
Abstract
Thrombin is a potent platelet activator, acting through proteinase-activated receptors −1 and −4 (PAR1 and PAR4). Of these, PAR-1 is activated more rapidly and by lower thrombin concentrations. Consequently, PAR-1 has been extensively investigated as a target for anti-platelet drugs to prevent myocardial infarction. Q94 has been reported to act as an allosteric modulator of PAR1, potently and selectively inhibiting PAR1-Gαq coupling in multiple cell lines, but its effects on human platelet activation have not been previously studied. Platelet Ca2+ signaling, integrin αIIbβ3 activation and α-granule secretion were monitored following stimulation by a PAR1-activating peptide (PAR1-AP). Although Q94 inhibited these responses, its potency was low compared to other PAR1 antagonists. In addition, αIIbβ3 activation and α-granule secretion in response to other platelet activators were also inhibited with similar potency. Finally, in endothelial cells, Q94 did not inhibit PAR1-dependent Ca2+ signaling. Our data suggest that Q94 may have PAR1-independent off-target effects in platelets, precluding its use as a selective PAR1 allosteric modulator.
Introduction
Platelets are the key cell in arterial thrombosis, the underlying trigger of most heart attacks and ischemic strokes, and responsible for many deaths worldwide [Citation1]. Thrombin is a potent platelet activator, acting through proteinase-activated receptor-1 and −4 (PAR1 and PAR4) [Citation2,Citation3]. Of these, PAR-1 is activated more rapidly and by lower thrombin concentrations. PAR1 cleavage by thrombin reveals a new N-terminal tethered ligand that triggers signaling through Gαq, Gα12/13 and, in some cells, Gαi [Citation4]. However, the PAR1 N-terminus can be cleaved by other proteases to reveal different tethered ligands with biased signaling compared to the ligand revealed by thrombin [Citation5]. Activated protein C (APC) is anti-thrombotic, anti-inflammatory and pro-fibrinolytic [Citation6]. In endothelial cells, APC cleaves PAR1 to promote barrier integrity and reduces endothelial cell death in inflammation [Citation7], in a β-arrestin-dependent but Gαq-independent manner [Citation8]. These opposing roles of PAR1 have important consequences for inhibition of PAR1.
Vorapaxar (SCH530348) is the only PAR1 antagonist yet approved for prevention of myocardial infarction [Citation9,Citation10]. Vorapaxar is a competitive, reversible non-peptide antagonist, with long receptor off-rate [Citation11]. However, vorapaxar increases intracranial hemorrhage risk in some patients, which may be due to inhibition of the cytoprotective effects of APC in the endothelium [Citation10,Citation12]. An alternative strategy is to inhibit pro-thrombotic Gαq signaling downstream of PAR1 but leave Gα-independent cytoprotective signaling intact [Citation4,Citation10]. Parmodulins (ML161, JF5) are allosteric modulators of PAR1, acting at the intracellular face of PAR1 to inhibit PAR1’s Gαq-dependent Ca2+ signaling in platelets without inhibiting APC-mediated cytoprotection [Citation12–14]. Alternatively, parmodulins may directly stimulate cytoprotective signaling from PAR1, acting as allosteric agonists [Citation15]. The intracellular face of PAR1 therefore represents an intriguing target for drug action.
Q94 has also been reported to inhibit PAR1-Gαq coupling. It is presumed to act at the intracellular face of PAR1 as it was identified in competitive screen between a Gαq peptide mimic and the PAR1 C-terminus [Citation16]. Q94 inhibited thrombin-induced responses in mouse lung fibroblasts, A549 cells and human microvascular endothelial cells (HMECs) [Citation16–18]. Q94 also reduced doxorubicin-induced albuminuria in a mouse model of nephropathy [Citation19], and activated factor X (FXa)-induced myofibroblast differentiation in lung injury [Citation20]. We had initially planned to characterize the interaction between Q94 and PAR1 in platelets with the aim of exploring related molecular scaffolds as potential allosteric modulators. However, we were unable to repeat in platelets the inhibitory effect reported in other cells with Q94 from two sources. Our data also suggest that Q94 has PAR1-independent off-target effects in platelets, precluding its use as a selective PAR1 allosteric modulator.
