
Abstract
Pulmonary hypertension (PH) is defined by increased mean pulmonary artery pressure, and the clinical classification includes five etiologies, of which we investigated subgroup 1, pulmonary arterial hypertension (PAH) and subgroup 4, chronic thrombotic and/or embolic disease (CTEPH). Platelets participate in both innate and adaptive immune responses and could possibly contribute to the suggested systemic inflammation associated with PAH. In this study, we utilized flow cytometry to analyze platelet activation and platelet-monocyte (PMA) and granulocyte (PGA) aggregates in PAH and CTEPH patients and healthy control subjects. The plasma concentration of proinflammatory cytokines was measured by multiplex electrochemiluminescence. Our main finding is that circulating platelets are activated in the circulation and form aggregates with both monocytes and granulocytes in patients with idiopathic PAH (IPAH), associated PAH (APAH) and pulmonary hypertension due to CTEPH. There was a strong correlation between the platelet activation, assessed as P-selectin, and the number of aggregates formed. IL-6, IL-8, IL-10 and TNF-α were increased in all PH subgroups as compared to healthy controls, and PMAs were associated with circulating IL-6, IL-8 and IL-10, whereas PGAs were associated with IL-6. The increased concentrations of platelet-leukocyte aggregates found in PAH/CTEPH patients might thus contribute to the inflammatory state in PH.
Introduction
Pulmonary hypertension (PH) is a progressive disease associated with poor outcome. PH can be divided into five categories, each with different subgroups with category-specific treatments [Citation1–3]. The most common PH diagnoses in Sweden 2019 were pulmonary arterial hypertension (PAH), PH subgroup 1, further subgrouped into idiopathic PAH (IPAH) and associated PAH (APAH), and pulmonary hypertension due to chronic thrombotic and/or embolic disease (CTEPH), PH subgroup 4 [Citation4]. Both IPAH and APAH have an incidence of 3–4 per million inhabitants in Sweden, whereas the incidence for CTEPH is 2–3 per million. Data from other western countries look similar [Citation5]. Despite the development of advanced therapies, survival prospects remain poor for both conditions with right ventricular failure as the main cause of early death [Citation6].
The key pathologic change observed in PAH is remodeling of precapillary resistance pulmonary arteries, characterized by thickening of the intima, media, and adventitia. As the disease progresses, intimal fibrosis occurs, which, along with in situ thrombosis, leads to the development of plexiform lesions [Citation7]. CTEPH is secondary to the obstruction of the pulmonary arteries by thrombus after a pulmonary embolism. A combination of defective angiogenesis, impaired fibrinolysis and impaired endothelial function leads to the vascular remodeling [Citation8].
A common observation in patients with PAH and CTEPH is the presence of inflammation found in histopathologic specimens and blood samples [Citation9–13], often combined with signs of highly activated platelets [Citation14]. Besides their involvement in hemostasis, platelets also participate in innate and adaptive immune responses [Citation15] and can physically interact with all leukocyte subtypes, with the highest affinity for monocytes/macrophages and neutrophils [Citation16–18]. Platelets can bind to leukocytes by a great number of different surface receptors such as P-selectin (CD62P), that bind to its leukocyte counterpart PSGL-1, and GPIbα binding to CD11b [Citation19,Citation20]. Circulating platelet-monocyte aggregates (PMA) as well as platelet-granulocyte aggregates (PGA) are increased in coronary artery disease [Citation21–23]. The increase in PMAs induces a proinflammatory plasma profile, and the formation of P-selectin dependent PMAs in whole blood leads to production of e.g. tissue factor (TF) and IL-8 [Citation24]. An inverse correlation between IL-10 and PMA formation has also been found [Citation24].
There is evidence for monocyte activation in PAH patients [Citation25–27] and platelet-neutrophil formation has been described in a rat model [Citation28], but data on circulating PMA and PGA levels is lacking in a human PH cohort. It is therefore not known if these aggregates differ across patients and controls and if they can contribute to the systemic inflammation in PAH. In this study, we have therefore analyzed circulating platelet-leukocyte aggregates and proinflammatory cytokines in a cohort of IPAH, APAH, and CTEPH patients and compared the results in this group and to a group of healthy control subjects. Our findings suggest that PMAs play a role in the PAH pathology but do not differ in any apparent way between the subgroups in our cohort.
