
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Platelet microparticles (PMPs) are vesicles that are released by platelets into the extracellular space and play a role in antiphospholipid antibody syndromes. PMPs have recently been recognized as a new and viable cell. There is growing evidence that the anti-β2 glycoprotein (GPI)/β2GPI complex is associated with aberrant activation of PMPs. Although studies suggest that aberrant activation of PMPs may lead to inflammatory necrosis of endothelial cells, the underlying mechanisms remain unclear. We found that although the difference in the number of PMPs was not statistically significant, NLR family pyrin domain containing 3 (NLRP3) within PMPs was increased during stimulation of anti-β2GPI/β2GPI complexes. Furthermore, we demonstrated that anti-β2GPI/β2GPI complex-induced PMPs effectively stimulated endothelial cell pyroptosis via the NLRP3/nuclear factor (NF)-κB/gasdermin D (GSDMD) signaling pathway as well as the NLRP3/Caspase-1 signaling pathway. Additionally, inhibition of NLRP3 expression in PMPs effectively reduced the inflammatory response and pyroptosis in endothelial cells. Our data suggest that PMPs aberrantly activated by anti-β2GPI/β2GPI complexes play a vital role in endothelial cell pyroptosis, and these studies provide major insights into the mechanisms of thrombosis during the treatment of antiphospholipid antibody syndrome.
Plain Language Summary
What is the context?
Antiphospholipid syndrome (APS), an acquired autoimmune disease of unknown etiology. Clinical manifestations include arteriovenous thrombosis, recurrent miscarriages and thrombocytopenia. Endothelial cell damage is common in APS
Anti-β2 glycoprotein I antibody, one of the most common APS antibodies, is the main target antigen of anti-β2GPI. Studies have shown that the anti-β2GPI/β2GPI complex accelerates inflammatory cell necrosis.
Pyroptosis, also known as inflammatory cell necrosis, is a new form of cell death. Pyroptosis is caused by the activation of the NLRP3 inflammasome, which manifests itself as swelling, lysis and perforation of the cell membrane.
Platelet micro-particles (PMPs) are vesicular components that are released extracellularly by platelet activation and are the most abundant and common type of circulating particles in the blood, causing an inflammatory response in the endothelium. There is limited evidence that anti-β2GPI/β2GPI complexes can accelerate endothelial cell pyroptosis by mediating platelet activation to produce PMPs. However, more research is needed to investigate the specific mechanisms by which PMPs cause endothelial cell pyroptosis.
What is new?
This is the first study on the role of NLRP3 in PMPs. NLRP3 expression in PMPs was increased by stimulation of anti-β2GPI/β2GPI complexes.
NLRP3 in PMPs is closely associated with GSDMD-N, a protein involved in endothelial pyroptosis.
Anti-β2GPI/β2GPI stimulated PNPs induce pyroptosis via NLRP3/NF-κB/GSDMD and NLRP3/Caspase-1/IL-1β axis.
What is the impact?
The aim of this study was to investigate the specific mechanism of endothelial cell pyroptosis induced by platelet-released PMPs activated by anti-β2GPI/β2GPI complexes. This finding provides new ideas on the mechanism of endothelial cell scorching in APS and provides a new drug target for the clinical treatment of APS.
Introduction
Antiphospholipid syndrome (APS) is an acquired autoimmune disease of unknown etiology [Citation1]. This disease is characterized by positive serum antiphospholipid antibodies as well as clinical manifestations including arteriovenous thrombosis, recurrent pregnancy loss, and thrombocytopenia. Vascular endothelial cell injury is a common pathological feature of antiphospholipid syndrome [Citation2]. Although endothelial cell injury is multifactorial, anti-β2 glycoprotein I antibodies play an important role [Citation3].
Anti-β2 glycoprotein I (β2GPI) antibodies are among the most frequent antiphospholipid antibodies. β2GPI is a five-structural domain phospholipid-binding protein and is the main target antigen of anti-β2GPI [Citation4,Citation5]. Anti-β2GPI/β2GPI complexes (IC) in APS patients directly promote monocyte adhesion to the endothelium, leading to increased tissue factor synthesis and inflammatory damage of endothelial cells, which is a key process in thrombosis [Citation6].