Materials and Methods
Materials
Chemicals were obtained from Sigma Aldrich (UK) unless otherwise stated. Q94 was obtained from Tocris (Bristol, UK) and ChemDiv (San Diego, USA). Vorapaxar and ML161 were from Generon (Slough, UK). PAR1-AP (SFLLRN-amide) and PAR4-AP (AYPGKF-amide) were from Bachem (Weil am Rhein, Germany). U46619 was from ApexBio (Stratech, Cambridge, UK). Cal520-AM was from AAT Bioquest (Stratech, Cambridge, UK). Fluorescein isothiocyanate (FITC)-conjugated PAC-1 and phycoerythrin (PE)-conjugated anti-CD62P antibodies were from BD Biosciences (Wokingham, UK).
Isolation of Platelets from Human Blood
The use of blood from healthy drug-free volunteers was approved by the Human Biology Research Ethics Committee, University of Cambridge. Volunteers gave written informed consent in accordance with the Declaration of Helsinki. Blood was collected by venepuncture into vacutainers containing sodium citrate (3.2% v/v). Washed platelets were prepared as described previously [Citation21] in HEPES-buffered saline (HBS; pH 7.4, 135 mM NaCl, 10 mM HEPES, 3 mM KCl, 1 mM MgCl2, and 0.34 mM NaH2PO4 supplemented with 5 mM D-glucose) with apyrase (0.02 U/ml). Washed platelets were rested (30°C; 30 minutes) before experiments took place. CaCl2 (2 mM) was added immediately prior to stimulation.
Platelet Ca2+ Signaling
Platelets were loaded with Cal520 as previously described[Citation22]. Cal520-loaded washed platelets were incubated with antagonists or vehicle control for 30 minutes prior to stimulation, then stimulated in 96-well plates (black-walled, clear-bottomed; 180 μl/well; 5 × 107 platelets/mL; 37°C) with PAR1-AP dispensed by plate reader on-board injectors (FluoStar Omega, BMD Labtech). Fluorescence (excitation, 485 nm; emission, 520 nm) was recorded every 5 seconds for 300 seconds, with 20 seconds recorded prior to stimulation.
Flow Cytometry
Platelets (1 x 108/ml) were incubated with antagonists (30 minutes) prior to stimulation with agonists (5 minutes; 37°C), stained with antibodies (2 minutes; room temperature), then fixed with 1% paraformaldehyde. Samples were analyzed by flow cytometry (BD Accuri C6). Compensation was performed using OneComp eBeads (ThermoFisher). Antibodies used were FITC-conjugated PAC-1 antibody, which binds activated integrin αIIbβ3, and PE-conjugated anti-human CD62P antibody, as a marker of α-granule secretion, at dilutions 1:20 and 1:25 respectively.
Human Umbilical Vein Endothelial Cell (HUVEC) Ca2+ Signaling
HUVEC were cultured as previously described [Citation23]. For experiments, HUVEC was seeded in 96-well plates (2 x 104 cells/well) and grown overnight (5% CO2 at 37°C) to confluency. Media was removed and cells were loaded with Cal520 (2 μM in Hanks’ Balanced Salt Solution [HBSS]; 30 minutes) then washed twice with HBSS. HUVECs were incubated with antagonists (30 minutes; room temperature) prior to stimulation. PAR1-AP or control was dispensed by on-board injectors of a fluorescence plate reader (FluoStar Omega; BMG Labtech). Fluorescence was recorded every 3 seconds for 180 seconds, with 15 seconds recorded prior to stimulation.
Data Analysis
Cal520 fluorescence was normalized to the mean fluorescence prior to agonist addition to give F/F0. Where shown, average data are mean ± standard error of mean (s.e.m.). The number of independent biological replicates is indicated in the figure legends. Inhibition curves were fitted with a variable slope model with the equation Y = 100/(1 + 10^((LogIC50-X)*HillSlope))), where Y is the response as % of control and X is the Log[inhibitor], using Prism (Graphpad, v9). Where this model did not give a good fit, data are presented without curve fitting. For these data, 1-way ANOVA followed by Dunnett’s multiple comparison test was used (* p < .05; ** p < .01, compared to control).