Materials and methods
Patient cohort
Patients admitted to the outpatient clinic for PAH at Uppsala University Hospital, Uppsala, Sweden, were asked to participate in this cross-sectional study. A total of 70 patients with PAH/CTEPH were included. In the present study, blood samples from 48 randomly selected patients out of the 70 were analyzed. Information of other medical diagnoses and prescribed medications was gathered together with data from right heart catheterization at diagnosis. The results of a 6-min walk test were collected within two months of study inclusion. Healthy individuals, similar numbers of females and males, without any medical treatment were asked to participate as healthy controls (HC) and included during the same time period as the PH patients (n = 20). The study was approved by the local ethics committee (ref. no. 2010/343) and was conducted in accordance with the ethical principles of the Declaration of Helsinki.
Blood collection
Whole blood from PH patients and the HC group was collected in citrate tubes (3.8%) by direct puncture without stasis. The preparation of whole blood started within 30 minutes (min) of collection. Part of the blood sample was used for whole blood flow cytometry analysis. The rest was centrifuged at 2000 × g for 15 min and plasma aliquots were frozen and stored at −80°C.
Whole blood flow cytometry
Flow cytometry was performed on whole blood using a FC500 MPL flow cytometer with 488 nm laser (Beckman Coulter). P-selectin was detected on platelets with anti-CD62P (clone AK-6; AbD Serotec, Oxford, UK) with an irrelevant isotype antibody as control (DakoCytomation, Glostrup Denmark). The blood was incubated with antibodies for 20 minutes in the dark at room temperature and thereafter fixed with cold 0.2% paraformaldehyde/hepes saline. No washing steps were used. The samples were diluted 1:20 in PBS before analysis. Single platelets were identified by their forward and side scatter characteristics, and the percentage of positive cells was determined. Platelet-leukocyte aggregate formation was determined with antibodies against CD45 (DakoCytomation, Glostrup Denmark) and CD62P. The CD62P was switched to an irrelevant isotype antibody for control. The samples were incubated for 20 minutes in the dark at room temperature and fixed in cold 0.5% formaldehyde in 0.9% NaCl and gated as described previously [Citation29]. Briefly, leukocytes were selected based on their CD45 expression which also helped to exclude single platelets. The CD45+ events were then discriminated based on their forward and side scatter characteristics where mainly the side scatter (i.e. the granularity) differed between monocytes and granulocytes. CD14 (DakoCytomation, Glostrup Denmark) then confirmed the gating of the monocytes, whereas CD16 (DakoCytomation) allowed division of the granulocytes into eosinophils and neutrophils. For analysis, at least 5000 leukocytes were counted and the number of monocytes and granulocytes with conjugated platelets was determined by the mean fluorescence intensity (MFI) of CD62P-positive cells in the respective population.
Analysis of plasma proteins
Plasma samples from 48 patients and 20 healthy controls were analyzed by electrochemiluminescence using the V-PLEX Inflammatory Panel 1 Human Kit (Meso Scale Discovery, Gaithersburg, MD) on a MESO QuickPlex SQ 120 instrument at the SciLifeLab Affinity Proteomics Uppsala Unit (Uppsala University, Sweden). Dynamic ranges (LLOD-ULOQ, pg/ml): IFN-γ 0.2–1500; IL-10 0.02–365; IL-12p70 0.04–487; IL-13 0.75–465; IL-1β 0.02–589; IL-2 0.12–1490; IL-4 0.01–263; IL-6 0.10–721; IL-8 0.05–553; TNF-α 0.11–388. The included positive controls were detected as expected for all cytokine assays (data not shown).
Statistics
The baseline characteristics were described as frequencies, mean (SD). The hemodynamic variables, PMAs, PGAs and markers for inflammation were described using median (25th–75th percentile). Differences were calculated by the two-tailed Mann-Whitney test or Kruskal-Wallis, whereas Spearman rank correlation coefficient was used for correlation between platelet-leukocyte aggregates and inflammatory plasma biomarkers. Statistical tests were performed using SPSS version statistics 23 and GraphPad Prism 8. A p-value of ≤ 0.05 was considered to be statistically significant.
Results
Patient characteristics
In total, 48 patients, with a mean age of 67 years, whereof 27 (56.2%) women, were included in the cross-sectional study (). The majority of the patients were diagnosed with IPAH (n = 26), 11 had APAH and 11 had CTEPH. Hypertension and diabetes mellitus were more common in IPAH and CTEPH compared to APAH. The CTEPH group had the lowest levels of NTproBNP and performed best in the 6-min walking test. In total 31 (64.5%), 18 (39.1%) and 8 (16.7%) were treated with PDE inhibitor, endothelin inhibitor, and prostacyclin, respectively, upon inclusion.
Table I. Patient characteristics.
