
Abstract
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder with a related enzyme deficiency involved in the adrenal corticosteroid synthesis pathway due to genetic mutations. 17α-hydroxylase deficiency(17α-OHD) is a rare form of CAH. Herein, we reported clinical data on diagnosis and treatment regimens for a 17α-hydroxylase-deficient patient. A 24-year-old female patient was admitted to the hospital with limb numbness for 7 days and sudden limb weakness. Full laboratory and radio-imaging investigations showed hypokalemia and abdominal occupation. Abnormal rhythm of cortisol(Cor) and adrenocorticotrophic hormone (ACTH)was observed. The diagnosis was confirmed by molecular mutation detection, which showed a homozygous mutation of c.987del in the 17-hydroxylase/17,20-lyase deficiency (17OHD) lease-related CYP17A1 from both biological parents. The patient was treated with prednisone acetate and estradiol valerate. After one year of treatment with predisoone acetate and estradiol valerate, the patient had normal menstruation, increased blood potassium, estradiol and 24h-UFC, and decreased ACTH level. There is no significant change in large adrenal hyperplasia lesions although sexual characteristics and menstrual cycles have recovered. Through this case and literature review, it can be concluded that CAH with 17α-OHD can be diagnosed according to the genetic detection.
Introduction
Congenital adrenal hyperplasia (CAH) [Citation1] is one of the most common autosomal recessive metabolic disorders caused by teensy me deficiency, which involved in the adrenal cortical hormone synthesis pathway. The blockade of Cor biosynthesis results in excessive secretion of Corticotropin-Releasing Hormone (CRH) and adrenocorticotrophic hormone (ACTH), thereby leading to adrenal and pituitary hyperplasia. CAH is an autosomal recessive disorder, and approximately 95% of CAH cases are caused by 21-hydroxylase deficiency, with the remaining 5–8% mainly due to 11b-hydroxylase deficiency. 17α-hydroxylase deficiency (17α-OHD) is a less common form of CAH, accounting for about 1% of cases, with an estimated annual incidence ranging from1/5000 to 1/100000 [Citation2–4]. The 17 α-OHD form sin patients are associated with sex hormone generation CYP17A1 gene mutation, glucocorticoid and disorder with excess mineralocorticoid synthesis. Herein, a case report of CAH with 17 α-OHD was described including the diagnosis and treatment of patient.
Case presentation
A 24-year-old female was admitted to the local hospital due to limb weakness. Blood biochemistry and abdominal computerized tomography(CT) results indicated hypokalemia and abdominal occupation due to left adrenal area and prerenal space tumors, and limb weakness was not improved after potassium supplementation treatment for 5 days. Therefore, she was transferred to the emergency department of the First Hospital of Lanzhou University and complained of no menarche and no secondary sex characteristics. The patient was 170 cm in height, weight 70 kg, upper 76 cm, lower 94 cm, and lower/upper 1.236. Blood pressure was 133/93 mmHg, and the heart rate was 95 times/min. Physical examination patients had no axillary and pubic hairs, thoracic deformity, scoliosis, breast stage Tanner I, the vulva was of the female infantilism type. No beard, vellus hair growth increased.
Results of medium dose dexamethasone test for congenital adrenal hyperplasia showed in . Androstenedione was less than 0.3 ng/ml and serum dehydroepiandrosterone (DHEA) was less than 15.0 μg/dL. The 24-h urinary free cortisol (24h-UFC) was less than 10 μg/24h. Primary aldehyde screening showed 0.24 pg/ml of renin. The horizontal position test showed that renin levels were 0.19 pg/ml in the supine position and 0.29 pg/ml in the standing position. Cor rhythm demonstrated Cor(8-16-24) <1.00-<1.00-<1.00 μg/dL. ACTH rhythms showed ACTH(8-16-24) 71.8-85.0 pg/ml-59 pg/ml. Abdominal enhancement CT revealed bilateral abnormal adrenal enhancement of nodules and mass shadow, internal fat density shadow and uneven enhancement scan, with the right size of about 17 × 12mm and the left size of about 98 × 83mm (). X-ray of chest indicated the thoracic vertebra bent slightly to the right (). Additionally, pituitary magnetic resonance imaging (MRI) horizontal sweep showed the pituitary form is slightly full and the height was about 0.9 cm (). X-ray of hand showed the epiphyseal of the distal ulna appeared and the epiphyseal and distal radius were not closed and wrist bones with 13-year-old bone age was observed (). Gynecology B ultrasound showed uterine body size was about 22x8x4mm and bilateral ovaries were not detected. B ultrasound showed diffuse lesions of left thyroid lobe, with hypoechoic TI-RADS III nodules on both sides of the thyroid lobe.
