKD with no proven benefit.Citation77 Thalidomide has also reportedly been used with success in four patients with Blau syndrome.Citation78
Other conventional medications
There is evidence from case reports of the use of azathioprine, methotrexate, cyclosporine, leflunomide, mycophenolate mofetil, dapsone, thalidomide, sulfasalazine, statins, cimetidine, and antihistamines in patients suffering from the autoinflammatory diseases.Citation65
Targeting the molecular mechanism: Biologic therapies
With understanding of the disease pathogenesis of the monogenic autoinflammatory diseases and further evidence from new clinical trials, newer more targeted approaches have been developed.
IL-1 blockade
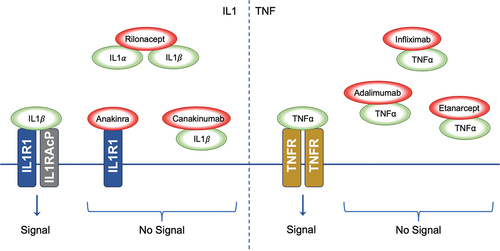
The inflammatory cascade is triggered when the IL-1 receptor (IL-1 R) binds to the ligands IL-1α and IL-1β (). IL-1 R is expressed on most human cells. IL-1 inhibitory molecules were first discovered in the urine of patients with monocytic leukaemia, and their use was hypothesised to be beneficial in treatment of patients with inflammatory conditions.Citation79 This has been confirmed with substantial evidence supporting the use of IL-1 inhibitor therapy in inflammasome-driven diseases in particular.Citation65,Citation79,Citation80 There are three commercially available IL-1 inhibitory therapies: anakinra, canakinumab, and rilonacept.
Figure 2. Inhibition of Cytokine Signalling by Therapeutic Agents. The left panel demonstrates the signalling cascade initiated by IL1 binding to the IL1R1-IL1RACP complex. IL1α and IL1β interaction with their receptor results in downstream signalling. The biologic agents Anakinra, Rilonacept, and Canakinumab disrupt IL1 signalling; Anakinra by antagonising receptor interaction, Rilonacept by sequestering IL1α and IL1β, and Canakinumab by neutralising IL1β, thus inhibiting the signal. The right panel shows TNFα signalling through the TNFR. The biologics Infliximab, Adalimumab, and Etanercept impede TNFα interaction with TNFR, thereby preventing signal transduction; IL1: Interleukin-1, IL1α: Interleukin-1 alpha, IL1β: Interleukin-1 beta, IL1R1: Interleukin-1 Receptor Type 1, IL1RACP: Interleukin-1 Receptor Accessory Protein, TNFα: Tumour Necrosis Factor-alpha, TNFR: Tumour Necrosis Factor Receptor.
Anakinra
Anakinra is a competitive inhibitor of IL-1 R agonists which mimics the activity of endogenous IL-1 R antagonist (IL-1RA). It is a daily subcutaneous injection, with a half-life of approximately 6 h. It has received FDA approval for the treatment of NOMID since 2012, and EMA approval for all types of CAPS since 2013.Citation65 It is 80% renally excreted hence requires preserved renal function when administered, and there is evidence from pre-clinical models it can cross the blood–brain barrier.Citation81 Given long-standing usage in the treatment of patients with rheumatoid arthritis, there are good longitudinal safety data for its usage with no evidence of increase in both opportunistic infections and malignancies.Citation82 Biologic registers do identify higher rates of serious skin infections and respiratory tract infections in patients prescribed anakinra, however.Citation83 The most frequent adverse reaction is an injection site skin reaction, which tends to decline with time without the need for discontinuation. From limited registry data, there is no documented increase in congenital malformations or miscarriages when anakinra is administered in pregnant patients.
Canakinumab
Canakinumab is a fully humanised IgG1 monoclonal antibody specific for IL-1β.Citation80 It is a bi-monthly subcutaneous injection, with an elimination half-life of 26 days. In 2016 based on the results of the CLUSTER trial, both the FDA and EMA approved the use of canakinumab in crFMF, TRAPS, and MKD.Citation84 Canakinumab has a longer half-life than anakinra but does not appear to penetrate the blood–brain barrier. It is not influenced by renal function.Citation85 There are limited data on the impact of canakinumab on pregnancy and breastfeeding. Adverse events relate to mild urinary and respiratory infections, although rarely serious infections have been reported in a CAPS registry; there is also a rare occurrence of neutropenia and thrombocytopenia in patients prescribed canakinumab.Citation86
Rilonacept
Rilonacept is a dimeric fusion protein of the Fc portion of human IgG1 and of the human IL-1 receptor extracellular domain-binding IL-1/IL-1. It is a weekly subcutaneous injection, with a half-life of 7 days.Citation80 It received FDA approval in 2008 for the treatment of FCAS/MWS in patients over the age of 12.Citation87 As a large molecule is speculated not to cross the blood–brain barrier, it is likely excreted via the reticuloendothelial system as opposed to renally with dose adjustment not required in renal disease.Citation80 Adverse events relate to local injection site reactions, headache, urinary and respiratory infections.