Microparticles (MPs) have gained a fair amount of attention because of their important role in inflammation, prothrombotic states, and autoimmune diseases [Citation7–11]. MPs are ultramicroscopic membranous vesicles with a diameter of approximately 0.05–1 μm. Several types of MPs exist in the human body, including platelet, endothelial cells, and erythrocyte-derived particles [Citation12]. Platelet-derived microparticles (PMPs) are the most abundant, accounting for approximately 70–90% of circulating MPs [Citation13]. PMPs are very small membranous vesicles released outside the cell after platelet stimulation. These vesicles can carry a markers of the parent cell, such as phosphatidylserine, lipids, and membrane proteins on their surface, and participate in intracellular communication by transferring proteins, mRNAs, and microRNAs to target cells [Citation14,Citation15]. PMPs can cause inflammatory responses directly by promoting neutrophil activation and adhesion to the endothelium [Citation16]. Increasing evidence indicates that PMPs may have efficient pro-coagulant and pro-inflammatory activities in autoimmune diseases [Citation9,Citation17]. Nonetheless, our team and others have demonstrated that IC can stimulate aberrant PMPs activation through the toll-like receptor (TLR)4/p38 signaling pathway by binding to TLR4 on platelets, which releases PMPs [Citation18]. However, the possibility that PMPs stimulated by IC (IC-PMPs) may lead to inflammatory damage of APS endothelial cells has should be further investigated.
Pyroptosis has been increasingly referred to in the context of disease development mechanisms. Pyroptosis, also known as cellular inflammatory necrosis, is different from other forms of cell death such as apoptosis [Citation19]. Pyroptosis is cell death via NLR family pyrin domain containing 3 (NLRP3) inflammasome activation, which manifests as swelling, lysis and perforation of cell membranes [Citation20]. Specifically, activation of inflammasomes induces the cleavage of gasdermin D (GSDMD), which releases the N-terminal structural domain (GSDMD-N) and causes perforation of the cell membrane [Citation21]. Inflammatory factors such as interleukin (IL)-1β and IL-18 can be released after pyroptosis [Citation22]. NLRP3 inflammatory vesicles generate GSDMD, pro-IL-1β and pro-IL-18 by activating nuclear factor (NF)-κB, and induce GSDMD cleavage, IL-1β and IL-18 maturation by activating Caspase-1, leading to pyroptosis [Citation23,Citation24]. During inflammation, IC can induce increased NLRP3 in platelets. Recent studies have found that increased Caspase-1 protein expression in MPs directly activates endothelial cell pyroptosis [Citation25]. However, the relationship between IC-PMPs and endothelial cell pyroptosis remains unclear.
Therefore, the aim of this study was to determine the potential biological mechanisms by which IC-PMPs may affect endothelial cell pyroptosis in APS.
Methods
Inclusion criteria
Healthy individuals without autoimmune diseases and not taking any medication for at least 14 days before the experiment start date were enrolled. Patients with other inflammatory diseases, infections, or malignant diseases were excluded as they may have high numbers of PMPs. Blood samples were collected from healthy subjects with the approval of the Institutional Ethics Committee of Harbin Medical University.
PMPs preparation and stimulation
Blood was collected with sodium citrate anticoagulant. 30 tubes blood from healthy individuals were centrifuged at 150 × g for 15 min at 20°C. The supernatant was collected and further centrifuged at 1000 × g for 10 min to collect platelets. After washing the precipitate once with phosphate buffered saline (PBS), the platelets were resuspended in serum-free RPMI-1640 medium (Sigma, Saint Louis, USA). Stimulation with anti-β2GPI (10 μg/ml)/β2GPI (100 μg/ml) (Sino Biological, Beijing, China) was performed at 37°C (from 0.5 to 8 h). The samples were incubated with thrombin (10 μg/ml; Solarbio, Beijing, China), lipopolysaccharide (LPS) (100 ng/mL, Sigma, Saint Louis, USA), anti-β2GPI (10 μg/ml)/β2GPI (100 μg/ml) (Sino Biological, Beijing, China), PBS, Mouse normal IgG (10 µg/mL; Santa Cruz Biotechnology, Dallas, USA) at 37°C for 30 min and with the NLRP3 inhibitor MCC950 (10 µM; MCE, New Jersey, USA) at 37°C for 10 min. The residual platelets were removed by centrifugation at 3200 × g for 10 min. The supernatant was collected and centrifuged at 20 000 × g and 18°C for 90 min to collect the PMPs. Finally, the precipitate containing PMPs was resuspended in serum-free RPMI-1640 medium. PMPs were stored at −80°C in a refrigerator.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Biotechnology Co (iCell Bioscience Inc, Shanghai, China). Cells were cultured in RPMI-1640 medium (Sigma, Saint Louis, USA) supplemented with 10% fetal bovine serum, at 37°C, 5% CO2, and 95% O2. In experiments involving pharmacological interventions, HUVECs were pretreated with inhibitors (10 µM PDTC, 10 µM VX-765) for 3 h and then treated with appropriate concentrations of PMPs for 6 h in the presence of these inhibitors.
