
Abstract
Objective
ERα (estrogen receptor alpha) exerts nuclear genomic actions and membrane-initiated non-genomic effects. The mutation of aspartic acid into alanine in vitro revealed the critical role of aspartic acid 258 (corresponding to mouse amino acid site 262) of ERα for non-nuclear function. Our previous in vitro study revealed that this mutation blocked estrogen’s non-genomic effects on vascular endothelial H2S release. Here, we studied the in vivo role of the aspartic acid 262 of ERα in the reproductive system and in the vascular tissue.
Approach and Results
We generated a mouse model harboring a point mutation of the murine counterpart of this aspartic acid into alanine (ERαD262A). Our results showed that the ERαD262A females are fertile with standard hormonal serum levels, but the uterine development and responded with estrogen and follicular development are disrupted. In line with our previous study, we found that the rapid dilation of the aorta was abrogated in ERαD262A mice. In contrast to the previously reported R264-ERα mice, the classical estrogen genomic effector SP1/NOS3/AP1 and the nongenomic effectors p-eNOs, p-AKT, and p-ERK were disturbed in the ERαD262A aorta. Besides, the serum H2S concentration was decreased in ERαD262A mice. Together, ERαD262A mice showed compromised both genomic and non-genomic actions in response to E2.
Conclusions
These data showed that aspartic acid 262 of ERα are important for both genomic and non-genomic effects of E2. Our data provide a theoretical basis for further selecting an effective non-genomic mouse model and provide a new direction for developing estrogen non-genomic effect inhibitors.
Introduction
ERα (estrogen receptor alpha), one of the nuclear receptor family, is involved in many physiological processes, including cardiovascular shield, metabolism, and reproduction [Citation1]. Estrogen, particularly of 17β-estradiol (E2), either interacts with nuclear ERα and exerts direct effects by binding to DNA sequences via estrogen-responsive elements or activates intracellular signaling cascades via protein-protein tethering with transcriptional factors [Citation2]. In vivo studies suggest that the two activation functions (AF), AF-1 and AF-2, are the main regulators for gene transcription of ERα in cell- and tissue-specific manner [Citation3–5]. However, in addition to these “genomic” actions, ERα can mediate membrane-initiated steroid signaling [Citation6], term “nongenomic” or “extracellular” actions. Several decades ago, studies showed that estrogen rapidly mobilized intracellular calcium and generated cAMP [Citation7, Citation8].
Recently in vitro studies described that estrogen activates ERα to modulate potassium currents, activate phospholipase C, increase nitric oxide production in endothelial, and stimulate protein kinase pathways (PI3K/Akt, ERK) [Citation9–13]. In particular, human ERα Cys-447 is a site of palmitoylation that promotes caveolin-1 interaction and plasma membrane localization [Citation14–16]. While the mouse model mutated for non-palmitoylatable Cys447Ala of ERα (ERα-C451A) showed the importance of membrane actions for fertility and some rapid vascular effects, such as acceleration of reendothelialization and endothelial NO synthase phosphorylation and activation [Citation14].
Previous studies reported that, besides palmitoylation, a sequence in the C-domain of human ERα (aa251-260) allowed the segregation between non-nuclear and nuclear actions of estrogen [Citation17, Citation18]. Studies revealed that this domain is involved in rapid NO production in transfected monkey kidney COS cells [Citation19]. Nonetheless, point mutations within this region, such as R256A, K257, and R260A, all lost the activation of endothelial NO synthase (eNOS) and ERK phosphorylation in response to estrogen and preserved the estrogen-induced transcriptional response [Citation18]. A recent study showed that the knock-in mouse model of ERα mutated for the arginine 264 (the mouse counterpart of human arginine 260) into alanine (ESR1R264A), the mice lost the rapid vascular effects of estrogens, such as rapid endothelial repairment, eNOS activation, and arteries dilatation. However, ESR1R264A female mice are fertile [Citation20]. Our previous study showed that human ERα mutated for the aspartic acid 258 into alanine prevents the nongenomic effect of estrogen on classical ERK and Akt pathways and the estrogen-induced PKG activation [Citation21]. Nevertheless, the specific roles of this aspartic acid 258 of ERα in reproduction and vascular system remained to investigate in vivo.