Figure 1. CT and MRI test for patient. (A) Enhanced CT of the patient’s abdomen, right adrenal hyperplasia by the black arrow, left adrenal hyperplasia by the white arrow; (B) Rabat; (C) Patient’s head MRI and hyperplastic pituitary by the white arrow. (D) Left-hand flat piece and bone age
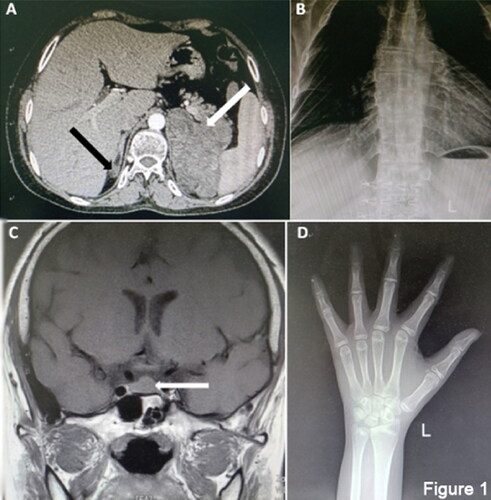
Table 1. Congenital adrenal hyperplasia test.
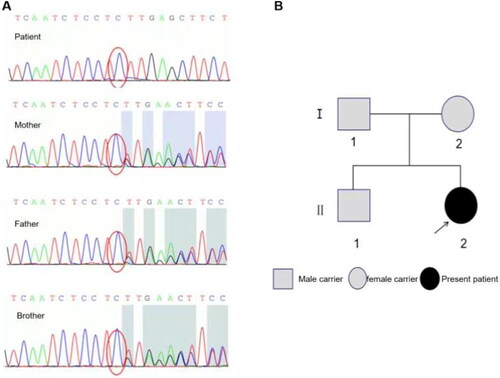
Chromosome analysis showed chromosome karyotype 46, XX. Using the Illumina second-generation sequencing platform, exon sequencing of CHA related genes CYP17A1 was performed, and patient samples had a homozygous mutation of c.987del in the 17-OHD lease-related gene CYP17A1. Pedigree verification confirmed that the mutation was a heterozygous mutation of c.987delC from both parents (). Meanwhile, the genetic map of 7α-OHD was mapped by sequencing (). The patients were diagnosed as CAH with 17 α-OHD. After oral administration of Prednisone acetate 5 mg at noon and estradiol valerate 0.5 mg in the morning, the limb weakness was completely recovered. The level of blood potassium, estradiol (E2), and 24h-UFC were increased at 3 months after discharge. Although the level of ACTH decreased, the rhythm has not appeared. The dose of prednisone acetate was adjusted to 7.5 mg/d. One year later, the patient developed regular vaginal bleeding with menstrual cycle of 28–30 days and periods lasting 5-6 days. The uterine size was larger than that obtained before treatment. Blood potassium estradiol, 24h-UFC, Cor and ACTH rhythm decreased. Cor(8Am)0.37 μg/dL was higher compared to the one before treatment. ACTH(8Am)2.54 pg/ml and 17α-hydroxy progesterone (17-αOHP) <0.04 ng/mL were significantly lower than that before treatment.
Figure 2. CYP17A1 sequencing and genetic map of Patient’s family. (A) Sequencing results of CYP17A1 of the patient, the patient’s mother, the patient’s father and the patient’s brother from top to bottom. The patient had a homozygous mutation of c.987del, while her parents and elder brother all had heterozygous mutations of c.987del; (B) Patient’s genetic map of 17α-OHD.