PMPs extraction and flow cytometry analysis
The corresponding antibodies and resuspended PMPs were equilibrated at 37°C for 10 min. Subsequently, 30 μL of freshly thawed PMPs, 5 μL of mouse anti-human CD41a-phaeohemoglobin (PE) (BD Pharmingen, San Jose, USA) and 5 μL of fluorescein isothiocyanate (FITC)-Annexin V antibody (wanleibio, Shenyang, China) were incubated at 37°C for 30 min in the dark. The mixtures were thinned with 420 μL Annexin V-binding buffer. For gating and counting, 0.8 and 3 μm beads were added to the diluted samples. Isotype controls (PE Mouse IgG1, κ Isotype control) were utilized to differentiate between true positive events and background noise, and to improve the specificity of the MPs assay. PMPs were detected as Annexin V+/CD41a+ particles using a FACSCalibur cytometer (BD Biosciences, New Jersey, USA). In flow cytometry analysis, parameter data are displayed as a two-dimensional dot matrix graph. The horizontal coordinate indicates FITC and the vertical coordinate indicates PE. Three micrometer standard microspheres were used for particle counting and 0.8 μm standard microspheres were used for particle localization gating. Forward scattered light log (FSC-LOG) and side scattered light log (SSC-LOG) scatter plots were established. The detection gaps of the instrument were adjusted according to the measurement results of the blank positioning tubes, and the double positive regions of PMPs surface marker CD41a and Annexin V were selected as PMPs. The concentration of PMPs was calculated based on the number of standard fluorescent microspheres out of 10 000. The number of PMPs was computed using the equation:
PMPs coincubation with HUVECs
One day before the experiment, 1 × 105 HUVECs in total were inoculated into 24-well plates, and membranes were marked with PKH67 (Sigma, Saint Louis, USA) for 5 min. Dil (Solarbio, Beijing, China) was used to tag 1 × 107 PMPs for 10 min. PMPs were filtered by centrifugation at 1500 × g through a 100,000 kDa Millipore filter for 10 min, and mixed with 100 μL medium using a pipette before being added to the wells. An equivalent volume of complete medium containing 10% fetal bovine serum was added to the negative control. PMPs and HUVECs were analyzed after 6 h of incubation. The ratio of HUVECs to PMPs was 1:100 and the same ratio was used for all subsequent co-culture experiments.
Western blot analysis
The total proteins in HUVECs were extracted using the RIPA lysis buffer. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) 10% was performed to separate the proteins. Primary antibodies against NLRP3 (1:1000, wanleibio, Shenyang, China), NF-κB, p-NF-κB (1:1000, wanleibio, Shenyang, China), Caspase-1, Caspase-1 p20 (1:1000, wanleibio, Shenyang, China), GSDMD (1:1000, Santa Cruz Biotechnology, Dallas, USA), GSDMD-N (1:1000, abcam, Cambridge, UK), and β-actin (1:1000, ZSGB-BIO, Beijing, China) were used to detect the corresponding proteins. The bands were incubated with specific HRP-conjugated secondary antibodies (1:5000, ZSGB-BIO, Beijing, China). Fluorescence/Chemiluminescence Imaging System (CLINX Science Instruments, Shanghai, China) was used to detect specific bands using ECL Western blotting substrate (Beyotime, Shanghai, China).
ELISA analysis
The supernatants from HUVECs in culture were collected and the expression of IL-1β and IL-18 in the supernatants was measured using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Jianglaibio, Shanghai, China). All operating steps were performed according to the manufacturer’s instructions.