In the present study, we generated a homologous recombination knock-in mouse model of ERα mutated for the aspartic acid 262 (the mouse counterpart of human aspartic acid 258) into alanine (designated ESR1D262A). In contrast to several other mouse models mutated for ERα, the D262A female mice are fertile, with normal serum E2 (estrogen), FSH (follicle stimulating hormone), LH (luteinizing hormone), T (testosterone) and PG (progesterone) levels. However, the uterine responses to estrogen and follicular development are disrupted. Meanwhile, the rapid vascular effects of estrogens initiated at the plasma membrane, such as rapid endothelial repair, eNOS activation, and dilatation of arteries, were lost in ESR1D262A mice.
Results
Generation of ESR1D262A mice
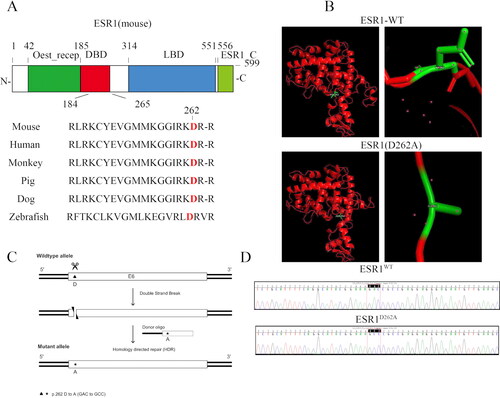
To study the potential role of aspartic acid 262 of ERα in the rapid signaling in vivo, we generated mutant mice using a targeting construct that contained two base-pair changes in exon, which mutated the aspartic acid 262 into alanine, where all six species conserved (). The mutated ERα protein had a similar structure to the wild-type ERα (). The D262A mutation was confirmed using DNA sequencing after a polymerase chain reaction using tail DNA as a template ().
Figure 1. Generation of ESR1D262A mice. (A) Representation of the structure domain within Murine ERα and conservative analysis of amino acids in ESR1 genes across six species. (B) Representation cartoon of the 3D structure of ESR1WT and ESR1D262A mutation type. (C) Schematic illustration of the targeting strategy used to introduce the mutation. (D) Genotyping sequencing of ESR1 gene result of ESR1WT and ESR1D262A mice.
Impaired uterine development and responded with chronic to 17β-estradiol in ESR1D262A mice
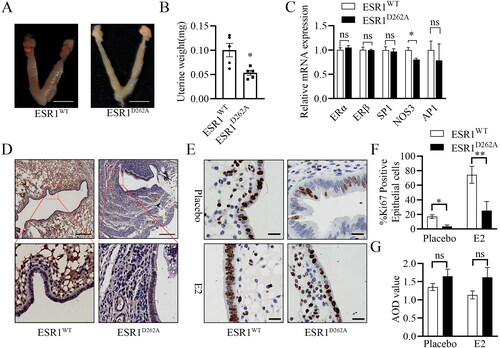
Since the mice mutated for ERα are usually infertility, including those explicitly mutated for the palmitoylation site [Citation14], we evaluated the reproductive system of ESR1D262A mice for the first time. The uterus tissue of ESR1D262A mice had a long and thin appearance compared to that of ESR1WT mice, ESR1D262A mice also had lower uterine weight than ESR1WT mice (). Interestingly, ESR1D262A female mice were fertile when continuously mated with ESR1D262A males (6.3 pups per litter). Previous studies showed that in transfected HeLa cells, this specific human counterpart mutant ERα-D258A has an average capacity to activate gene transcription via either direct, ERE-mediated binding to DNA or indirect DNA binding via the transcription factors SP-1 or AP-1 [Citation18]. Furthermore, O’Brien et al. suggested that estrogen-induced proliferation of uterine epithelial cells is independent of ERα binding to classical ERE [Citation22]. In detached uterine tissue, there is a similarity level of ERα, ERβ, SP1, and AP1 mRNA between WT and ESR1D262A mice, we also found a decrease level of NOS3 (eNOS protein coding gene) mRNA in ESR1D262A mice (). Immunodetection of ERα shows a decreased level of nuclear component in ESR1D262A mouse uterus epithelial cells (). We also performed Ki-67 immunodetection in transverse uterus sections following E2 treatment, our result demonstrated that E2 induced a significant decrease in epithelial proliferation in ESR1D262A mice compared to ESR1WT mice ().