IL-1 blockade in autoinflammatory diseases
FMF
Anakinra is recommended for crFMF or experiencing protracted febrile myalgia. This is based on Eurofever registry data, and RCTs of anakinra demonstrating clear benefit.Citation70 This was seen in patients prescribed canakinumab, and there is promising evidence of efficacy for rilonacept as well.
Although it may be possible to treat ocular manifestations of FMF through increasing Colchicine dosing alone,Citation88 the use of anti-IL-1 therapies can help resolve resistant cases. Case reports also describe the use of IL-1 inhibitors to treat ocular complications of FMF; a case series described FMF-associated uveitis of unspecified subtype in two patients treated with canakinumab, one patient achieved remission whilst another had recurrence of uveitis after 10 months of therapyCitation89; a Turkish case report describes control of optic neuritis in a paediatric patient with crFMF with anakinra, which was then switched to canakinumab.Citation90
TRAPS
IL-1 inhibitors have demonstrated efficacy in TRAPS and are superior to etanercept based on retrospective data.Citation65 Anakinra has shown benefit in 90% of patients with TRAPS in registry data, and complete remission was observed in 67% of these and it is recommended for use in patients with TRAPS not controlled by NSAIDs or steroid therapy.Citation75 Canakinumab has also shown significant efficacy with 19/20 patients exhibiting complete remission in an open-label trial,Citation91 but similar to FMF patients on canakinumab, there were more adverse events than in placebo for these patients. The randomised placebo-controlled CLUSTER study trial further supported the use of canakinumab in the treatment of TRAPS with 45% of patients assigned treatment experiencing complete response compared to 8% in the placebo arm, which increased to 73% of patients on dose escalation in the trial.Citation84 However, the specific treatment of ocular complications of TRAPS has not been described with IL-1 inhibitors.
MKD
IL-1 inhibitors can control or relieve symptoms in most patients. From registry data of 62 patients with MKD treated with anakinra, there was 84% response.Citation65 A phase II study of canakinumab showed reduced frequency of attacks, normalised inflammatory markers, and complete clinical response in all patients. Based on the CLUSTER trial,Citation84 FDA and EMA approval for canakinumab in MKD were granted with 35% achieving complete remission vs 6% on placebo, escalating to 57% of patients on an escalated dosing regimen. There is no specific reporting of the impact of IL-1 therapies on treating ocular manifestations of MKD; however, they are recommended as first line for both flares and long-term management of EULAR/ACR evidence-based expert consensus.Citation68
CAPS
Anakinra, canakinumab, and rilonacept have all been approved by the FDA and EMA for CAPS and are typically first line therapy for all age groups of patients.Citation65 For anakinra, a long-term open-label study of CINCA patients was the basis for approval by the FDA with symptoms and inflammatory markers improved.Citation92 There was also evidence of improved leptomeningeal and cochlear involvement in these patients indicative of anakinra being able to cross the blood–brain barrier. Canakinumab was also effective in inducing remission in 75–90% of patients treated with it in the EuroFever registry, with similar levels of control to anakinra.Citation65 An RCT of Rilonacept has also provided evidence of efficacy.Citation87 While conjunctivitis is the more common ocular feature of CAPS, IL-1 inhibitors have also reported control of more severe ocular manifestations such as uveitis, with reported control of a severe granulomatous uveitis with stromal keratitis in a patient with CINCA after starting canakinumabCitation93 and rapid control of a posterior uveitis in a patient with CINCA after starting anakinra.Citation94 Four patients with MWS also had resolution of their uveitis after starting anakinra therapy in separate case reports.Citation95,Citation96
Blau syndrome
Isolated case reports have reported rapid remission of uveitis in the context of treatment-resistant Blau syndrome with IL-1 inhibitors including anakinraCitation97 and canakinumab.Citation98
TNF blockade
Anti-TNFs have been used to treat the monogenic inflammatory diseases, however there is poorer efficacy of these biologics compared to the IL-1 inhibitors.Citation65 The agents used have included:
Etanercept – dimeric human TNF receptor p75-Fc fusion protein
Infliximab – chimeric monoclonal antibody against TNF
Adalimumab – fully human mAb against TNF
None of these therapies have received either FDA or EMA approval for their use in the monogenic autoinflammatory diseases.
Anti-TNFs in autoinflammatory diseases
FMF
There is evidence from case series reporting some benefit when used to treat CRFMF, especially when other autoimmune comorbidities are present, e.g. ankylosing spondylitis or psoriasis.Citation99,Citation100 This has also been identified in registry data with more benefit in patients with co-incident arthritis.Citation65
TRAPS
Etanercept has reported some benefit in reducing severity of attacks and helping reduce ongoing steroid dosing in patients with TRAPS.Citation65 However, it is often discontinued due to lack of effect over time, which is the basis for consensus recommendations for use in some TRAPS patients.Citation75 The other anti-TNFs infliximab and adalimumab have been associated with severe paradoxical reactions and are not recommended for use in patients suffering from TRAPS.Citation101 Anti-TNFs have efficacy in managing the systemic disease, which should guide management given that the main ocular manifestations of TRAPS are conjunctivitis or periorbital oedema. However, a single case report does describe a patient presenting with fever and bilateral panuveitis diagnosed with TRAPS with genetic testing who achieved sustained control of inflammation for 16 months follow-up whilst on adalimumab therapy.Citation28 Similar to patients with JIA continuing to experience new or ongoing flares of uveitis on etanercept,Citation102 etanercept has also been linked to the incidence of anterior uveitis in patients with TRAPS treated with itCitation103; its use should therefore be re-evaluated to consider stopping or switching to an alternative biologic in patients with TRAPS that suffer uveitis whilst prescribed etanercept. Compared to patients treated with anakinra, TRAPS patients are less likely to evidence complete remission on anti-TNFs.