Immunofluorescence
Caspase-1 p20, and GSDMD-N expression was measured in HUVECs following immunofluorescence staining. Briefly, the cells were fixed with 4% paraformaldehyde for 30 min, permeabilized using 0.5% Triton X-100 for 15 min, and then blocked using goat serum. Wet boxes were placed into fixed cells to react with Caspase-1 p20 (1:200, wanleibio, Shenyang, China), and GSDMD-N (1:200, abcam, Cambridge, UK) antibodies. The samples were incubated overnight at 4°C and stored in the refrigerator. The next day, the primary antibody was discarded, and the samples were incubated with FITC/TRITC-conjugated secondary antibody (1:200, ZSGB-BIO, Beijing, China) for 1 h, strictly ensuring light-proof operation throughout the process. The nuclei were stained using 4,’6-diamidino-2-phenylindole (DAPI) (1 μg/mL, Invitrogen, Carlsbad, CA, USA) for 7 min. The cells were imaged using an immunofluorescence inverted microscope (Ti-U, NiKon, Japan).
Caspase-1 activity detection assay
Caspase-1 activity was measured using the caspase-1 activity assay kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. The sample turbidity was measured at 405 nm in a microplate reader. The total protein of the cell lysates was also normalized to construct a calibration curve, and Caspase-1 activity was calculated accordingly.
Statistical analyses
All data were collected and data were expressed as mean ± standard deviation (SD) for three independent experiments and analyzed using SPSS 26.0 software. Specifically, for continuous variables, normally distributed data were expressed as mean ± standard error by Student’s t-test, while non-normally distributed data were assessed by Mann-Whitney U-test and expressed as median. Data from three and more groups were compared between groups by one-way analysis of variance (ANOVA) with Dunnet’s post-hoc test. These data analyses were performed using GraphPad Prism version 7 (GraphPad Software, Inc.) software. p < .05 was considered to be statistically significant.
Result
Number of PMPs did not change significantly, but NLRP3 expression was increased by stimulation of IC
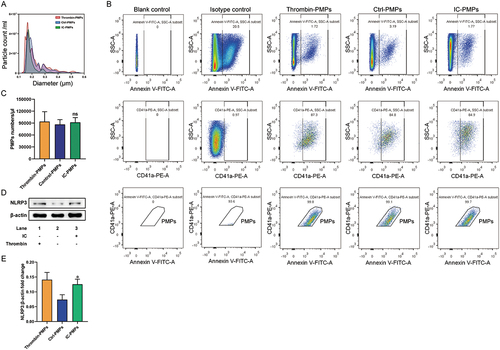
NLRP3 induces cleavage of GSDMD into an active fragment that mediates pyroptosis through activation of Caspase-1. Our recent work confirms that IC can stimulate platelets to produce increased NLRP3 expression. Given the similarity between PMPs and platelets in terms of composition [Citation26], we investigated whether NLRP3 acts as a key component of PMPs with a possible role in endothelial cell pyroptosis. First, we stimulated 30 platelet samples collected from healthy individuals with IC for 30 minutes and identified the extracted PMPs using a PSS nanoparticle size analyzer and a FACSCalibur cytometer. Nanoparticle size analysis demonstrated that > 99% of PMPs in the sample had 0.1–1.0 μm in diameter ( and Supplementary Table 1). To determine the level of PMPs, PMPs were detected using the platelet-specific markers CD41a and Annexin V. Flow cytometry showed that most of these MPs were CD41a and Annexin V-positive PMPs (). The levels of PMPs in the thrombin-stimulated positive group (102289.00 ± 12856.13/µl), negative control group (85844.00 ± 18416.52/µl) and IC-stimulated group (84293.00 ± 12352.59/µl) were not significantly different (P > .05) (). For the first time, we found a significant increase in NLRP3 expression in PMPs after thrombin and IC stimulation (). These results suggest that PMPs produced by abnormal platelet activation are unchanged in number and increased in intrinsic NLRP3 expression by stimulation with IC.