Figure 2. Impaired uterine development and responded with chronic to 17β-estradiol in ESR1D262A mice. (A-D) Mice were sacrificed at the age of 12 weeks, and tissue was dissected. (A) Representative images of ESR1WT and ESR1D262A female reproductive organs. Scale bar = 1 cm. (B) Quantification of uterine wet weight. n = 5. (C) qRT-PCR of ERα, ERβ, SP1, NOS3 and AP1 mRNA transcripts. n = 3. (D) Representative images of ERα immunodetection in transverse uterus sections in WT and ESR1D262A. Scale bar = 200 μM. (E–G) WT and ESR1D262A mice underwent sham or OVX surgery at 8 weeks, with or without E2 treatment for 4 weeks. (E) Representative Ki-67 immunodetection in transverse uterus sections and quantification of percentage of Ki-67 positive cells in the epithelium (F) and average optical density stromal and epithelial compartments from transverse uterine sections (G). Scale bar = 20 μM. n = 3. Data presented mean ± SEM. *p < 0.05 vs. placebo in ESR1WT and ESR1D262A mice respectively. ns. not significant.
Adult ovarian phenotype is disturbed in the ESR1D262A mice
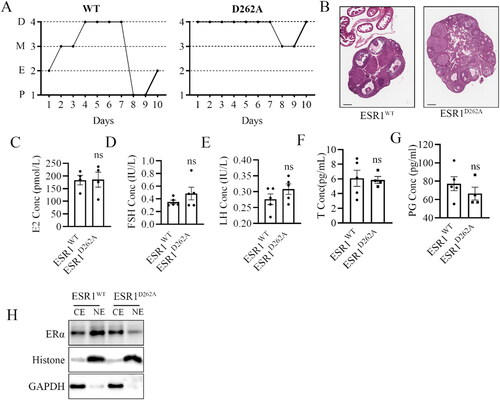
As mentioned previously, we observed regular litters in ESR1D262A mice. To further investigate ovarian function, we performed estrous cyclicity and histomorphology analysis on the ovaries of 12-week-old female mice. The result of vaginal cytology revealed that ESR1WT mice were in a consecutive stage (diestrus, metestrus, estrus, proestrus), whereas ESR1D262A mice were in the diestrus for seven consecutive days (). Histomorphology analysis of the ovaries revealed an excess of mature corpora lutea and atretic follicles in ESR1D262A ovaries, indicating abnormal development of follicles (). We also measured steroid sex hormone levels in intact 12-week-old female mice. Interestingly, E2, FSH, LH, T and PG have no difference between the genotypes (). To further characterized this mouse model, we evaluated the ERα expression in the cell nuclear and cytoplasmic components of mouse ovarian tissue. ERα protein was reduced in nuclear fractions ().
Figure 3. Adult ovarian phenotype is disturbed in the ESR1D262A mice. Representative images of estrous cycles (A) and Histological observation (B) of ovarian in ESR1WT and ESR1D262A mice. Scale bar = 200 μM. (C–G) Serum E2 (C), FSH (D), LH (E) T(F), PG (G) concentration in 12-weeks-old ESR1WT and ESR1D262A mice. n = 4-5. (H) ERα expression in the cell nuclear (NE) and cytoplasmic components (CE) of mouse ovarian tissue. Data presented mean ± SEM. *p < 0.05, ns. not significant. D, diestrus; M, metestrus; E, estrus; P, proestrus.
ESR1D262A mice attenuated E2-induced vasodilation
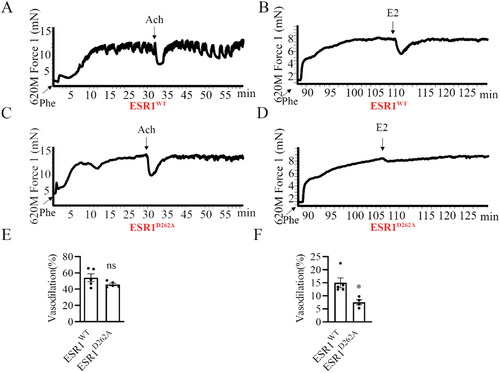
The protective vascular effects of E2 implicated the non-genomic actions of estrogens, that is, rapid dilation and acceleration of reendothelialization [Citation6, Citation23, Citation24]. So, we evaluated the vasodilatory effects of E2 (10−5 M) in ESR1WT and ESR1D262A mice by measuring dilatory responses of the isolated phenylephrine-pre-constricted aorta. There was no difference in vasodilation among the genotypes (), whereas ESR1D262A mice aorta presented significantly decreased vasodilation response to E2 compared with ESR1WT mice aorta (), revealing the critical role of ESR1D262A in the estrogen-regulated rapid vasodilatation.