Increased levels of activated platelets in patients with PAH/CTEPH
A total platelet count showed no differences between the HC and the PAH/CTEPH groups regarding the number of circulating platelets (). In contrast, there was a significant discrepancy between control and PAH/CTEPH subjects for the platelet activation marker CD62P, with higher levels in the PH patients (). By analyzing the patient group with or without the patients standing on different treatments, we found no impact of PDE inhibitor, endothelin inhibitors, prostacyclin, or oral anticoagulant treatment on the CD62P levels in our cohort (data not shown).
Figure 1. Circulating platelets are activated in pulmonary arterial hypertension patients. A) The number of platelets and B-C) their activation state were assessed in healthy controls (HC, N = 20) and PH patients (N = 48) by flow cytometry as described in Methods. A two-tailed Mann-Whitney test was used to calculate p-values. MFI = Mean fluorescence intensity. ** = p < .01 *** p < .001.
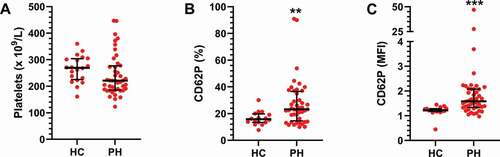
In a search for differences within the PH cohort, the PH patients were divided into subgroups based on their clinical diagnosis (). No differences were found between the three subgroups for the platelet count (). As for the CD62P expression, the increase was most pronounced in the IPAH and APAH patients, but there were no significant inter-group differences (). This indicates that there is an increase of activated platelets within the PH cohort regardless of PH subgroup.
Figure 2. Circulating platelets do not differ across pulmonary hypertension subgroups. The cohort was divided into subgroups based on clinical diagnosis: IPAH (N = 26), APAH (N = 11), CTEPH (N = 11) and healthy controls (HC, N = 20). A) The number of platelets and B-C) their activation state were then assessed by flow cytometry, as described in Methods, and compared across the patient groups using a Kruskal–Wallis test. MFI = Mean fluorescence intensity. ns = not significant.
Activated platelets form aggregates with monocytes and granulocytes in PAH/CTEPH patients
All samples were then analyzed for CD62P positive PMAs and PGAs by flow cytometry. The presence of PMAs with CD62P expression was increased by about 5 times, whereas the PGAs were up approximately 2.5 times in the PAH/CTEPH group as compared to the HC subjects (). There were no differences between the PH subgroups for any of the aggregate types or the expression of CD62P within aggregates ().
Figure 3. Platelet monocyte and platelet granulocyte aggregates are increased in PH patients. The numbers of A) platelet monocyte aggregates (PMA) and B) platelet granulocyte aggregates (PGA) were assessed in healthy controls (HC, N = 20) and PH patients (N = 48) by flow cytometry as described in Methods. A two-tailed Mann–Whitney test was used to calculate p-values. *** p < .001.
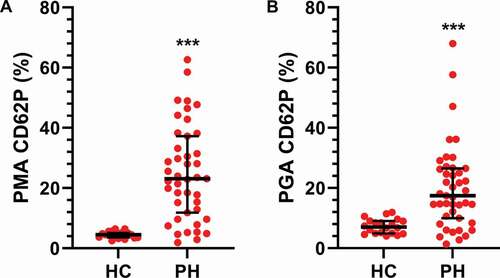
Figure 4. Platelet monocyte- and platelets granulocyte aggregates do not differ across PH subgroups. The cohort was divided into subgroups based on their clinical diagnosis: IPAH (N = 26), APAH (N = 11), CTEPH (N = 11) and healthy controls (HC, N = 20). The numbers of A-B) platelet monocyte aggregates (PMA) and C-D) platelet granulocyte aggregates (PGA) were assessed by flow cytometry, as described in Methods, and compared between the patient groups using a Kruskal–Wallis test. MFI = Mean fluorescence intensity. ns = not significant.
The increase in activated platelets is thereby reflected in increased PMA and PGA formation in the PH patients, but there were no apparent differences across the different diagnoses. By analyzing the patient group with or without the patients standing on different treatments, we found no impact of PDE inhibitor, endothelin inhibitors, prostacyclin, or oral anticoagulant treatment on the numbers of PMAs and PGAs in our cohort (data not shown).