MKD
Anti-TNFs have been documented in case reports to improve symptoms and reduce the frequency of attacks of MKD.Citation75 Anti-IL1 therapies remain first-line with greater control of symptoms reported in case series,Citation104 however anti-TNFs can be considered in cases where anti-IL-1 therapies are ineffective.Citation68 Etanercept is the most prescribed anti-TNF for this purpose based on registry data.Citation65 The use of anti-TNFs to treat uveitis related to MKD has been described in a case report of a 2-month-old boy followed up for 7 years after presenting with bilateral panuveitis, which was managed with steroids, methotrexate, and adalimumab but continued to experience ongoing uveitis flare-ups.Citation33 However, a patient with corneal inflammation secondary to MKD, with a recurrent nummular keratitis, experienced significant improvement of their symptoms and cessation of keratitis flare-ups following a regimen including infliximab and methotrexate therapy.Citation105
CAPS
There is no documented evidence to support the efficacy of treatments for CAPS syndrome.
Blau syndrome
Anti-TNF therapies have demonstrated efficacy from multiple case reports and case series of patients diagnosed with BS and have also been identified as effective in the control of uveitis secondary to BS. Based on a review of 38 published case reports of BS,Citation106 62 patients were treated with good disease control on anti-TNF therapy in 27 of 31 patients treated with infliximab, 21 of 24 patients treated with adalimumab, and 5 of 7 patients treated with etanercept.
IL-6 blockade
Tocilizumab is a humanised anti-IL6 receptor antibody which has been approved for use in RA, JIA, GCA, and as a treatment for the CAR-T therapy associated cytokine release syndrome.Citation107 There is currently limited experience of its use in the treatment of the MAISs; hence, there are no current approvals. However, IL-6 inhibition may provide additional benefits, especially in cases of monogenic autoinflammatory disease not well controlled with other therapies. The use of Tocilizumab has been reported in the treatment of crFMF with evidence of symptom relief and control of amyloidosis.Citation108 It has also been used in case series of TRAPS and HDS/MKD patients with some evidence of improvement.Citation107,Citation109 There have been two reported negative results with patients treated with Tocilizumab for CAPS with initial control followed by rapid relapse in both.Citation110,Citation111
There is also a single-case report from China of a 13-year-old patient diagnosed with BS suffering treatment-resistant uveitis, which was managed with tocilizumab, helping taper their steroid therapy to a much lower ongoing dose,Citation112 although wider usage of IL-6 inhibition in BS has not since been reported.
JAK inhibitors
JAK kinase inhibitors suppress the STAT1 transcription factor pathway which blocks the induction of IFN stimulated genes and hence reduces the production of IFN. The JAK inhibitors include tofacitinib, baricitinib, and ruxolitinib and have been approved for the treatment of rheumatoid arthritis, psoriatic arthritis, IBD, myelofibrosis, and polycythaemia rubra vera.Citation113 There are reports of use of the JAK inhibitor tofacitinib in the treatment of crFMF patients who failed IL-1 inhibitor, anti-TNF, and IL-6 inhibition.Citation114 JAK inhibitors have been used to treat patients with BS with tofacitinib inducing remission in three patients unresponsive to initial therapies including anti-TNF therapy.Citation115 Another case report of a patient with anti-TNF refractory BS described remission once prescribed tofacitinib, which was then switched to baricitinib for maintenance therapy due to tofacitinib-induced lymphopenia.Citation116
Future directions
As our understanding of the inflammasome grows, new avenues for disease treatment are emerging. Specifically, we can now target oxidative stress, autophagy, and the complement cascade to alter the course of the disease.Citation117
There remains a need for higher quality evidence from clinical trials to inform management of patients with MAIs. To this end, the development of shared clinical databases to further characterise response in affected patients to newer biologic therapy regimens will continue to provide valuable real-world data of the impact of therapies for patients with these rare diseases.
Acknowledgments
The authors have no specific acknowledgements to make.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Federici S, Sormani MP, Ozen S, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74(5):799–805. doi:10.1136/ANNRHEUMDIS-2014-206580.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/J.CELL.2010.01.040.
- Kaneko N, Kurata M, Yamamoto T, Morikawa S, Masumoto J. The role of interleukin-1 in general pathology. Inflamm Regen. 2019;39(1). doi:10.1186/S41232-019-0101-5.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol. 2009;124(6):1141–1149. doi:10.1016/J.JACI.2009.11.016.
- Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019;78(8):1025–1032. doi:10.1136/ANNRHEUMDIS-2019-215048.
- Onen F. Familial Mediterranean fever. Rheumatol Int. 2006;26(6):489–496. doi:10.1007/S00296-005-0074-3.