Figure 4. E2-induced vasodilation was attenuated in ESR1D262A mice. (A-D) Representative images of wall tension measurement and vasodilation of ESR1WT (A, B) and ESR1D262A (C, D) mice Aortae. (A, C) Aortae were dissected out and pretreated for 30 min with 200 mM L-NNA (NO inhibitor), and 10 mM indo (prostaglandin synthesis inhibitors) and pre-constricted with phenylephrine (Phe, 10−6 M) before testing with acetylcholine (Ach, 10−5 M), and quantitative analysis was present in the bottom panel (E). (B, D) mutation 262 site in ESR1 gene significantly attenuated E2-induced vasodilation, and quantitative analysis was present in the bottom panel (F). n = 5. Data presented mean ± SEM. *p < 0.05, ns. not significant.
Both genomic and non-genomic effect of E2 phenotype in the aorta is disturbed in ESR1D262A mice
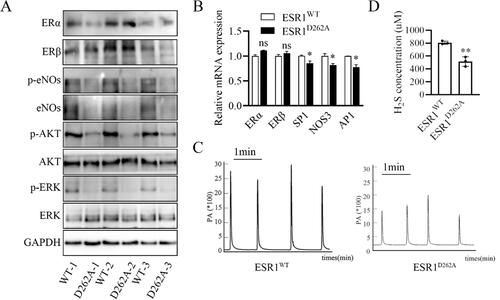
Point mutation of ESR1D262A increased ERα protein level in the thoracic aorta but decreased the genomic effector eNOs protein level in vascular tissues (). Decreased level of gene-effect molecular SP1, NOS3 and AP1 mRNA was found in ESR1D262A mice (). Rapid dilatation in response to E2 is the consequence of stimulation of eNOS through Akt-mediated phosphorylation of eNOS at the S1177 site [Citation25], we then evaluated the phosphorylation of eNOS in the isolated thoracic aorta. Our results showed that a decreased phosphorylation level of eNOS, AKT, and ERK in ESR1D262A mice arteries (), we also evaluated ERα and ERβ levels in the aorta tissue, and the results show similar levels of protein and mRNA between WT and ESR1D262A mice (). Previous studies demonstrated that hydrogen sulfide (H2S) induced vasorelaxation, stimulated endothelial cell-related angiogenic properties, and protected against atherosclerosis [Citation26, Citation27]. Our previous study revealed that E2 non-genomic induced vascular endothelial H2S release [Citation21]. To investigate the role of ESR1D262A in inducing vascular endothelial H2S release, we measured the serum H2S concentration from ESR1WT and ESR1D262A mice. As shown in , ESR1D262A mice released significantly less serum H2S. Taken together, ESR1D262A mice presented corrupted phenotype of E2 induced both genomic and non-genomic effects.
Figure 5. Both genomic and non-genomic effect of E2 phenotype in the aorta is disturbed in ESR1D262A mice. (A) Representative western blotting images of ERα, ERβ, p-eNOs, eNOs, p-AKT, AKT, p-ERK and ERK expression in aorta from 3 paired ESR1WT and ESR1D262A mice. (B) qRT-PCR of ERα, ERβ, SP1, NOS3 and AP1 mRNA transcripts. n = 3. (C) Representative images of serum H2S concentration from ESR1WT and ESR1D262A mice. (D) Quantification of serum H2S concentration from ESR1WT and ESR1D262A mice. n = 3. Data presented mean ± SEM. **p < 0.01. ns. not significant.
Materials and methods
Animal
All experimental animal procedures were performed following the principles established by the Guangzhou Medical University and approved by the local Ethical Committee of Animal Care. The ESR1D262A knock-in mouse line was generated on a C57BL/6N background at the Cyagen Biosciences Inc (Guangzhou, China) by homologous recombination, as described in . Mouse-tail DNA was analyzed by PCR and DNA sequencing to determine the genotypes. Target region of the mouse ESR1 locus was amplified by PCR with specific primers (5′-CATTTGAAGAGGCAGTTACTCAACAC-3′ and 5′-TCCTTACCTGGCACTCTCTTTGC-3′). PCR products will be sequenced to confirm targeting (DNA Sequencing Primer: 5′-GGTCAATAAGCCCATCATTGAGG-3′). Vaginal smear and reproductive capacity were assessed using 8 weeks old female ESR1D262A and corresponding wild-type littermates. The uterine morphology, uterine weight, ovarian function, and tissue collection were accessed after sacrificed at age of 12 weeks.