Figure 1. IC stimulated platelets to produce similar numbers of PMPs with increased NLRP3. A. PSS nanoparticle size analysis of PMPs treated with IC for 30 min. B. Flow cytometry analysis of PMPs, which were double positive for CD41a-PE and Annexin V-FITC. C. Quantification of PMPs per μL. The PMP counts/μL between Thrombin-PMPs, Ctrl-PMPs and IC-PMPs. D. Western blot analysis of NLRP3 in PMPs treated with IC for 30 min. E. Quantification of NLRP3 protein levels. β-actin was used as an internal control. Data were presented as the means ± SD, n = 3. *P <.05, **P <.01, and ***P <.001 vs. the corresponding IC-PMPs group; ns, P >.05, no significant difference.
IC-PMPs are internalized by endothelial cells and expand cellular pyroptosis
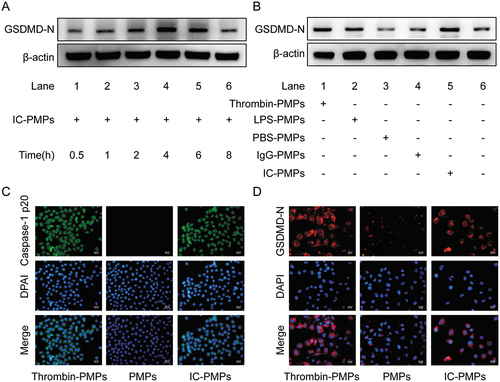
IC stimulate increased expression of NLRP3 in PMPs. This result indicates that NLRP3 activity within microparticles may interact with Caspase-1 p20 and GSDMD-N in endothelial cells to promote pyroptosis of target cells. To test this hypothesis, we performed immunofluorescence assays for the cell pyroptosis proteins GSDMD-N and Caspase-1 p20, as well as western blot for GSDMD-N to determine the role of IC-PMPs. Firstly, we analyzed the effect of PMPs on GSDMD-N expression in endothelial cells with different influencing factors. The degree of endothelial cell pyroptosis induced by IC-PMPs varied at different incubation times, GSDMD-N expression was most significant after 4–6 h (). In addition, with the influence of different stimulants, we added complexes to stimulate the production of PMPs to induce a significantly increased level of GSDMD-N expression in endothelial cells (). Thus, IC-PMPs play a major role in pyroptosis. To further confirm this observation, we selected endothelial cells of suitable density for immunofluorescence experiments to determine the expression of the pyroptosis indicators Caspase-1 p20 and GSDMD-N (). The results showed that IC-PMPs had a significant effect on the induction of endothelial cell pyroptosis compared with the control group. It was shown that the effect of PMPs on endothelial cells acts through the internalized form [Citation27], and we incubated HUVECs with Dil-labeled PMPs for 6 h. The same results were found by fluorescence microscopy (Supplementary Figure S1).
Figure 2. Expression of pyroptosis protein was detected in different stimulations and the phenomenon of internalized PMPs. A. Western blot analysis of GSDMD-N in endothelial cells treated with PMPs produced by stimulation with IC for 30 min, 1 h, 2 h, 4 h, 6 h and 8 h. B. Western blot analysis of GSDMD-N in endothelial cells treated with PMPs stimulated by LPS, thrombin, PBS, IgG, and IC for 6 h. β-actin was used as an internal control. C. Immunofluorescence analysis of Caspase-1 p20 (green) in endothelial cells as observed through each group. D. Immunofluorescence analysis of GSDMD-N (red) in endothelial cells as observed through each group. Scale bar is 50 µm.