Results
Q94 Inhibits PAR1-dependent Platelet Activation with Low Potency
To investigate the effect of different PAR1 antagonists on platelet Ca2+ signaling, Cal520-loaded platelets were treated with different concentrations of antagonist, or DMSO (control), and stimulated with PAR1-AP (10 μM). Several orthosteric antagonists of PAR1 (vorapaxar, SCH-79797, and FR171113) inhibited the increase in Cal520 fluorescence in a concentration-dependent manner, with an IC50 of 0.02 μM (vorapaxar), 0.18 μM (SCH-79797), and 0.36 μM (FR171113), respectively (). Similarly, ML161 and JF5, allosteric modulators of PAR1, also inhibited the increase in fluorescence in a concentration-dependent manner. The IC50 of ML161 was 1.10 μM, whereas JF5 was less potent, with an IC50 of 12.8 μM (). Although Q94 (Tocris) also inhibited fluorescence increase, its potency was low compared to most other drugs tested (IC50 16.4 μM; Q94 is shown in both to aid comparison.) Representative traces are shown in . To confirm the relatively low potency of Q94, we also purchased Q94 from ChemDiv, the source of Q94 in Deng et al [Citation16]. ChemDiv Q94 showed lower potency to Q94 from Tocris (IC50 of 35.9 μM; ).
Figure 1. Effects of Q94 and other PAR1 antagonists on PAR1-dependent Ca2+ signaling. Cytosolic Ca2+ signals were monitored in Cal520-loaded human platelets in response to PAR1-AP (10 μM). Platelets were pre-treated with various concentrations of the indicated drugs or DMSO as vehicle control. Cal520 fluorescence was quantified as area under curve (AUC) as described in the Methods and is expressed as the % of DMSO-treated platelets (control). (a) shows orthosteric antagonists, whereas (b) shows intracellular allosteric modulators. Q94 (Tocris) is repeated in (a and b) to aid comparison with other drugs. Representative fluorescence signals in the presence of various concentrations of Q94 (Tocris) are shown in (c). The black arrowhead indicates the addition of PAR1-AP. In (d), platelets were pre-treated with various concentrations of Q94 (ChemDiv). In (e-f), platelets were pre-treated with the indicated drugs (or DMSO as vehicle control), stimulated with PAR1-AP, then stained with PAC1-FITC and anti-CD62P-PE. The median fluorescence intensity of bound antibody is expressed as a % of DMSO-treated platelets (control). Data are presented as mean ± standard error of mean (n = 5).
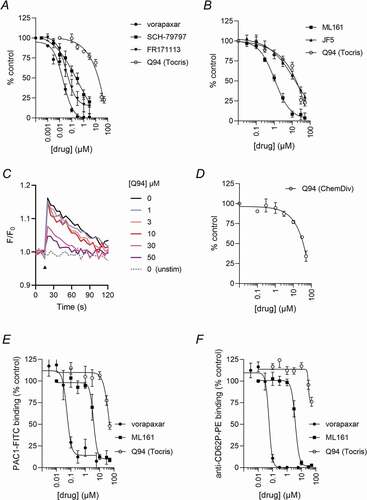
To further investigate the effect of Q94 (Tocris), PAR1-AP-induced αIIbβ3 activation and α-granule secretion were measured. Q94 displayed some inhibitory action but again with low potency (). Vorapaxar and ML161 inhibited both platelet functions, demonstrating that this assay is sensitive to orthosteric antagonists and intracellular allosteric modulators.
Q94 Inhibits PAR1-dependent Ca2+ Signaling in HUVECs with Low Potency
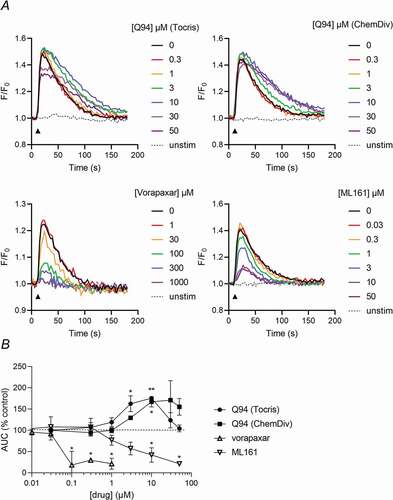
Q94 has been previously described in human microvascular endothelial cells (HMECs) as inhibiting PAR1-dependent Ca2+ signaling. However, we could not see a similar inhibitory effect of Q94 (Tocris or ChemDiv) in human umbilical vein endothelial cells (HUVECs). Q94 (Tocris) partially inhibited the peak increase in F/F0 at high concentrations, but both Q94 (Tocris) and Q94 (ChemDiv) increased AUC at concentrations greater than 1 μM (). In contrast, PAR1-dependent Ca2+ signaling was inhibited by vorapaxar or ML161, demonstrating that this assay is sensitive to orthosteric antagonists and intracellular allosteric modulators.