- Yazici A, Ozdal P, Yuksekkaya P, Elgin U, Teke MY, Sari E. Ophthalmic manifestations in familial Mediterranean fever: a case series of 6 patients. Eur J Ophthalmol. 2014;24(4):593–598. doi:10.5301/EJO.5000398.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum. 2009;61(10):1447–1453. doi:10.1002/ART.24458.
- Wan CK, He C, Sun L, Egwuagu CE, Leonard WJ. Cutting edge: IL-1 receptor signaling is critical for the development of autoimmune uveitis. J Immunol. 2016;196(2):543–546. doi:10.4049/JIMMUNOL.1502080.
- Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The pyrin inflammasome in health and disease. Front Immunol. 2019;10:1745. doi:10.3389/FIMMU.2019.01745.
- Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17(8):914–921. doi:10.1038/NI.3457.
- Lancieri M, Bustaffa M, Palmeri S, et al. An update on familial Mediterranean fever. Int J Mol Sci. 2023;24(11):9584. doi:10.3390/IJMS24119584.
- Sari I, Birlik M, Kasifoglu T. Familial Mediterranean fever: an updated review. Eur J Rheumatol. 2014;1(1):21–33. doi:10.5152/EURJRHEUM.2014.006.
- Majeed HA, Rawashdeh M, El-Shanti H, Qubain H, Khuri-Bulos N, Shahin HM. Familial Mediterranean fever in children: the expanded clinical profile. QJM. 1999;92(6):309–318. doi:10.1093/QJMED/92.6.309.
- Özçakar ZB, Yalçinkaya F, Çakar N, et al. Application of the new pediatric criteria and tel hashomer criteria in heterozygous patients with clinical features of FMF. Eur J Pediatr. 2011;170(8):1055–1057. doi:10.1007/S00431-011-1404-Y.
- Yalçinkaya F, Özen S, Özçakar ZB, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford). 2009;48(4):395–398. doi:10.1093/RHEUMATOLOGY/KEN509.
- Petrushkin H, Stanford M, Fortune F, Jawad AS. Clinical review: familial Mediterranean Fever-An Overview of Pathogenesis, symptoms, ocular manifestations, and treatment. Ocul Immunol Inflamm. 2016;24(4):422–430. doi:10.3109/09273948.2015.1010012.
- Georgakopoulos CD, Antonopoulos I, Makri OE, Vasilakis P, Liossis SNC, Andonopoulos AP. Acute posterior multifocal placoid pigment epitheliopathy in a patient with familial Mediterranean fever. Clin Exp Optom. 2016;99(4):385–387. doi:10.1111/CXO.12401.
- Maccora I, Marrani E, Mastrolia MV, et al. Ocular involvement in monogenic autoinflammatory disease. Autoimmun Rev. 2021;20(11):102944. doi:10.1016/J.AUTREV.2021.102944.
- Gundogan FC, Akay F, Uzun S, Ozge G, Toyran S, Genç H. Choroidal thickness changes in the acute attack period in patients with familial Mediterranean fever. Ophthalmologica. 2016;235(2):72–77. doi:10.1159/000442216.
- Thapa S, Kharel R, Shrestha J. Role of choroidal thickness assessment in unilateral acute anterior uveitis. Indian J Ophthalmol. 2020;68(9):1869–1874. doi:10.4103/IJO.IJO_688_20.
- Mkrtchyan GM, Boyajyan AS, Ayvazyan AA, Beglaryan AA. Classical pathway complement activity in familial Mediterranean fever. Clin Biochem. 2006;39(7):688–691. doi:10.1016/J.CLINBIOCHEM.2006.02.016.
- Cudrici C, Deuitch N, Aksentijevich I. Revisiting TNF receptor-associated periodic syndrome (TRAPS): Current perspectives. Int J Mol Sci. 2020;21(9):3263. doi:10.3390/IJMS21093263.
- Zegarska J, Wiesik-Szewczyk E, Hryniewiecka E, et al. Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) with a new pathogenic variant in TNFRSF1A gene in a family of the adult male with renal AA amyloidosis-diagnostic and therapeutic challenge for Clinicians. J Clin Med. 2021;10(3):1–10. doi:10.3390/JCM10030465.
- Lachmann HJ, Papa R, Gerhold K, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the eurofever/EUROTRAPS international registry. Ann Rheum Dis. 2014;73(12):2160–2167. doi:10.1136/ANNRHEUMDIS-2013-204184.
- Van Gijn ME, Ceccherini I, Shinar Y, et al. New workflow for classification of genetic variants’ pathogenicity applied to hereditary recurrent fevers by the international study group for systemic autoinflammatory diseases (INSAID). J Med Genet. 2018;55(8):530–537. doi:10.1136/JMEDGENET-2017-105216.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97(1):133–144. doi:10.1016/S0092-8674(00)80721-7.
- Cocho L, Urbaneja E, Herreras JM. Vision-threatening bilateral panuveitis and TRAPS in a child: an uncommon association. Int Ophthalmol. 2019;39(1):219–223. doi:10.1007/S10792-017-0785-Y.
- Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016;9:101–110. doi:10.2147/TACG.S93933.