IC-PMPs promote pyroptosis via the NLRP3/NF-κB/GSDMD signaling axis
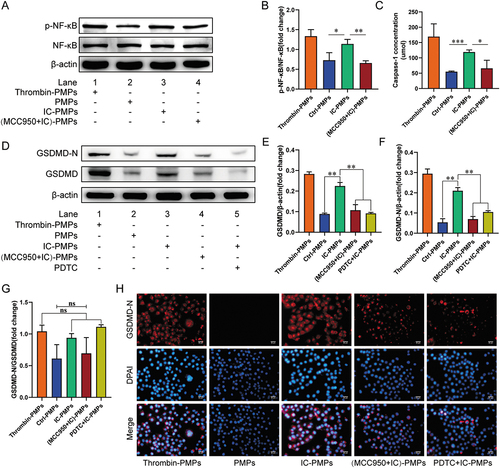
We next investigated the potential mechanism of endothelial cell pyroptosis induced by IC stimulated PMPs. NF-κB, a protein complex, is released from the cytoplasmic NF-κB/NF-κB inhibitor (IκB)α complexes and activated when cells are stimulated, thereby initiating the expression of the pyroptosis protein GSDMD [Citation23]. Hyperactivation of NF-κB in endothelial cells is associated with pyroptosis. To determine whether NLRP3 in PMPs regulates NF-κB to induce endothelial cell pyroptosis, we first analyzed the absence of NF-κB protein in IC-PMPs (Supplementary Figure S2). Then Western blot analysis showed that IC-PMPs significantly increased the phosphorylation of NF-κB protein in endothelial cells, whereas unstimulated PMPs failed to significantly increase the expression of NF-κB phosphorylation (). This result indicates that NLRP3 within IC-PMPs may increase the overactivation of NF-κB. To test this hypothesis, we incubated IC-PMPs with or without the specific NLRP3 inhibitor MCC950 and observed the effect of (MCC950+IC)-PMPs on NF-κB protein phosphorylation in endothelial cells in the presence of MCC950-specific inhibition of NLRP3 in IC-PMPs. Secondly, to confirm whether the NF-κB signaling pathway is involved in PMPs-mediated pyroptosis, we detected that IC-PMPs not only increased the production of GSDMD but also the GSDMD cleavage component, GSDMD-N. This indicates that IC-PMPs may induce endothelial cell pyroptosis by promoting the production of GSDMD. When MCC950 specifically inhibited NLRP3 expression in IC-PMPs and pyrrolidine dithiocarbamate (PDTC) inhibited the phosphorylation of NF-κB protein in endothelial cells, we observed a significant decrease in GSDMD and GSDMD-N expression (). This suggests that IC-PMPs may induce endothelial cell pyroptosis through activation of the NLRP3/NF-κB/GSDMD signaling pathway. Notably, the expression profile of NLRP3 in IC-PMPs had a greater impact on GSDMD-N expression compared to GSDMD. When NLRP3 expression was suppressed in (MCC950+IC)-PMPs, GSDMD-N expression was reduced more relative to that of GSDMD (). This may be due to the fact that the specific inhibition of NLRP3 contained in IC-PMPs inhibited not only the phosphorylation of NF-κB in endothelial cells, but also the activation of caspase-1 (). Thus, the cleavage of GSDMD into GSDMD-N was significantly inhibited. In contrast, PDTC specifically inhibited only the phosphorylation of NF-κB in endothelial cells, reducing GSDMD-N production in a manner that reduced GSDMD protein expression. Thus, in the presence of PDTC, GSDMD expression and GSDMD-N expression were reduced synergistically (). To better observe the expression of pyroptosis-related proteins in the presence of different inhibitors, we performed immunofluorescence staining experiments after co-incubation of PMPs with HUVECs and verified that the expression of red fluorescently labeled GSDMD-N protein was consistent with the previous experiments (). This further confirms that IC-PMPs may induce pyroptosis by activating the NLRP3/NF-κB/GSDMD signaling pathway.
Figure 3. Detection of NF-κB, GSDMD and GSDMD-N in endothelial cells stimulated by PMPs for 6 h. A. Western blot analysis of NF-κB and p-NF-κB in endothelial cells treated with PMPs produced by stimulation with IC for 6 h. B. Quantification of p-NF-κB/NF-κB protein levels. C. Measurement of extracellular Caspase-1 activity in endothelial cells. D. Western blot analysis of GSDMD and GSDMD-N in endothelial cells treated with PMPs produced by stimulation with IC for 6 h. E-G. Quantification of GSDMD and GSDMD-N protein levels. β-actin was used as an internal control. H. Immunofluorescence analysis of GSDMD-N (red) in endothelial cells as observed through each group. Scale bar, 50 μm. Data were presented as the means ± SD, n = 3.*P <.05; **P <.01; ***P <.001; ns, P >.05, no significant difference.