In another experiment, Female ESR1D262A and corresponding wild-type littermates at four weeks of age were anesthetized using isoflurane (4% for induction, 2% for maintenance), then underwent Sham and ovariectomized (OVX), where two sides of ovaries were surgically excised with a single surgical incision as previously described [Citation28]. After two weeks of the surgery, mice were subcutaneously implanted pellets releasing either Placebo or E2 (0.18 mg 17β-estradiol, 21-days release, Innovative Research of America, Sarasota, Florida). The mice were sacrificed at the age of 12 weeks, the uterine tissue sections were used for immunohistochemistry.
Estrous cycle detection
The vaginal cytology method [Citation29] was used to determine the estrous cycle in 2-month-old adult female mice for 10 consecutive days. Briefly, 100 μL of sterile saline was gently flushed into the vagina using soft plastic pipettes, and the vaginal smear was taken every day morning (9 am). The cells were stained with crystal violet (Beyotime, Shanghai, China) for 1 min. According to the features of nucleated epithelial cells, cornified squamous epithelial cells, and leukocytes, proestrus, estrus, metestrus, and diestrus was identify.
Immunohistochemistry
Female ESR1D262A and corresponding wild-type littermates were euthanized by CO2 inhalation at 3 months of age, the uterus and ovarian were dissected, weighed, then formalin-fixed and paraffin-embedded. Uterus and ovarian tissue paraffin blocks were then sectioned at 4 µm thickness (RM2016, Leica biosystems) and placed on a glass slide. Tissue sections were deparaffined using xylene and rehydrated in serial ethanol dilution from 100% to 70%. Ovarian tissue then stained by hematoxylin and eosin. Rehydrated uterus tissue sections were incubated with 0.3% H2O2 to quench the endogenous peroxidase and then blocked with 3% BSA (#G5001, Servicebio). Primary antibodies of Ki-67 (1:200, #12202S, Cell Signaling Technology) were used to treat the tissue section at 4 °C overnight, followed by the corresponding HRP-conjugated secondary antibodies. After washing, the protein signal was visualized using DAB (#GK500710, Gene Tech). The percentage of Ki-67 was evaluated by Ki-67 positive epithelial cell/total epithelial cell from 3 microscopic fields at bright field microscope (Leica CS2, Leica Biosystems). Meanwhile, image J software was used to quantify the average optical density (AOD).
Tissue protein extractions and western blotting
Dissected uteri and ovarian were homogenized by a tissue homogenizer (KZ-III-FP, Servicebio) in cold RIPA buffer (#P0013C, Beyotime) and phosphatase inhibitor (#P0044, Sigma). Total nuclear and cytoplasmic fractions were isolated using the NE-PER nuclear and cytoplasmic extraction reagents (#78833, Thermo Scientific). The supernatant was collected, and BCA assay (#23235, Thermo Scientific) was used to determine protein concentrations. 25 μg proteins were separated on a 10% SDS/PAGE gel for separation by electrophoresis. Separated protein was then transferred to the PVDF membrane, and protein was detected and visualized using enhanced chemiluminescence reagent (#WBKLS0500, Millipore) and Imaging Systems (Amersham Imager 600, GE). All antibodies used in this study were as follows: anti-ERα (#AB24155) was obtained from Abcam, anti-ERβ (#sc-390243) was obtained from Santa Cruz, anti-eNOs (#9572), anti-p-eNOS (#9570), anti-AKT (#4691), anti-p-AKT (#4060), anti-ERK (#4695), anti-p-ERK (#4370), anti-histone H3 (#4499) were obtained from Cell Signaling Technology. anti-GAPDH (#60004-1-Ig) were obtained from Proteintech.
RNA isolation and RT-qPCR
The aorta was homogenized by a tissue homogenizer (KZ-III-FP, Servicebio), and total RNA was prepared using Fast Tissue RNA Purification Kit (#EZB-RN5, EZBioscience) according to the manufacturer’s instruction. 0.5 μg RNA was reverse using Evo M-MLV reverse transcriptase (#AG11706, Accurate Biology) following the user’s manual. RT-PCR was performed on thermal cycler (CFX96, Bio-Rad) using the SYBR Green probe (#AG11701, Accurate Biology). Results were normalized by relative to actin using the 2-△△Ct method as described in previous publication [Citation21]. The detailed information of PCR primers was list in .
Table 1. detailed information PCR primers.
Hormone assays
At 12 weeks of age, blood was collected via cardiac puncture from euthanized female ESR1D262A and wild-type littermates. Heparin treatment of the blood prevented coagulation. Serum was separated after centrifuging for 5 min. Serum levels of E2 (estrogen), FSH (follicle stimulating hormone), and LH (luteinizing hormone) were determined using the automatic biochemistry analyzer (Alinity, #07P5074, #07P4974, #07P9174). Serum levels of T (testosterone) and PG (progesterone) determined using the Elisa kit (Blue Gene Biotech, #E03A0019, #E03P0200) according to the manufacturer’s directions.