Inflammatory markers are increased in PAH/CTEPH patient plasma
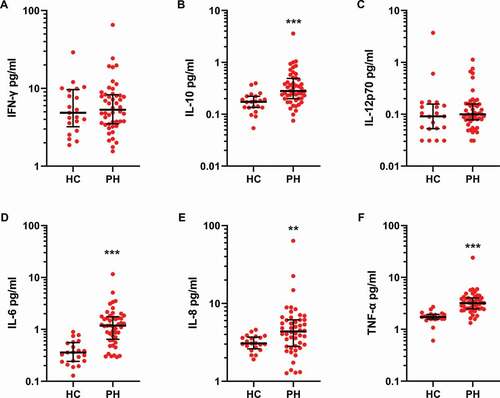
Ten inflammatory proteins were quantified in control and PAH/CTEPH patient plasma. A majority of samples were below the LoD (see Methods) for IL-13, IL-1β, IL-2 and IL-4, and these analytes were therefore not analyzed further. The levels of IFN-γ (median IQR, pg/ml HC 4.8 (3.2–9.7) vs PH 5.3 (3.5–8.3)) and IL-12p70 (HC 0.09 (0.05–0.16) vs PH 0.10 (0.08–0.16)) did not differ between the controls and patients, whereas significant differences were recorded for IL-10 (HC 0.17 (0.13–0.17) vs PH 0.28 (0.20–0.49), p < .001), IL-6 (HC 0.36 (0.24–0.55) vs PH 1.2 (0.64–1.8), p < .001), IL-8 (HC 3.1 (2.6–3.7) vs PH 4.4 (2.8–6.1), p < .01) and TNF-α 1.7 (HC 1.6–1.9) vs PH 3.2 (2.5–4.0), p < .001 ().
Figure 5. Inflammatory proteins are increased in PH patient plasma. Proinflammatory proteins were analyzed by electrochemiluminescence in plasma from healthy controls (HC, N = 20) and PH patients (N = 48) as described in Methods. Group differences were determined using a two-tailed Mann–Whitney test. ** = p < .01 *** p < .001.
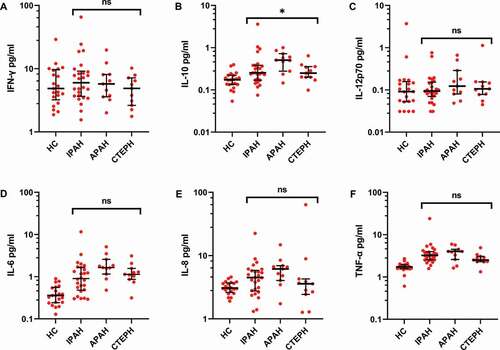
When comparing the PH subgroups there was a difference in IL-10 levels, with the highest values in the APAH group (). IL-6, IL-8 and TNF-α in the APAH group showed a tendency to have higher median levels compared to the IPAH and CTEPH groups.
Figure 6. Subgroup analysis of inflammatory proteins in PH patient plasma shows small differences. The cohort was divided into subgroups based on their clinical diagnosis: IPAH (N = 26), APAH (N = 11), CTEPH (N = 11) and healthy controls (HC, N = 20). Proinflammatory proteins were analyzed by electrochemiluminescence in plasma, as described in Methods, and compared across the patient groups using a Kruskal–Wallis test. * = p < .05.
Correlations of platelets markers and inflammatory cytokines
There was an association of platelet CD62P expression and CD62P in PMAs (r = 0.66, p < .001) and PGAs (r = 0.63, p < .001) (supplemental figure 1). We next analyzed the four cytokines found upregulated in the PH group (shown in ) in relation to the aggregate levels. In the PMA group there was an association to IL-10 (−0.39, p ≤ .01), IL-6 (−0.29, p ≤ .01) and IL-8 (0.29, p ≤ .05) and in the PGA group to IL-6 (−0.44, ≤0.01) (supplemental figure 2).
Discussion
In the present study our main findings were that platelets from PAH/CTEPH patients are activated in the circulation and form aggregates with both monocytes and granulocytes. A number of proinflammatory proteins were upregulated and associated with the number of circulating PMAs expressing P-selectin. These findings confirm that pulmonary arterial hypertension is closely correlated to inflammation also on a systemic level and indicate that PMAs could contribute to this inflammation.
Platelets play an important role in the pathophysiology of pulmonary arterial hypertension, but how and when they contribute is disputed. There is an association of thrombocytopenia with poor PH prognosis [Citation30,Citation31]. The reasons behind the low platelet count are unknown, but progressive right heart failure leading to hypertension in the hepatic portal system, enlarged spleen, and platelet sequestration is one plausible theory [Citation32]. In the present cohort, all patients were well above the limit for grade 1 thrombocytopenia (<150 × 109/L), and we saw no differences between healthy and PH patients or in the PH patient group regarding the total number of platelets.
Platelet activation, described as P-selectin/CD62P expression, includes a number of processes that are critical for hemostasis and inflammation and requires intracellular signal transduction initiated by surface receptors for adhesion proteins and soluble agonists. These ligands are generated or exposed during inflammation and cause damage to the vascular endothelium. Increased platelet activation has been found to contribute to the thrombotic state found in CTEPH patients, and upregulation of soluble P-selectin was described in a cohort of IPAH patients [Citation33]. We have furthermore shown an increase in P-selectin on circulating platelet microvesicles isolated from PAH patients [Citation14]. In contrast, other studies described no differences of platelet activation in PAH compared to healthy subjects [Citation34,Citation35].