IC-PMPs increase the release of IL-1β and IL-18 from endothelial cells via the NLRP3/Caspase-1 axis
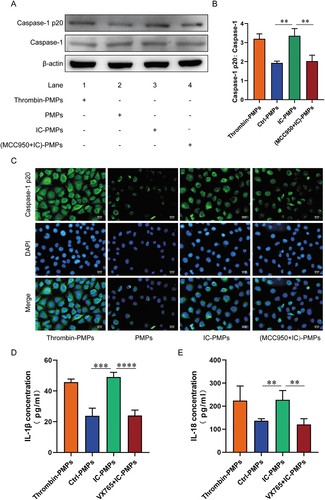
Pyroptosis is a form of programmed cell death mediated primarily by activation of Caspase-1, and this form of cell death is accompanied by cell swelling, osmotic lysis, and release of the pro-inflammatory cytokines IL-1β and IL-18 [Citation28,Citation29]. We sought to test whether active Caspase-1 is essential in the induction of endothelial cell pyroptosis by IC-PMPs. Therefore, we observed the effect of PMPs under different stimuli on Caspase-1 activation. IC-PMPs had a significant effect on Caspase-1 activation in endothelial cells, and the activation effect was significantly diminished in the presence of MCC950-inhibited PMPs (). This suggested that IC-PMPs may induce endothelial cell pyroptosis by stimulating Caspase-1 activation. To better observe the effect of PMPs stimulated by IC on Caspase-1 expression in endothelial cells, we performed immunofluorescence experiments and observed results consistent with previous experiments (). We then examined the secretion of IL-1β and IL-18 when PMPs under different stimuli induced endothelial pyroptosis. IC-PMPs induced endothelial cells to secrete large amounts of the inflammatory factors IL-1β and IL-18, whereas untreated PMPs induced endothelial cells to secrete a minimal amount of IL-1β and IL-18 (). IC-PMPs induced a significant reduction in the secretion of the inflammatory factors IL-1β and IL-18 by endothelial cells when Caspase-1 activation was inhibited by VX765 (). This phenomenon confirms the role of Caspase-1 activation in regulating IC-PMPs to stimulate the secretion of IL-1β and IL-18 and induce endothelial pyroptosis.
Figure 4. Caspase-1, Caspase-1 p20, il-1β and IL-18 were measured in endothelial cells stimulated by PMPs. A. Western blot analysis of Caspase-1 and Caspase-1 p20 in endothelial cells treated with PMPs produced by stimulation with IC for 6 h. B. Quantification of the Caspase-1 p20/caspase-1 fold change. β-actin was used as an internal control. C. Immunofluorescence analysis of Caspase-1 p20 (green) in endothelial cells as observed through each group. Scale bar, 50 μm. D-E. ELISA analysis of the levels of secreted IL-1β and IL-18. Data were presented as the means ± SD, n = 3.*P <.05; **P <.01; ***P <.001; ns, P >.05, no significant difference.
Discussion
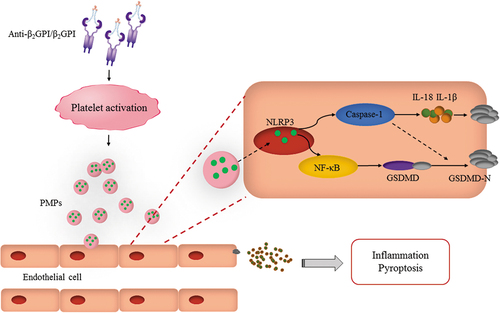
Antiphospholipid syndrome (APS) is a non-organ-specific autoimmune disease. Clinical patients mainly present with arteriovenous, microvascular thrombosis and obstetric complications [Citation30,Citation31]. Platelet-derived microparticles (PMPs) released after platelet activation are considered novel effectors that may play a key role in the development of APS. Previous studies have reported that PMPs stimulated by APS-related antibodies can cause inflammatory responses and trophoblast dysfunction [Citation27]. However, there is little indication in the current study whether a correlation exists between the substances contained in PMPs and endothelial cell pyroptosis. Here, we report that IC-PMPs are involved in the progression of APS thrombosis through delivery of NLRP3, leading to endothelial cell pyroptosis ().
Figure 5. Schematic diagram of the role and mechanism of IC-PMPs on the induction of endothelial cell pyroptosis. In APS, PMPs released from activated platelets are internalized by endothelial cells during hyper-IC stimulation. NLRP3 within PMPs is released, and endothelial cell pyroptosis and inflammatory responses are induced via the NLRP3/NF-κB/GSDMD and NLRP3/Caspase-1 signaling pathways, which are involved in the mechanism of thrombosis in patients with APS.