Artery preparation and wall tension measurement
As previously described [Citation21], 8 mm-long thoracic aorta segments were dissected and cut into four segments (2 mm per segment) in an ice-cold physiological salt solution. After pretreated with L-NAME (200 μM) and indomethacin (10 μM), the artery rings were mounted on a needle holder. The pressure was controlled by a DMT620 multi-channel isolated vascular tone measurement system (World Precision Instruments, Sarasota, FL, USA), and isometric contractions were recorded by a force transducer connected to an analog-to-digital converter system. Thirty minutes after equilibrium in the organ bath, KPSS solution (Kcl, 60 nM) was used to assess artery viability, and the endothelial integrity was determined by adding acetylcholine (Ach, 10 μM) after phenylephrine (Phe, 1 μM) was treated to plateau. E2 (10 μM) dependent dilation was assessed after the rings were contracted for a second time by adding Phe for 30 min.
Measurement of H2S level
H2S concentration in serum was assessed by Amperometric H2S sensors (ISO-H2S-100, World Precision Instruments, Sarasota, FL, USA). Before each experiment, the amperometric H2S sensor was calibrated with Na2S.9H2O stock solution, and a calibration curve was created. The current output was measured while the tip of the H2S sensor was immersed in the medium for 10–15 millimeters. The concentration of dissolved H2S in the solution was calculated based on the calibration curve.
Statistical analyses
Data are presented as means ± SEM. All experiments were repeated at least 3 times. Unpaired Student’s t-tests and Mann-Whitney tests were used in comparing two groups. Multiple comparisons used one-way ANOVA or Mann-Whitney tests where the data was not normally distributed with SPSS software. The difference was considered significant if p < 0.05. *p < 0.05, **p < 0.01, ***p < 0.001, ns. not significant ().
Table 2. Body weight increase of ESR1D262A mice.
Discussion
In the present study, we used the ESR1D262A mice to investigate the effect of aspartic acid 262 into alanine on the steroid receptor physiological function in vivo. Our findings revealed that mutated for the aspartic acid 262 into alanine on ERα have no effect on female fertilities. However, the mutant 262 site of ERα reveals an important role for the estrogen receptor in uterine and ovarian development and vasodilation in non-classical target organs. With the first constructed ESR1D262A mice, our study revealed that the ERα D262 site is involved in both the development of the reproductive system and the rapid vasodilation process of estrogen.
The aspartic acid at position 258 (corresponding to mouse amino acid site 262) of ERα has a high homology among several species. However, when the aspartic acid is mutated to alanine, the tertiary structure of the protein does not change. Meanwhile, both studies by Qian Wu and our group confirmed at the cellular level that site 258 of ERα is a binding site for GPCRs, and that the signaling pathway of estrogen non-genomic effect transduction is blocked after replacing the aspartic acid at this site to alanine [Citation18, Citation21]. Previous studies revealed that the non-genomic effects of estrogen could avoid the negative effects caused by genomic effects [Citation23, Citation30]. Thus, mouse models of non-genomic effects have gradually become a hot research topic. Taken together, we constructed C57BL/6N mice with ERα point mutation ERαD262A mice.
The impact of mice’s reproductive capacity on the non-genomic effects of ERα remains controversial. Mouse models of the non-genomic effects of ERαAF-1° [Citation3] and ERαAF-2° [Citation31] are infertile, DPM (disrupting peptide mice) mouse model (32) and the ESR1R264A mouse model (20) reported remained fertile. By observing the reproductive phenotype of ESR1D262A mice, we found ESR1D262A mice are totally fertile even with a changed ovarian phenotype and uterine development. LH serum levels, which are typically affected in all previous ERα mutants, are normal in ESR1D262A mice. Moreover, circulating ovarian hormones levels such as E2, FSH, T and PG are completely normal as compared to WT mice. Blocked nonnuclear function of ERα by point mutants in 258 sites in vivo shows a decrease in the mRNA level of eNOs and impaired estrogen response in uterine proliferation. All these results reveal the complexity of ERα-D258A subsignaling in the control of female reproductive phenotype.