- Houten SM, van Woerden CS, Wijburg FA, Wanders RJA, Waterham HR. Carrier frequency of the V377I (1129G>A) MVK mutation, associated with hyper-IgD and periodic fever syndrome, in the Netherlands. Eur J Hum Genet. 2003;11(2):196–200. doi:10.1038/SJ.EJHG.5200933.
- Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J. 2016;14(1). doi: 10.1186/S12969-016-0091-7.
- Çağlayan Ş, Mardinoğlu G, Yarar MH, et al. The assessment of autoinflammatory disease classification criteria (eurofever/PRINTO) in a real-life cohort. Clin Rheumatol. 2023;42(6):1645–1653. doi:10.1007/S10067-023-06557-0.
- Agarwal N, Kothari M. Uveitis, glaucoma, and cataract with mevalonate kinase deficiency. J Aapos. 2022;26(2):93–95. doi:10.1016/J.JAAPOS.2021.11.009.
- Shinkai K, McCalmont TH, Leslie KS. Cryopyrin-associated periodic syndromes and autoinflammation. Clin Exp Dermatol. 2008;33(1):1–9. doi:10.1111/J.1365-2230.2007.02540.X.
- Yu JR, Leslie KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11(1):12–20. doi:10.1007/S11882-010-0160-9.
- Kuemmerle-Deschner JB. CAPS–pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol. 2015;37(4):377–385. doi:10.1007/S00281-015-0491-7.
- Mehr S, Allen R, Boros C, et al. Cryopyrin-associated periodic syndrome in Australian children and adults: Epidemiological, clinical and treatment characteristics. J Paediatr Child Health. 2016;52(9):889–895. doi:10.1111/JPC.13270.
- Tran TA. Muckle-Wells syndrome: clinical perspectives. Open Access Rheumatol. 2017;9:123–129. doi:10.2147/OARRR.S114447.
- Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014;12(1): doi: 10.1186/1546-0096-12-33.
- Chiu B, Chan J, Das S, Alshamma Z, Sergi C. Pediatric sarcoidosis: a review with emphasis on early onset and high-risk sarcoidosis and diagnostic challenges. Diagnostics (Basel). 2019;9(4):160. doi:10.3390/DIAGNOSTICS9040160.
- Kaufman KP, Becker ML. Distinguishing blau syndrome from systemic sarcoidosis. Curr Allergy Asthma Rep. 2021;21(2): doi: 10.1007/S11882-021-00991-3.
- Sfriso P, Caso F, Tognon S, Galozzi P, Gava A, Punzi L. Blau syndrome, clinical and genetic aspects. Autoimmun Rev. 2012;12(1):44–51. doi:10.1016/J.AUTREV.2012.07.028.
- Rosé CD, Pans S, Casteels I, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford). 2015;54(6):1008–1016. doi:10.1093/RHEUMATOLOGY/KEU437.
- Sarens IL, Casteels I, Anton J, et al. Blau syndrome-associated uveitis: preliminary results from an international prospective interventional case series. Am J Ophthalmol. 2018;187:158–166. doi:10.1016/J.AJO.2017.08.017.
- Carreño E, Guly CM, Chilov M, et al. Optic nerve and retinal features in uveitis associated with juvenile systemic granulomatous disease (Blau syndrome). Acta Ophthalmol. 2015;93(3):253–257. doi:10.1111/AOS.12544.
- Williams LB, Javed A, Sabri A, et al. ALPK1 missense pathogenic variant in five families leads to ROSAH syndrome, an ocular multisystem autosomal dominant disorder. Genet Med. 2019;21(9):2103–2115. doi:10.1038/S41436-019-0476-3.
- Zhou P, She Y, Dong N, et al. Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature. 2018;561(7721):122–126. doi:10.1038/S41586-018-0433-3.
- Kozycki CT, Kodati S, Huryn L, et al. Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome. Ann Rheum Dis. 2022;81(10):1453–1464. doi:10.1136/ANNRHEUMDIS-2022-222629.
- Huryn LA, Kozycki CT, Serpen JY, et al. Ophthalmic manifestations of ROSAH (retinal dystrophy, optic nerve edema, splenomegaly, anhidrosis, and headache) syndrome, an Inherited NF κB-Mediated autoinflammatory disease with retinal dystrophy. Ophthalmology. 2023;130(4):423–432. doi:10.1016/J.OPHTHA.2022.10.026.
- Aeschlimann FA, Batu ED, Canna SW, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. 2018;77(5):728–735. doi:10.1136/ANNRHEUMDIS-2017-212403.
- Wartz IE, O’Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. doi:10.1038/NATURE02794.
- Berteau F, Rouviere B, Delluc A, et al. Autosomic dominant familial behçet disease and haploinsufficiency A20: a review of the literature. Autoimmun Rev. 2018;17(8):809–815. doi:10.1016/J.AUTREV.2018.02.012.
- Ohnishi H, Kawamoto N, Seishima M, Ohara O, Fukao T. A Japanese family case with juvenile onset Behçet’s disease caused by TNFAIP3 mutation. Allergol Int. 2017;66(1):146–148. doi:10.1016/J.ALIT.2016.06.006.