Figure 2. Q94 does not potently inhibit PAR1-dependent Ca2+ signaling in HUVEC. Cytosolic Ca2+ signals were monitored in Cal520-loaded HUVEC following pre-treatment with the indicated drugs (or DMSO as vehicle control, where drug concentration is indicated as 0), then stimulation with PAR1-AP. Representative traces are shown in (a) for each drug. The point of PAR1-AP addition is indicated by the black arrowheads. In (b), AUC expressed as % DMSO-treated control. Data are presented as mean ± standard error of mean (n = 3).
Q94 Inhibits Platelet Activation Downstream of Several Platelet Receptors
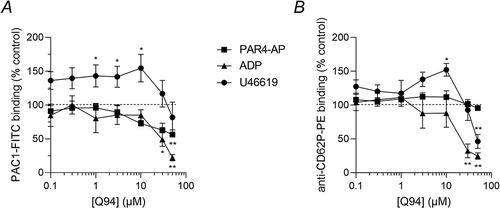
To investigate the receptor selectivity of Q94, platelets were stimulated with PAR4-AP, the thromboxane analogue, U46619, or the P2Y1/P2Y12 agonist, ADP. Each agonist trigged αIIbβ3 activation and α-granule secretion. Q94 inhibited these functional responses to each agonist (), indicating that its effect is not selective to PAR1. PAR4-AP-stimulated platelets were least affected, with inhibition of αIIbβ3 activation but no significant inhibition of α-granule secretion. The responses to ADP were more strongly inhibited by Q94 greater than 10 μM. The effect on U46619-induced platelet activation was more complex, with Q94 enhancing αIIbβ3 activation and α-granule secretion at 10 μM or below, an effect that reversed above 10 μM. Despite this more complex pattern, it is clear that Q94 is not selective for PAR1-dependent platelet activation.
Figure 3. Q94 inhibits platelet activation downstream of several platelet receptors. Platelets were pre-treated with various concentrations of Q94 (or DMSO as vehicle control), stimulated for 10 minutes with PAR4-AP (1 mM), ADP (100 μM) or U46619 (10 μM), then stained with PAC1-FITC (a) and anti-CD62P-PE (b). The median fluorescence intensity of bound antibody is expressed as a % of DMSO-treated platelets (control). Data are presented as mean ± standard error of mean (n = 5; * p < .05, ** p < .01 compared to vehicle control for that agonist).
Discussion
Platelet PAR1 has attracted extensive attention as an anti-thrombotic target [Citation3], with vorapaxar approved for the prevention of myocardial infarction in patients without a history of stroke or bleeding [Citation9]. However, vorapaxar blocks cytoprotective signaling downstream of APC in addition to pro-thrombotic signaling downstream of thrombin [Citation12]. In contrast, allosteric modulators may inhibit pro-thrombotic signaling, particularly that mediated by Gαq, without blocking the cytoprotective signals, providing an alternative therapeutic approach. Unfortunately, although Q94 is reported to be a potent, selective allosteric inhibitor of PAR1-Gαq coupling, we were unable to observe potent inhibition of PAR1-dependent Ca2+ signaling in platelets and found off-target inhibition of other platelet activators at similar concentrations.
PAR1-dependent Ca2+ signaling was inhibited by a range of orthosteric antagonists and allosteric modulators. Q94 showed concentration-dependent inhibition of PAR1 signaling in this study, with an IC50 of 16 μM (Q94 Tocris). We were surprised by the high IC50 of Q94. In a previous study, Q94 potently inhibited thrombin-induced Ca2+ signaling in HMECs (IC50 0.01 μM) and was approximately 5-times more potent that SCH-79797 [Citation18]. In contrast, in our study Q94 (Tocris) was 91-times less potent than SCH-79797. Although the IC50 itself will depend on experimental conditions, the difference in relative potency compared to SCH-79797 suggested that Q94 (Tocris) was not acting in a potent manner in platelets. We therefore obtained Q94 from ChemDiv, the original source in Deng et al [Citation16]. Q94 (ChemDiv) inhibition of PAR1 signaling also had a high IC50 (36 μM). We observed similar low potency at inhibiting key platelet function responses, integrin αIIbβ3 activation and α-granule secretion. These data demonstrate that Q94 is not a potent modulator of PAR1-dependent platelet activation. Similarly, in a Doctoral thesis, Q94 (synthesized in house or purchased from ChemDiv) was reported to be ineffective on thrombin-induced Ca2+ release in lung fibroblasts and of poor solubility [Citation24]. It is possible that Q94 is unstable and rapidly degrades, in which case it is an unreliable modulator of PAR1. Whether lacking in potency, solubility or stability, Q94 is not a useful tool to probe PAR1-Gαq signaling in platelets.