Qian Wu et al. found that ER-D258A mutations inhibited estrogenic non-genomic signaling without affecting the genomic effects at cellular level [Citation18]. Our previous study showed that in cultured human umbilical vein endothelial cells (HUVECs), the ERα-D258A mutation blocked estrogen induction of cystathionine-gamma-lyase (CSE) phosphorylation and H2S release through non-genomic effects. We next explored the membrane-initiated vascular actions of ERα in ESR1D262A mice. Our study showed that locus 262 is critical in maintaining rapid vascular diastole by estrogen in vascular tissues, and serum H2S levels was also decreased in ESR1D262A mice. Further study is needed to investigate the upstream signaling that precedes the decreased level of serum H2S.
The activation of intracellular protein kinases by the non-genomic effects of estrogen extensively phosphorylated a range of downstream molecules. eNOs, as a downstream signaling molecule of non-genomic effects, its protein level responds to the estrogen genomic effects in ESR1R264A mice vascular and uterus tissues. Our study examined the phosphorylation levels of eNOs/AKT/ERK in vascular tissues, suggesting that ERα D262 is critical in the non-genomic effects of estrogen.
In contrast to previous report that ERα D262A mutation had no effect in the genomic effect of estrogen in vitro [Citation21], the results of plasma membrane separation experiments on mouse ovarian tissues showed that mutations at ERα D262 inhibited the entry of ERα into the nucleus, resulting in the suppression of NOS3 mRNA in uterus tissues, decrease level of SP1 and AP1 mRNA in vascular tissue, suggesting that this is not a perfect model for genetic segregation of nuclear and membrane effects of ERα in the vasculature.
In conclusion, our study denied ESR1D262A mice as a tool to study non-genomic effects alone. Mutations at locus 258 inhibited genomic effects in the ovary, uterus, and vasculature by decreasing the distribution of ERα in the nucleus, thus inhibited estrogen target organs, disrupting the ovary and uterine development and follicular development cycles. In vascular tissues, the rapid vasodilatory effect of estrogen was blocked. The present study provides a theoretical basis for further selecting an effective non-genomic mouse model and provides a new direction for developing estrogen non-genomic effect inhibitors.
Ethics approval
Animal experiments was approved by experiments Animal Ethics Committees of Guangzhou Medical University (Protocol number: 2017-093).
Consent for publication
All authors read and approved the final manuscript for publication.
Author contributions
H.P. L. conceived and supervised the study. X.Y. X. wrote the original draft of the manuscript. H.P. L. and S.T. revised the manuscript. X.Y. X., H.W. Y., S.H. Z., P. L., X.S. L., Y.Q. G., Y.X. X., and G.J. Z. performed the experiments and analyzed the data.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87(3):1–8. doi: 10.1152/physrev.00026.2006.
- Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–170. doi: 10.1016/bs.apcsb.2019.01.001.
- Billon-Gales A, Fontaine C, Filipe C, et al. The transactivating function 1 of estrogen receptor alpha is dispensable for the vasculoprotective actions of 17beta-estradiol. Proc Natl Acad Sci U S A. 2009;106(6):2053–2058. doi: 10.1073/pnas.0808742106.
- Billon-Gales A, Krust A, Fontaine C, et al. Activation function 2 (AF2) of estrogen receptor-alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A. 2011;108(32):13311–13316. doi: 10.1073/pnas.1105632108.
- Abot A, Fontaine C, Raymond-Letron I, et al. The AF-1 activation function of estrogen receptor alpha is necessary and sufficient for uterine epithelial cell proliferation in vivo. Endocrinology. 2013;154(6):2222–2233. doi: 10.1210/en.2012-2059.
- Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111(2):E283–90. doi: 10.1073/pnas.1322057111.
- Pietras RJ, Szego CM. Specific binding sites for oestrogen at the outer surfaces of isolated endometrial cells. Nature. 1977;265(5589):69–72. doi: 10.1038/265069a0.
- Szego CM, Davis JS. Adenosine 3′,5′-monophosphate in rat uterus: acute elevation by estrogen. Proc Natl Acad Sci U S A. 1967;58(4):1711–1718. doi: 10.1073/pnas.58.4.1711.
- Hammes SR, Levin ER. Minireview: recent advances in extranuclear steroid receptor actions. Endocrinology. 2011;152(12):4489–4495. doi: 10.1210/en.2011-1470.
- Simoncini T. Mechanisms of action of estrogen receptors in vascular cells: relevance for menopause and aging. Climacteric. 2009;12(Suppl 1):6–11. doi: 10.1080/13697130902986385.
- Kim KH, Bender JR. Membrane-initiated actions of estrogen on the endothelium. Mol Cell Endocrinol. 2009;308(1–2):3–8. doi: 10.1016/j.mce.2009.03.025.