- Shigemura T, Kaneko N, Kobayashi N, et al. Novel heterozygous C243Y A20/TNFAIP3 gene mutation is responsible for chronic inflammation in autosomal-dominant Behçet’s disease. RMD Open. 2016;2(1). doi:10.1136/RMDOPEN-2015-000223.
- Takagi M, Ogata S, Ueno H, et al. Haploinsufficiency of TNFAIP3 (A20) by germline mutation is involved in autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2017;139(6):1914–1922. doi:10.1016/J.JACI.2016.09.038.
- Duncan CJA, Dinnigan E, Theobald R, et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann Rheum Dis. 2018;77(5):783–786. doi:10.1136/ANNRHEUMDIS-2016-210944.
- Shaheen ZR, Williams SJA, Binstadt BA. Case report: a novel TNFAIP3 mutation causing haploinsufficiency of A20 with a lupus-like phenotype. Front Immunol. 2021;12:629457. doi:10.3389/FIMMU.2021.629457.
- Zanatta L, Biscaro F, Bresolin S, et al. Case report: an early-onset inflammatory colitis due to a variant in TNFAIP3 causing A20 haploinsufficiency. Front Pediatr. 2022;10:1044007. doi:10.3389/FPED.2022.1044007.
- Aslani N, Asnaashari K, Parvaneh N, et al. TNFAIP3 mutation causing haploinsufficiency of A20 with a hemophagocytic lymphohistiocytosis phenotype: a report of two cases. Pediatr Rheumatol Online J. 2022;20(1). doi:10.1186/S12969-022-00735-1.
- Chen Y, Ye Z, Chen L, et al. Association of clinical phenotypes in haploinsufficiency A20 (HA20) with disrupted domains of A20. Front Immunol. 2020;11:574992. doi:10.3389/FIMMU.2020.574992.
- Bousfiha A, Moundir A, Tangye SG, et al. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42(7):1508–1520. doi:10.1007/S10875-022-01352-Z.
- Moghaddas F, Masters SL. The classification, genetic diagnosis and modelling of monogenic autoinflammatory disorders. Clin Sci (Lond). 2018;132(17):1901–1924. doi:10.1042/CS20171498.
- Gupta V, Ramam M. Monogenic autoinflammatory syndromes in children: through the dermatologist’s lens. Indian J Pediatr Dermatol. 2018;19(3):194. doi:10.4103/IJPD.IJPD_9_18.
- Awad F, Assrawi E, Louvrier C, et al. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol Ther. 2018;187:133–149. doi:10.1016/J.PHARMTHERA.2018.02.011.
- Ter HN, Lachmann H, Özen S, et al. Treatment of autoinflammatory diseases: results from the eurofever registry and a literature review. Ann Rheum Dis. 2013;72(5):678–685. doi:10.1136/ANNRHEUMDIS-2011-201268.
- Soriano A, Soriano M, Espinosa G, et al. Current therapeutic options for the main monogenic autoinflammatory diseases and PFAPA syndrome: evidence-based approach and proposal of a practical guide. Front Immunol. 2020;11:531851. doi:10.3389/FIMMU.2020.00865/BIBTEX.
- Hansmann S, Lainka E, Horneff G, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J. 2020;18(1). doi:10.1186/S12969-020-0409-3.
- Romano M, Arici ZS, Piskin D, et al. The 2021 EULAR/American college of rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Ann Rheum Dis. 2022;81(7):907–921. doi:10.1136/ANNRHEUMDIS-2021-221801.
- Slobodnick A, Shah B, Pillinger MH, Krasnokutsky S. Colchicine: old and new. Am J Med. 2015;128(5):461–470. doi:10.1016/j.amjmed.2014.12.010.
- Ozen S, Demirkaya E, Erer B, et al. EULAR recommendations for the management of familial Mediterranean fever. Ann Rheum Dis. 2016;75(4):644–651. doi:10.1136/ANNRHEUMDIS-2015-208690.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516.
- Indraratna PL, Virk S, Gurram D, Day RO. Use of colchicine in pregnancy: a systematic review and meta-analysis. Rheumatology. 2018;57(2):382–387. doi:10.1093/RHEUMATOLOGY/KEX353.
- Hardy RS, Raza K, Cooper MS. Therapeutic glucocorticoids: mechanisms of actions in rheumatic diseases. Nat Rev Rheumatol. 2020;16(3):133–144. doi:10.1038/s41584-020-0371-y.
- Rom E, Amarilyo G, Levinski Y, et al. Protracted febrile myalgia syndrome treated with pulse of corticosteroids. Semin Arthritis Rheum. 2018;47(6):897–899. doi:10.1016/J.SEMARTHRIT.2017.10.008.
- Ter Haar NM, Oswald M, Jeyaratnam J, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636–1644. doi:10.1136/ANNRHEUMDIS-2015-207546/-/DC1.
- Millrine D, Kishimoto T. A brighter side to thalidomide: its potential use in immunological disorders. Trends Mol Med. 2017;23(4):348–361. doi:10.1016/j.molmed.2017.02.006.