The increased number of P-selectin-presenting platelets indicates highly activated platelets in PH patients. By e.g. releasing serotonin and thromboxane A2 there is a link from platelet activation to vasoconstriction of pulmonary vessels, and both proteins indeed have dysregulated signaling in PAH patients [Citation36,Citation37]. In the present study, we found no differences regarding the amount of P-selectin comparing IPAH, APAH and CTEPH patients, neither on single platelets nor in platelet-leukocyte aggregates. The IPAH patients tended to have higher levels of P-selectin, but larger studies are needed to confirm this finding.
Platelets can aggregate with immune cells, and this complex formation is seen as a very sensitive marker of platelet activation [Citation38,Citation39]. PMA formation appears, however, to be disease specific and is e.g. increased in stable angina pectoris [Citation40] and acute myocardial infarction [Citation21,Citation22], whereas an inverse correlation was found between inflammation in ulcerative colitis and PMAs [Citation41]. To the best of our knowledge, this is the first time PMAs and PGAs have been measured in PH patients, and the results clearly show increased levels as compared to healthy controls and a correlation between activated platelets and PMAs/PGAs. Our understanding of this phenomenon in physiological and pathological processes is limited, however, and more studies are needed to uncover common mechanisms.
It is clear that systemic markers of inflammation can be found in the circulation of PH patients. Several studies have shown increased levels of proinflammatory cytokines in IPAH cohorts among others IL-6, IL-8, IL-10, IL-12p70 and TNF-α [Citation42–45]. IL-1α, IL-1β, IL-6, TNF-α and IL-13 were furthermore shown to be associated with mortality in male patients [Citation46]. In the present study, IL-13, IL-1β, IL-2 and IL-4 were below the limit of detection in all samples. IL-12p70 levels were similar between PH patients and controls, but we could confirm the increase in IL-6, IL-8, IL-10 and TNF-α similar to previous reports. The differences might be explained by diverse study cohorts in terms of age, sex, and diagnoses as well as sample handling and the method used. Currently, it is unclear how inflammation may contribute to the pathogenesis of PAH i.e. if it initiates vascular remodeling or its propagation or if it is just a reactive response to ongoing remodeling as indicated in an animal study [Citation47].
Apart from gaining insight into the mechanisms of PAH, understanding the link between inflammation, activated platelets, and PAH may help identify future therapeutic targets. Since these four confirmed markers are part of different proinflammatory systems, a targeted approach to each of these could provide some clues of their relative importance and causality.
The different aggregates were associated with some of the cytokines in the total PH group. All but one cytokine vs aggregate correlation was negative meaning that the patients with the most circulating complexes had the lowest levels of theses circulating cytokines. The binding of activated platelets to e.g. monocytes can increase their adhesiveness to inflamed endothelial cells or exposed subendothelial cells (reviewed in for instance [Citation48]) and the platelets would serve as bridges for the leukocytes for binding to the vessel wall [Citation49–51]. Applying this theory on our data would mean that the PMAs/PGAs in the most activated patients are cleared from the circulation and not detected by the flow cytometer. The leukocytes would still be able to produce cytokines prior binding to the vessel wall or even after the initial attachment and could also trigger and endothelial release. Additional experiments are, however, needed in order to prove this theory in the present settings. It is also interesting to note that IL-6 was associated to both PMAs and PGAs, this especially since blocking of the IL-6/IL6-R/GP130 signaling complex has shown promise as a therapeutic strategy in PAH [Citation52,Citation53].
There are some limitations of the present study. The study cohort was collected at one center and consists of a limited number of patients within each group, which might influence the results of e.g the differences regarding PMAs or PGAs among different subgroups. The majority of the patients were treated with PAH-specific treatment, but only a crude analysis could be done regarding their effects on the number of activated platelets, platelet-leukocyte aggregates or inflammatory cytokines. Again, a larger study material is needed to clarify this. Finally, our focus on cytokine release and platelet leukocyte aggregation made us choose P-selectin as a marker for platelet activation. It is possible that another approach, such as measuring PAC-1 instead, could have shifted the results somewhat.
In conclusion, patients with PAH/CTEPH have increased amounts of circulating PMAs and PGAs expressing P-selectin together with higher concentrations of inflammatory cytokines. There is an association of PMAs and selected cytokines in the patients that needs further evaluation to increase our knowledge of the involvement of activated platelets in inflammation in pulmonary hypertension.