Discussion
This study reported a genetically proven case with combined 17-hydroxylase/17,20-lyase deficiency (17OHD). Abnormal rhythm of Cor and ACTH was observed. Cor(8-16-24) < 1.00– < 1.00– <1.00 μg/dL were consistently below the normal lower limit of 5 μg/dL; ACTH(8-16-24) 71.8–85.0 pg/ml-59 pg/ml were consistently above the normal upper limit of 46 pg/dL (). Deletion mutations at the c.987del site of CYP17A1 gene was observed in this CAH case (rs764919516). After treated with prednisone acetate and estradiol valerate, the patient has developed normal vaginal bleeding. Around 1% of CAH patients have 17 α-OHD that was first reported by Biglieri in 1966 [Citation4]. CYP17A1 is the pathogenic gene of this disease, composed of 7 introns and 8 exons, and expressed in both adrenal and gonadal glands. Therefore, CYP17A1 gene defect can lead to the biosynthesis of adrenal and gonadal steroids, and patients show dual defects of adrenal and gonadal functions [Citation5,Citation6].
There are more than 100 mutations in the CYP17A1 gene including point mutation, insertion, deletion, and frameshift mutation, which are mostly located at the C terminus [Citation7]. Genetic mutation studies in 26 Chinese 17-αOHD patients showed that c.985_987del and c.1460_1469del were the most common types of mutations with 60.8% and 21.7% of the mutant alleles, respectively [Citation8]. It was already demonstrated that CYP17A1 encoded a cytochrome P450c17 protein which includes 17α-hydroxylase acting on 17-hydroxylation of progesterone and pregnenolone and 17,20-lyase catalyzing the conversion of 17-hydroxyenolone and 17-hydroxyprogesterone to DHEA and androstenedione, respectively. Moreover, 17 α-OHD contains two forms. One is a deficiency of 17-OHD and another one is the isolated 17,20-lyase deficiency (ILD) [Citation9]. In this case, a homozygous mutation was found in the exon region of CYP17A1 gene: c.987del deletion mutation, resulting in a translational p.Tyr329fs encoding mutation, which is also the most common mutation site in 17-αOHD patients. The homozygous mutation of CYP17A1 in this patient was found to come from her parents, which was in line with the autosomal recessive inheritance rule.
This case was diagnosed as 17-OHD and the deficiency of both enzymes resulted in limited biosynthesis of Cor, androgen, and estrogen, while the level of corticosterone, 11-deoxycorticosterone (11-deoxycorticosterone, DOC) was increased [Citation10]. Disorder of Cor synthesis can suppress the negative feedback inhibition on the pituitary, thereby inducing the ACTH generation and adrenal hyperplasia. In addition, decreased estrogen was contributed to the menstruation, breast, and female genital development, while low androgens might suppress the growth of pubic and axillary hair and reduce estrogen generation as a precursor substance. Furthermore, increased DOC can cause water and sodium retention and low renin hypertensive [Citation11], which accompanied by hypokalemia, alkalosis and weak limbs. The majority of patients are associated with low potassium and hypertension as this case report [Citation12]. A similar case was reported by Lee [Citation13], which mainly involved in abdominal pain and hypertension with a massive adrenal cortical adenoma and a left mass size was about 100x63x86mm that was rare in the reported cases. Wang reported that imaging characteristics of nine patients with 17 α-OHD, which showed that the average maximum area of 640.1 mm2 on the transverse axis of the adrenal gland was only about 3 times greater than the normal value (150 mm2), while the size of the adrenal mass increased more than 10 times the normal value [Citation14].
Treatment of 17α-OHD mainly involves glucocorticoid supplementation and adrenal steroid replacement. Glucocorticoids help reduce 11-DOC and ACTH. In addition, calcium antagonists, spironolactone, angiotensin receptor blockers, and cortisone are commonly used to control blood pressure. Steroid hormone replacement is done to preserve a woman’s sexual characteristics. For patients with karyotype 46 and XX, estrogen and progesterone replacement therapy can be used to induce the menstrual cycle. In this case, after oral administration of prednisone acetate and estradiol valerate, the patient had complete recovery of limb weakness, and increased blood potassium, estradiol (E2), and 24h-UFC, which was consistent with literature reports [Citation15,Citation16].