- Drenth J, Vonke A, Simon A, et al. Limited efficacy of thalidomide in the treatment of febrile attacks of the hyper-IgD and periodic fever syndrome: a randomized, double-blind, placebo-controlled trial. J Pharmacol Exp Ther. 2021;298(3): 1221–1226. https://pubmed.ncbi.nlm.nih.gov/11504824/
- Yasui K, Yashiro M, Tsuge M, et al. Thalidomide dramatically improves the symptoms of early-onset sarcoidosis/Blau syndrome: its possible action and mechanism. Arthritis Rheum. 2010;62(1):250–257. doi:10.1002/ART.25035.
- Cavalli G, Dinarello CA. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology. 2015;54(12):2134–2144. doi:10.1093/RHEUMATOLOGY/KEV269.
- Malcova H, Strizova Z, Milota T, et al. IL-1 inhibitors in the treatment of monogenic periodic fever syndromes: from the past to the future perspectives. Front Immunol. 2021;11:619257. doi:10.3389/FIMMU.2020.619257/BIBTEX.
- Sjöström EO, Culot M, Leickt L, et al. Transport study of interleukin-1 inhibitors using a human in vitro model of the blood-brain barrier. Brain Behav Immun Health. 2021;16:100307. doi:10.1016/J.BBIH.2021.100307.
- Lopalco G, Rigante D, Giannini M, et al. Safety profile of anakinra in the management of rheumatologic, metabolic and autoinflammatory disorders. Clin Exp Rheumatol. 2016;34(3):531–538. https://www.clinexprheumatol.org/abstract.asp?a=9859. Accessed September 8, 2023
- Ramírez J, Cañete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. 2018;17(7):727–732. doi:10.1080/14740338.2018.1486819.
- De Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. 2018;378(20):1908–1919. doi:10.1056/NEJMOA1706314/SUPPL_FILE/NEJMOA1706314_DISCLOSURES.PDF.
- Chakraborty A, Tannenbaum S, Rordorf C, et al. Pharmacokinetic and pharmacodynamic properties of canakinumab, a human anti-interleukin-1β monoclonal antibody. Clin Pharmacokinet. 2012;51(6):e1–e18. doi:10.2165/11599820-000000000-00000.
- Brogan PA, Hofer M, Kuemmerle-Deschner JB, et al. FRI0503 Efficacy and safety of canakinumab in patients with cryopyrin associated periodic syndromes: an open-label, phase-III, extension study. Ann Rheum Dis. 2016;75(Suppl 2):620–621. doi:10.1136/ANNRHEUMDIS-2016-EULAR.4865.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58(8):2443–2452. doi:10.1002/ART.23687.
- Petrushkin H, Karagiannis DA, Bird A, Jawad ASM. Intermediate uveitis associated with familial Mediterranean fever. Clin Exp Rheumatol. 2015;33:170–170. https://www.clinexprheumatol.org/abstract.asp?a=9496. Accessed September 8, 2023.
- Yazılıtaş F, Aydoğ Ö, Özlü SG, et al. Canakinumab treatment in children with familial Mediterranean fever: report from a single center. Rheumatol Int. 2018;38(5):879–885. doi:10.1007/S00296-018-3993-5.
- Başaran Ö, Kavuncu S, Güven A, Uncu N, Acar-Çelikel B, Çakar N. Familial Mediterranean fever associated with optic neuritis, successfully treated with anti-interleukin 1 agents. Turk J Pediatr. 2016;58(3):327–330. doi:10.24953/TURKJPED.2016.03.018.
- Gattorno M, Obici L, Cattalini M, et al. Canakinumab treatment for patients with active recurrent or chronic TNF receptor-associated periodic syndrome (TRAPS): an open-label, phase II study. Ann Rheum Dis. 2017;76(1):173–178. doi:10.1136/ANNRHEUMDIS-2015-209031.
- Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med. 2006;355(6):581–592. doi:10.1056/NEJMOA055137/SUPPL_FILE/NEJM_GOLDBACH-MANSKY_581SA1.PDF.
- Hirano M, Seguchi J, Yamamura M, et al. Successful resolution of stromal keratitis and uveitis using canakinumab in a patient with chronic infantile neurologic, cutaneous, and articular syndrome: a case study. J Ophthalmic Inflamm Infect. 2015;5(1):1–5. doi:10.1186/S12348-015-0065-9.
- Teoh SCB, Sharma S, Hogan A, Lee R, Ramanan AV, Dick AD. Tailoring biological treatment: anakinra treatment of posterior uveitis associated with the CINCA syndrome. Br J Ophthalmol. 2007;91(2):263–264. doi:10.1136/BJO.2006.0101477.
- Oberg TJ, Vitale AT, Hoffman RO, Bohnsack JF, Warner JE. Cryopyrin-associated periodic syndromes and the eye. Ocul Immunol Inflamm. 2013;21(4):306–309. doi:10.3109/09273948.2013.765016.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011;63(3):840–849. doi:10.1002/ART.30149.
- Aróstegui JI, Arnal C, Merino R, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007;56(11):3805–3813. doi:10.1002/ART.22966.
- Simonini G, Xu Z, Caputo R, et al. Clinical and transcriptional response to the long-acting interleukin-1 blocker canakinumab in Blau syndrome-related uveitis. Arthritis Rheum. 2013;65(2):513–518. doi:10.1002/ART.37776.