Supplemental Material
Download PDF (289.9 KB)Acknowledgements
The authors would like to acknowledge support of Affinity Proteomics Uppsala at SciLifeLab Sweden for providing assistance in the protein analyses.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2087867
Additional information
Funding
References
- McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009;119:2250–2294. Epub 2009/ 04/01. doi:10.1161/CIRCULATIONAHA.109.192230.
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015;46:903–975. Epub 2015/ 09/01. doi:10.1183/13993003.01032-2015.
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53:1801913. Epub 2018/12/14. doi:10.1183/13993003.01913-2018.
- Swedish Pulmonary Arterial Hypertension Registry Annual Report. 2019. https://www.ucr.uu.se/spahr/arsrapporter/arsrapport-spahr-2020/viewdocument/148
- Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30:104–109. Epub 2007/ 03/16. doi:10.1183/09031936.00092306.
- Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, Olsson KM, Meyer K, Vizza CD, Vonk-Noordegraaf A, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 2017;50;1700740. Epub 2017/ 08/05. doi:10.1183/13993003.00740-2017.
- Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 1994;144:275–285. Epub 1994/ 02/01.
- Kim NH, Delcroix M, Jais X, Madani MM, Matsubara H, Mayer E, Ogo T, Tapson VF, Ghofrani HA, Jenkins DP. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2019;53:1801915. Epub 2018/12/14. doi:10.1183/13993003.01915-2018.
- Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 2014;115:165–175. Epub 2014/ 06/22. doi:10.1161/CIRCRESAHA.113.301141.
- Matthews DT, Hemnes AR. Current concepts in the pathogenesis of chronic thromboembolic pulmonary hypertension. Pulm Circ 2016;6:145–154. Epub 2016/ 06/03. doi:10.1086/686011.
- Bonderman D, Jakowitsch J, Adlbrecht C, Schemper M, Kyrle PA, Schonauer V, Exner M, Klepetko W, Kneussl MP, Maurer G, et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost 2005;93:512–516. Epub 2005/ 03/01. doi:10.1160/TH04-10-0657.
- Molossi S, Clausell N, Rabinovitch M. Reciprocal induction of tumor necrosis factor-alpha and interleukin-1 beta activity mediates fibronectin synthesis in coronary artery smooth muscle cells. J Cell Physiol 1995;163:19–29. Epub 1995/ 04/01. doi:10.1002/jcp.1041630104.
- Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, Bisserier M, Paulin R, Lambert C, Provencher S, et al. Kruppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res 2011;12:128. Epub 2011/ 09/29. doi:10.1186/1465-9921-12-128.
- Khandagale A, Aberg M, Wikstrom G, Bergstrom Lind S, Shevchenko G, Bjorklund E, Siegbahn A, Christersson C. Role of extracellular vesicles in pulmonary arterial hypertension: modulation of pulmonary endothelial function and angiogenesis. Arterioscler Thromb Vasc Biol 2020;40:2293–2309. Epub 2020/ 08/08. doi:10.1161/ATVBAHA.120.314152.
- Marcoux G, Laroche A, Espinoza Romero J, Boilard E. Role of platelets and megakaryocytes in adaptive immunity. Platelets 2021;32:340–351. Epub 2020/ 07/01. doi:10.1080/09537104.2020.1786043.
- Jungi TW, Spycher MO, Nydegger UE, Barandun S. Platelet-leukocyte interaction: selective binding of thrombin-stimulated platelets to human monocytes, polymorphonuclear leukocytes, and related cell lines. Blood 1986;67:629–636. Epub 1986/ 03/01. doi:10.1182/blood.V67.3.629.629.
- Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, Wagner DD, Furie B. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 1989;59:305–312. Epub 1989/ 10/20. doi:10.1016/0092-8674(89)90292-4.
- Rinder HM, Bonan JL, Rinder CS, Ault KA, Smith BR. Dynamics of leukocyte-platelet adhesion in whole blood. Blood 1991;78:1730–1737. Epub 1991/ 10/01. doi:10.1182/blood.V78.7.1730.1730.
- Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 2000;192:193–204. Epub 2000/ 07/19. doi:10.1084/jem.192.2.193.
- Finsterbusch M, Schrottmaier WC, Kral-Pointner JB, Salzmann M, Assinger A. Measuring and interpreting platelet-leukocyte aggregates. Platelets 2018;29:677–685. Epub 2018/ 02/21. doi:10.1080/09537104.2018.1430358.
- Loguinova M, Pinegina N, Kogan V, Vagida M, Arakelyan A, Shpektor A, Margolis L, Vasilieva E. Monocytes of different subsets in complexes with platelets in patients with myocardial infarction. Thromb Haemost 2018;118:1969–1981. Epub 2018/ 10/10. doi:10.1055/s-0038-1673342.