Conclusion
Congenital adrenal hyperplasia with 17 α-OHD can be diagnosed according to the genetic detection. There is no significant change in large adrenal hyperplasia lesions although sexual characteristics and menstrual cycles have recovered. Long-term administration of prednisone should pay attention to the prevention and treatment of bone loss.
Author contributions
All authors contributed to the conceptualization, writing, and editing of this work.
Ethics approval
This work has been carried out in accordance with the Declaration of Helsinki (2000) of the World Medical Association. This study was approved by the first hospital of Lanzhou University (LDYYLL-2022-384), and all participants provided written informed consent.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analyzed during this study are included in this published article.
Additional information
Funding
References
- Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Endocrinol Meta Clin North Am. 2017; 46(2):1–4. doi: 10.1016/j.ecl.2017.01.008.
- Oh YK, Ryoo U, Kim D, et al. 17α-hydroxlyase/17, 20-lyase deficiency in three siblings with primary amenorrhea and absence of secondary sexual development. J Pediatr Adolesc Gynecol. 2012;25(5):e103-5. Octdoi: 10.1016/j.jpag.2012.05.008.
- Podgórski R, Aebisher D, Stompor M, et al. Congenital adrenal hyperplasia: clinical symptoms and diagnostic methods. Acta Biochim Pol. 2018;65(1):25–33. doi: 10.18388/abp.2017_2343.
- Biglieri EG, Herron MA, Brust N. 17α-hydroxylation deficiency in man. J Clin Invest. 1966; 45(12):1946–1954. doi: 10.1172/JCI105499.
- Picado-Leonard J, Miller WL. Cloning and sequence of the human gene for P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): similarity with the gene for P450c21. DNA. 1987; 6(5):439–448. doi: 10.1089/dna.1987.6.439.
- Chung BC, Picado-Leonard J, Haniu M, et al. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc Natl Acad Sci USA. 1987;84(2):407–411. Jandoi: 10.1073/pnas.84.2.407.
- Human Gene Mutation Database Cardiff CYP17A1 gene. (Cited 2008 Oct 22). Available from: http://www.hgmd.cf. 258 ac.uk, 2020.
- Zhang M, Sun S, Liu Y, et al. New, recurrent, and prevalent mutations: clinical and molecular characterization of 26 Chinese patients with 17alpha-hydroxylase/17,20-lyase deficiency. J Steroid Biochem Mol Biol. 2015;150:11–16. doi: 10.1016/j.jsbmb.2015.02.007.
- Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017; 165(Pt A):71–78. doi: 10.1016/j.jsbmb.2016.02.002.
- Kota SK, Modi K, Jha R, et al. 17-α-Hydroxylase deficiency: an unusual case with primary amenorrhea and hypertension. Indian J Endocrinol Metab. 2011;15(2):127–129. Aprdoi: 10.4103/2230-8210.81945.
- Auchus ML, Auchus RJ. Human steroid biosynthesis for the oncologist. J Investig Med. 2012; 60(2):495–503. doi: 10.2310/JIM.0b013e3182408567.
- Kim YM, Kang M, Choi JH, et al. A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase deficiency, a case series of such mutations among koreans and functional characteristics of a novel mutation. Metabolism. 2014;63(1):42–49. doi: 10.1016/j.metabol.2013.08.015.
- Lee SJ, Song JE, Hwang S, et al. Untreated congenital adrenal hyperplasia with 17-α hydroxylase/17,20-Lyase deficiency presenting as massive adrenocortical tumor. Endocrinol Metab. 2015;30(3):408–413. doi: 10.3803/EnM.2015.30.3.408.
- W Y, Tx L, Sh R. Imaging characteristics of feminization induced by 17α-hydroxylase deficiency syndrome. Radiol Pract. 2018;33(09):923–926.
- Ammar R, Ramadan A. Incidental diagnosis of 17-alpha-hydroxylase deficiency: a case report. Oxf Med Case Reports. 2020; 2020(12):omaa108. doi: 10.1093/omcr/omaa108.
- Dos Santos LR, Heilbrun EP, Félix CS, et al. Congenital adrenal hyperplasia due to 17-α-hydroxylase deficiency: a case report. Rev Endocrinol. 2021;17(2):138–140. Novdoi: 10.17925/EE.2021.17.2.138.