- Wu Q, Chambliss K, Umetani M, et al. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286(17):14737–14743. doi: 10.1074/jbc.R110.191791.
- Ueda K, Karas RH. Emerging evidence of the importance of rapid, non-nuclear estrogen receptor signaling in the cardiovascular system. Steroids. 2013;78(6):589–596. doi: 10.1016/j.steroids.2012.12.006.
- Acconcia F, Ascenzi P, Bocedi A, et al. Palmitoylation-dependent estrogen receptor alpha membrane localization: regulation by 17beta-estradiol. Mol Biol Cell. 2005;16(1):231–237. doi: 10.1091/mbc.e04-07-0547.
- Acconcia F, Ascenzi P, Fabozzi G, et al. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. 2004;316(3):878–883. doi: 10.1016/j.bbrc.2004.02.129.
- Pedram A, Razandi M, Sainson RC, et al. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007;282(31):22278–22288. doi: 10.1074/jbc.M611877200.
- Krust A, Green S, Argos P, et al. The chicken oestrogen receptor sequence: homology with v-erbA and the human oestrogen and glucocorticoid receptors. Embo J. 1986;5(5):891–897. doi: 10.1002/j.1460-2075.1986.tb04300.x.
- Wu Q, Chambliss K, Lee WR, et al. Point mutations in the ERalpha galphai binding domain segregate nonnuclear from nuclear receptor function. Mol Endocrinol. 2013;27(1):2–11. doi: 10.1210/me.2011-1378.
- Hinshelwood MM, Corbin CJ, Tsang PC, et al. Isolation and characterization of a complementary deoxyribonucleic acid insert encoding bovine aromatase cytochrome P450. Endocrinology. 1993;133(5):1971–1977. doi: 10.1210/endo.133.5.8404644.
- Adlanmerini M, Febrissy C, Zahreddine R, et al. Mutation of arginine 264 on ERalpha (estrogen receptor alpha) selectively abrogates the rapid signaling of estradiol in the endothelium without altering fertility. Arterioscler Thromb Vasc Biol. 2020;40(9):2143–2158. doi: 10.1161/ATVBAHA.120.314159.
- Xu X, Yan Q, Liu X, et al. 17beta-Estradiol nongenomically induces vascular endothelial H(2)S release by promoting phosphorylation of cystathionine gamma-lyase. J Biol Chem. 2019;294(43):15577–15592. doi: 10.1074/jbc.RA119.008597.
- O’Brien JE, Peterson TJ, Tong MH, et al. Estrogen-induced proliferation of uterine epithelial cells is independent of estrogen receptor alpha binding to classical estrogen response elements. J Biol Chem. 2006;281(36):26683–26692. doi: 10.1074/jbc.M601522200.
- Chambliss KL, Wu Q, Oltmann S, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120(7):2319–2330. doi: 10.1172/JCI38291.
- Madak-Erdogan Z, Kim SH, Gong P, et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal. 2016;9(429):ra53. doi: 10.1126/scisignal.aad8170.
- Guo X, Razandi M, Pedram A, et al. Estrogen induces vascular wall dilation: mediation through kinase signaling to nitric oxide and estrogen receptors alpha and beta. J Biol Chem. 2005;280(20):19704–19710. doi: 10.1074/jbc.M501244200.
- Mani S, Li H, Untereiner A, et al. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;127(25):2523–2534. doi: 10.1161/CIRCULATIONAHA.113.002208.
- Wang Y, Zhao X, Jin H, et al. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009;29(2):173–179. doi: 10.1161/ATVBAHA.108.179333.
- Li X, Chen W, Li P, et al. Follicular stimulating hormone accelerates atherogenesis by increasing endothelial VCAM-1 expression. Theranostics. 2017;7(19):4671–4688. doi: 10.7150/thno.21216.
- McLean AC, Valenzuela N, Fai S, et al. Performing vaginal lavage, crystal violet staining, and vaginal cytological evaluation for mouse estrous cycle staging identification. J Vis Exp. 2012;15(67):e4389. doi: 10.3791/4389.
- Bernelot Moens SJ, Schnitzler GR, Nickerson M, et al. Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation. 2012;126(16):1993–2004. doi: 10.1161/circulationaha.112.124529.
- Arao Y, Hamilton KJ, Ray MK, et al. Estrogen receptor alpha AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc Natl Acad Sci U S A. 2011;108(36):14986–14991. doi: 10.1073/pnas.1109180108.