Since Q94 has been characterized in endothelial cells [Citation18], we examined whether it has an inhibitory effect in HUVEC. Whereas vorapaxar and ML161 both inhibited PAR1-AP-induced Ca2+ signaling, Q94 had no inhibitory effect. Surprisingly, a weak enhancement of Ca2+ signaling was seen at some concentrations of Q94 (both Tocris and ChemDiv). Although we do not know the mechanism that underlies this enhancement, it has also been reported in EA.hy926 cells [Citation14]. As this enhancement was not seen with either vorapaxar or ML161, it appears to be independent of PAR1.
Most problematically, Q94 also inhibited platelet αIIbβ3 activation and α-granule secretion independently of PAR1. This suggests that the inhibition of PAR1-dependent integrin αIIbβ3 activation and α-granule secretion may be unrelated to direct inhibition of PAR1 but instead be an effect on downstream intracellular signaling. Q94 most potently inhibited ADP-induced platelet activation, with relatively less effect on PAR4-AP-induced platelet activation. This may be because activation of PAR4 results in stronger activation of platelets than does activation of P2Y1 and P2Y12. Q94 also had a complex effect on U46619-induced platelet activation, with enhanced platelet activation at some concentrations of Q94. The underlying mechanism of this enhancement is not clear, but it further supports the conclusion that the effects of Q94 in platelets are not specific for PAR1 signaling.
In conclusion, Q94 is not a potent, selective modulator of platelet PAR1. Rather, it appears to inhibit platelet activation downstream of several receptors with similar, low potency. Q94 is therefore not a suitable tool for studying PAR1 in platelets and is unlikely to be a useful lead in developing PAR1 allosteric modulators.
Author Contributions
LRAF performed and analysed experiments and edited the manuscript. SLM performed experiments, supervised experiments, and edited the manuscript. TR directed research and edited the manuscript. MTH directed research, designed experiments and wrote the manuscript.
Disclosure Statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Raskob GE. ISTH Steering Committee for World Thrombosis Day. Thrombosis: a major contributor to the global disease burden. J Thromb Haemost 2014;12:1580–1590.
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature 2000;407:258–264. doi:10.1038/35025229.
- Hamilton JR. Protease-activated receptors as targets for antiplatelet therapy. Blood Rev 2009;23:61–65. doi:10.1016/j.blre.2008.06.002.
- Cunningham M, McIntosh K, Bushell T, Sloan G, Plevin R. Proteinase-activated receptors (PARs) as targets for antiplatelet therapy. Biochem Soc Trans 2016;44:606–612. doi:10.1042/BST20150282.
- Willis Fox O, Preston RJS. Molecular basis of protease-activated receptor 1 signaling diversity. J Thromb Haemost 2020;18:6–16. doi:10.1111/jth.14643.
- Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, Fernández JA, Martin G, Kupriyanov S, Mosnier LO, et al. PAR1 biased signaling is required for activated protein C in vivo benefits in sepsis and stroke. Blood 2018;131:1163–1171. doi:10.1182/blood-2017-10-810895.
- Schuepbach RA, Feistritzer C, Fernández JA, Griffin JH, Riewald M. Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thromb Haemost 2009;101:724–733. doi:10.1160/TH08-10-0632.
- Soh UJK, Trejo JA. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A 2011;108:E1372–E1380. doi:10.1073/pnas.1112482108.
- Magnani G, Bonaca MP, Braunwald E, Dalby AJ, Fox KAA, Murphy SA, Nicolau JC, Oude Ophuis T, Scirica BM, Spinar J, et al. Efficacy and safety of vorapaxar as approved for clinical use in the United States. J Am Heart Assoc 2015;4:e001505. doi:10.1161/JAHA.114.001505.
- Flaumenhaft R, De Ceunynck K. Targeting PAR1: now what? Trends Pharmacol Sci 2017;38:701–716. doi:10.1016/j.tips.2017.05.001.