- Seyahi E, Ozdogan H, Celik S, Ugurlu S, Yazici H. Treatment options in colchicine resistant familial Mediterranean fever patients: thalidomide and etanercept as adjunctive agents. Clin Exp Rheumatol. 2006;24(5 SUPPL. 42). https://www.clinexprheumatol.org/abstract.asp?a=2919.Accessed September 8, 2023.
- Bilgen SA, Kilic L, Akdogan A, et al. Effects of anti-tumor necrosis factor agents for familial Mediterranean fever patients with chronic arthritis and/or sacroiliitis who were resistant to colchicine treatment. J Clin Rheumatol. 2011;17(7):358–362. doi:10.1097/RHU.0B013E31823682F5.
- Nedjai B, Quillinan N, Coughlan RJ, et al. Lessons from anti-TNF biologics: infliximab failure in a TRAPS family with the t50M mutation in TNFRSF1A. Adv Exp Med Biol. 2011;691:409–419. doi:10.1007/978-1-4419-6612-4_43/COVER.
- Davies R, De Cock D, Kearsley-Fleet L, et al. The risk of uveitis in patients with JIA receiving etanercept: the challenges of analysing real-world data. Rheumatology (Oxford). 2020;59(6):1391–1397. doi:10.1093/RHEUMATOLOGY/KEZ449.
- Taban M, Dupps W, Mandell B, Perez V. Etanercept (enbrel)-associated inflammatory eye disease: case report and review of the literature. Ocul Immunol Inflamm. 2006;14(3):145–150. doi:10.1080/09273940600659393.
- Durel CA, Aouba A, Bienvenu B, et al. Observational study of a French and Belgian multicenter cohort of 23 patients diagnosed in adulthood with mevalonate kinase deficiency. Medicine. 2016;95(11):e3027. doi:10.1097/MD.0000000000003027.
- Kraus CL, Culican SM. Nummular keratopathy in a patient with Hyper-IgD Syndrome. Pediatr Rheumatol Online J. 2009;7(1):1–3. doi:10.1186/1546-0096-7-14.
- Chen J, Luo Y, Zhao M, et al. Effective treatment of TNFα inhibitors in Chinese patients with Blau syndrome. Arthritis Res Ther. 2019;21(1). doi:10.1186/S13075-019-2017-5.
- Rubbert-Roth A, Furst DE, Nebesky JM, Jin A, Berber E. A review of recent advances using tocilizumab in the treatment of rheumatic diseases. Rheumatol Ther. 2018;5(1):21–42. doi:10.1007/S40744-018-0102-X.
- Zheng J, Huang R, Huang Q, et al. Tocilizumab in the treatment of patients with AA amyloidosis secondary to familial Mediterranean fever. Rheumatology. 2015;54(3):564–565. doi:10.1093/RHEUMATOLOGY/KEU474.
- La Torre F, Muratore M, Vitale A, Moramarco F, Quarta L, Cantarini L. Canakinumab efficacy and long-term tocilizumab administration in tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Rheumatol Int. 2015;35(11):1943–1947. doi:10.1007/S00296-015-3305-2.
- Snegireva LS, Kostik MM, Caroli F, Ceccherini I, Gattorno M, Chasnyk VG. Failure of tocilizumab treatment in a CINCA patient: clinical and pathogenic implications. Rheumatology (Oxford). 2013;52(9):1731–1732. doi:10.1093/RHEUMATOLOGY/KET121.
- Matsubara T, Hasegawa M, Shiraishi M, et al. A severe case of chronic infantile neurologic, cutaneous, articular syndrome treated with biologic agents. Arthritis Rheum. 2006;54(7):2314–2320. doi:10.1002/ART.21965.
- Lu L, Shen M, Jiang D, et al. Blau syndrome with good reponses to Tocilizumab: a case report and focused literature review. Semin Arthritis Rheum. 2018;47(5):727–731. doi:10.1016/J.SEMARTHRIT.2017.09.010.
- Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–243. doi:10.1038/nrrheum.2017.23.
- Garcia-Robledo JE, Aragón CC, Nieto-Aristizabal I, Posso-Osorio I, Cañas CA, Tobón GJ. Tofacitinib for familial Mediterranean fever: a new alternative therapy? Rheumatology. 2019;58(3):553–554. doi:10.1093/RHEUMATOLOGY/KEY384.
- Zhang S, Cai Z, Mo X, Zeng H. Tofacitinib effectiveness in Blau syndrome: a case series of Chinese paediatric patients. Pediatr Rheumatol Online J. 2021;19(1): doi: 10.1186/S12969-021-00634-X.
- Álvarez-Reguera C, Prieto-Peña D, Herrero-Morant A, et al. Clinical and immunological study of Tofacitinib and Baricitinib in refractory Blau syndrome: case report and literature review. Ther Adv Musculoskelet Dis. 2022;14:1759720X221093211. doi:10.1177/1759720X221093211.
- Xu Q, Zhang J, Qin T, et al. The role of the inflammasomes in the pathogenesis of uveitis. Exp Eye Res. 2021;208:108618. doi:10.1016/J.EXER.2021.108618.