- Furman MI, Barnard MR, Krueger LA, Fox ML, Shilale EA, Lessard DM, Marchese P, Frelinger AL 3rd, Goldberg RJ, Michelson AD. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol 2001;38:1002–1006. Epub 2001/ 10/05. doi:10.1016/S0735-1097(01)01485-1.
- Schrottmaier WC, Mussbacher M, Salzmann M, Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis 2020;307:109–120. Epub 2020/ 05/23. doi:10.1016/j.atherosclerosis.2020.04.018.
- Christersson C, Johnell M, Siegbahn A. Tissue factor and IL8 production by P-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of Lyn and is inhibited by IL10. JThrombHaemost 2008;6:986–994. doi:10.1111/j.1538-7836.2008.02956.x.
- Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, Scheed A, Ritter C, Dahal BK, Vater A, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:897–908. Epub 2012/ 09/08. doi:10.1164/rccm.201202-0335OC.
- Gerasimovskaya E, Kratzer A, Sidiakova A, Salys J, Zamora M, Taraseviciene-Stewart L. Interplay of macrophages and T cells in the lung vasculature. Am J Physiol Lung Cell Mol Physiol 2012;302:L1014–1022. Epub 2012/ 03/06. doi:10.1152/ajplung.00357.2011.
- Yu YA, Malakhau Y, Yu CA, Phelan SJ, Cumming RI, Kan MJ, Mao L, Rajagopal S, Piantadosi CA, Gunn MD. Nonclassical monocytes sense hypoxia, regulate pulmonary vascular remodeling, and promote pulmonary hypertension. J Immunol 2020;204:1474–1485. Epub 2020/ 01/31. doi:10.4049/jimmunol.1900239.
- Hu XS, Du CQ, Zeng CL, Yao L, Zhang FR, Wang K, Chen JZ, Li N, Hu H. Systemic evaluation of platelet and leukocyte activation and interaction in a rat model of pulmonary arterial hypertension. Cardiology 2010;117:44–53. Epub 2010/ 10/07. doi:10.1159/000320107.
- Li N, Goodall AH, Hjemdahl P. Efficient flow cytometric assay for platelet-leukocyte aggregates in whole blood using fluorescence signal triggering. Cytometry 1999;35:154–161. Epub 1999/ 12/20. doi:10.1002/(SICI)1097-0320(19990201)35:2<154::AID-CYTO7>3.0.CO;2-Y.
- Taguchi H, Kataoka M, Yanagisawa R, Kawakami T, Tamura Y, Fukuda K, Yoshino H, Satoh T. Platelet level as a new prognostic factor for idiopathic pulmonary arterial hypertension in the era of combination therapy. Circ J 2012;76:1494–1500. Epub 2012/ 03/27. doi:10.1253/circj.CJ-11-1223.
- Mojadidi MK, Goodman-Meza D, Eshtehardi P, Pamerla M, Msaouel P, Roberts SC, Winoker JS, Jadeja NM, Zolty R. Thrombocytopenia is an independent predictor of mortality in pulmonary hypertension. Heart Lung 2014;43:569–573. Epub 2014/ 08/30. doi:10.1016/j.hrtlng.2014.07.006.
- Chin KM, Channick RN, de Lemos JA, Kim NH, Torres F, Rubin LJ. Hemodynamics and epoprostenol use are associated with thrombocytopenia in pulmonary arterial hypertension. Chest 2009;135:130–136. Epub 2008/ 08/23. doi:10.1378/chest.08-1323.
- Lapa M, Acencio MM, Farias AQ, Teixeira LR, Fernandes CJ, Jardim CP, Terra-Filho M. Selectins and platelet-derived growth factor (PDGF) in schistosomiasis-associated pulmonary hypertension. Lung 2014;192:981–986. Epub 2014/ 09/15. doi:10.1007/s00408-014-9643-0.
- Manakeng K, Prasertphol P, Phongpao K, Chuncharunee S, Tanyong D, Worawichawong S, Svasti S, Chaichompoo P. Elevated levels of platelet- and red cell-derived extracellular vesicles in transfusion-dependent beta-thalassemia/HbE patients with pulmonary arterial hypertension. Ann Hematol 2019;98:281–288. Epub 2018/ 10/20. doi:10.1007/s00277-018-3518-z.
- Maeda NY, Carvalho JH, Otake AH, Mesquita SM, Bydlowski SP, Lopes AA. Platelet protease-activated receptor 1 and membrane expression of P-selectin in pulmonary arterial hypertension. Thromb Res 2010;125:38–43. Epub 2009/ 05/19. doi:10.1016/j.thromres.2009.04.018.
- Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, Loyd JE. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992;327:70–75. Epub 1992/ 07/09. doi:10.1056/NEJM199207093270202.
- Launay JM, Herve P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, Etienne N, Drouet L, Humbert M, Simonneau G, et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med 2002;8:1129–1135. Epub 2002/ 09/24. doi:10.1038/nm764.
- Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 2001;104:1533–1537. Epub 2001/ 09/26. doi:10.1161/hc3801.095588.
- Gerrits AJ, Frelinger AL 3rd, Michelson AD. Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom 2016;78:6 15 11–16 15 10. Epub 2016/ 10/11.
- Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, Hechtman HB, Michelson AD. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol 1998;31:352–358. Epub 1998/ 02/14. doi:10.1016/S0735-1097(97)00510-X.
- Zamora C, Canto E, Nieto JC, Garcia-Planella E, Gordillo J, Ortiz MA, Suarez-Calvet X, Perea L, Julia G, Juarez C, et al. Inverse association between circulating monocyte-platelet complexes and inflammation in ulcerative colitis patients. Inflamm Bowel Dis 2018;24:818–828. Epub 2018/ 03/13. doi:10.1093/ibd/izx106.
- Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 1995;151:1628–1631. Epub 1995/ 05/01. doi:10.1164/ajrccm.151.5.7735624.
- Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010;122:920–927. Epub 2010/ 08/18. doi:10.1161/CIRCULATIONAHA.109.933762.
- Rhodes CJ, Wharton J, Howard LS, Gibbs JS, Wilkins MR. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart 2011;97:1054–1060. Epub 2011/ 05/12. doi:10.1136/hrt.2011.224857.
- Simpson CE, Chen JY, Damico RL, Hassoun PM, Martin LJ, Yang J, Nies M, Griffiths M, Vaidya RD, Brandal S, et al. Cellular sources of interleukin-6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur Respir J 2020;55;1901761. Epub 2020/ 02/08. doi:10.1183/13993003.01761-2019.
- Cracowski JL, Chabot F, Labarere J, Faure P, Degano B, Schwebel C, Chaouat A, Reynaud-Gaubert M, Cracowski C, Sitbon O, et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J 2014;43:915–917. Epub 2013/ 11/16. doi:10.1183/09031936.00151313.
- Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 2011;109:867–879. Epub 2011/ 08/27. doi:10.1161/CIRCRESAHA.110.236927.
- Ed Rainger G, Chimen M, Harrison MJ, Yates CM, Harrison P, Watson SP, Lordkipanidze M, Nash GB. The role of platelets in the recruitment of leukocytes during vascular disease. Platelets 2015;26:507–520. Epub 2015/ 07/22. doi:10.3109/09537104.2015.1064881.
- da Costa Martins P, van den Berk N, Ulfman LH, Koenderman L, Hordijk PL, Zwaginga JJ. Platelet-monocyte complexes support monocyte adhesion to endothelium by enhancing secondary tethering and cluster formation. Arterioscler Thromb Vasc Biol 2004;24:193–199. Epub 2003/ 11/15. doi:10.1161/01.ATV.0000106320.40933.E5.
- Kuckleburg CJ, Yates CM, Kalia N, Zhao Y, Nash GB, Watson SP, Rainger GE. Endothelial cell-borne platelet bridges selectively recruit monocytes in human and mouse models of vascular inflammation. Cardiovasc Res 2011;91:134–141. Epub 2011/ 02/03. doi:10.1093/cvr/cvr040.
- Schulz C, von Bruhl ML, Barocke V, Cullen P, Mayer K, Okrojek R, Steinhart A, Ahmad Z, Kremmer E, Nieswandt B, et al. EMMPRIN (CD147/basigin) mediates platelet-monocyte interactions in vivo and augments monocyte recruitment to the vascular wall. J Thrombosis Haemostasis 2011;9:1007–1019. Epub 2011/ 02/16. doi:10.1111/j.1538-7836.2011.04235.x.
- Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, Fadel E, Savale L, Huertas A, Humbert M, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest 2018;128:1956–1970. Epub 2018/ 04/10. doi:10.1172/JCI96462.
- Huang Z, Liu Z, Luo Q, Zhao Z, Zhao Q, Zheng Y, Xi Q, Tang Y. Glycoprotein 130 inhibitor ameliorates monocrotaline-induced pulmonary hypertension in rats. Can J Cardiol 2016;32:1356 e1351–1356 e1310. Epub 2016/ 10/30. doi:10.1016/j.cjca.2016.02.058.