- Bokoch MP, Jo H, Valcourt JR, Srinivasan Y, Pan AC, Capponi S, Grabe M, Dror RO, Shaw DE, DeGrado WF, et al. Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule. Biochemistry 2018;57:5748–5758. doi:10.1021/acs.biochem.8b00577.
- Aisiku O, Peters CG, De Ceunynck K, Ghosh CC, Dilks JR, Fustolo-Gunnink SF, Huang M, Dockendorff C, Parikh SM, Flaumenhaft R, et al. Parmodulins inhibit thrombus formation without inducing endothelial injury caused by vorapaxar. Blood 2015;125:1976–1985. doi:10.1182/blood-2014-09-599910.
- Dowal L, Sim DS, Dilks JR, Blair P, Beaudry S, Denker BM, Koukos G, Kuliopulos A, Flaumenhaft R. Identification of an antithrombotic allosteric modulator that acts through helix 8 of PAR1. Proc Natl Acad Sci U S A 2011;108:2951–2956. doi:10.1073/pnas.1014863108.
- Gandhi DM, Majewski MW, Rosas R, Kentala K, Foster TJ, Greve E, Dockendorff C. Characterization of protease-activated receptor (PAR) ligands: parmodulins are reversible allosteric inhibitors of PAR1-driven calcium mobilization in endothelial cells. Bioorganic Med Chem 2018;26:2514–2529. doi:10.1016/j.bmc.2018.04.016.
- De Ceunynck K, Peters CG, Jain A, Higgins SJ, Aisiku O, Fitch-Tewfik JL, Chaudhry SA, Dockendorff C, Parikh SM, Ingber DE, et al. PAR1 agonists stimulate APC-like endothelial cytoprotection and confer resistance to thromboinflammatory injury. Proc Natl Acad Sci U S A 2018;115:E982–E991. doi:10.1073/pnas.1718600115.
- Deng X, Mercer PF, Scotton CJ, Gilchrist A, Chambers RC. Thrombin induces fibroblast CCL2/JE production and release via coupling of PAR1 to Gαq and cooperation between ERK1/2 and Rho kinase signaling pathways. Mol Biol Cell 2008;19:2520–2533. doi:10.1091/mbc.e07-07-0720.
- Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Königshoff M, Howell DCJ, McAnulty RJ, Das A, Thorley AJ, Tetley TD, et al. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary Fibrosis. Am J Respir Crit Care Med 2009;179:414–425. doi:10.1164/rccm.200712-1827OC.
- Asteriti S, Daniele S, Porchia F, Dell’Anno MT, Fazzini A, Pugliesi I, Trincavelli ML, Taliani S, Martini C, Mazzoni MR, et al. Modulation of PAR1 signalling by benzimidazole compounds. Br J Pharmacol 2012;167:80–94. doi:10.1111/j.1476-5381.2012.01974.x.
- Guan Y, Nakano D, Zhang Y, Li L, Liu W, Nishida M, Kuwabara T, Morishita A, Hitomi H, Mori K, et al. A protease-activated receptor-1 antagonist protects against podocyte injury in a mouse model of nephropathy. J Pharmacol Sci 2017;135:81–88. doi:10.1016/j.jphs.2017.09.002.
- Scotton CJ, Krupiczojc MA, Königshoff M, Mercer PF, Lee YCG, Kaminski N, Morser J, Post JM, Maher TM, Nicholson AG, et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J Clin Invest 2009;119:2550–2563. doi:10.1172/JCI33288.
- Millington-Burgess SL, Harper MT. Epigallocatechin gallate inhibits release of extracellular vesicles from platelets without inhibiting phosphatidylserine exposure. Sci Rep 2021;11. doi:10.1038/s41598-021-97212-8.
- Millington-Burgess SL, Bonna AM, Rahman T, Harper MT. Ethaninidothioic acid (R5421) is not a selective inhibitor of platelet phospholipid scramblase activity. Br J Pharmacol 2020;177:4007–4020. doi:10.1111/bph.15152.
- Davies JE, Lopresto D, Apta BHR, Lin Z, Ma W, Harper MT. Using Yoda-1 to mimic laminar flow in vitro: a tool to simplify drug testing. Biochem Pharmacol 2019;168:473–480. doi:10.1016/j.bcp.2019.08.013.
- Knight E. The Development of Novel Vorapaxar Analogues as Topical Protease Activated Receptor-1 Antagonists for the Treatment of Idiopathic Pulmonary Fibrosis. [Dr. thesis]. London: UCL (University Coll); 2016.