Anti-β2GPI/β2GPI complexes (IC) bind to the surface membranes of monocytes and endothelial cells and promote tissue factor activity, thereby increasing the risk of thrombosis [Citation32,Citation33]. In 2018, reports showed that IC induced neutrophil extracellular trap formation and promoted thrombosis via activation of the TLR4/myeloid differentiation primary response 88 (MyD88)/mitogen-activated protein kinase (MAPK) axis [Citation34]. Furthermore, studies confirmed that the IC associated with APS can induce platelet activation and promote thrombosis via p38MAPK [Citation18]. This suggests that the IC promotes inflammatory responses and thrombosis during APS.
In recent years, research has shown a close relationship between PMPs and endothelial cell injury and their dysfunction. PMPs are ultra microproducts produced by platelets after stimulation, and different stimulating factors that cause platelet activation can lead to variability PMP formation [Citation35,Citation36]. Some MPs are highly efficient in promoting cellular inflammatory necrosis and blood coagulation [Citation8,Citation17]. Boudreau et al. demonstrated that PMPs can cause an inflammatory response by promoting neutrophil activation and adhesion to the endothelium, leading to endothelial dysfunction and thrombosis [Citation16]. Zhang et al showed that IC induced the release of PMPs to suppress the migration of HUVECs, resulting in endothelial cell dysfunction [Citation37]. Therefore, we hypothesized that IC-PMPs may affect endothelial cell behavior and cause pyroptosis. First, we performed a FACS double-positive analysis and a PSS particle size analysis to ensure the purity of PMPs. The results showed no significant difference in the counts of PMPs produced with the stimulation of IC. This result excludes the effect of the number of PMPs. Therefore, it can be assumed that the alteration of substances contained within PMPs exerts an important regulatory effect on endothelial cells. Second, to further test this, we observed the expression of NLRP3 in PMPs under different stimuli. The results indicated that PMPs stimulated by IC agonists contained a significant increase in NLRP3. Notably, there was no significant difference in the number of PMPs between the different stimulating factors. This result suggests that it is the abnormal activation of PMPs, rather than the change in the number of PMPs that may be involved in the pyroptosis of endothelial cells in APS. Previous studies have shown an increase in the relative levels of PMPs in APS [Citation38,Citation39], but corresponding studies have also observed no significant difference in the number of PMPs between patients with APS and healthy controls [Citation40,Citation41]. Among them, differences in patient selection (types of antibodies collected from patients and the high or low level of antibody potency), or technical differences in sample handling (method of collection, time of analysis, centrifugation speed and time), and whether analysis was performed on fresh or frozen samples may explain the variability of the findings. Therefore, further study is warranted. Controversy remains regarding the number of PMPs produced. Nonetheless, we confirmed that there was no significant difference in the number of microparticles produced by platelet stimulation with anti-β2GPI/β2GPI complexes associated with APS using PSS nanoparticle size analysis and flow cytometry.
PMPs are the essential reservoir units for diverse reactive proteins in platelets. Previous studies have verified the capacity of PMPs to induce or transmit a host of cytokines, chemokines, transcription factors and microRNAs to target cells [Citation14,Citation42]. Michael et al. demonstrated that PMPs infiltrate solid tumors and transfer platelet RNA to tumor cells [Citation43]. Zhang et al demonstrated that activation of platelets produces most of the cytokines released in PMPs [Citation44]. We have found that IC-PMPs are rich in NLRP3 using the western blot technique. The NLRP3 inflammasome is derived from the combination NLRP3 apoptosis-associated speck-like protein containing a caspase recruitment domain activation [Citation45,Citation46]. The NLRP3 inflammasome activates caspase-1 and IL-1β and IL-18. Furthermore, activation of caspase-1 transforms GSDMD into GSDMD-N and boosts the formation of membrane pores [Citation47,Citation48]. This process creates conditions for the cell to release large amounts of inflammatory cytokines, including IL-β and IL-18, and induce pyroptosis. We demonstrated that NLRP3 within PMPs has a vital role in endothelial cell pyroptosis. A substantial increase in Caspase-1 p20 and GSDMD-N protein levels was observed in endothelial cells, and increased levels of soluble forms of IL-1β and IL-18 were detected in the supernatants after treatment with IC-PMPs. This suggests that PMPs-derived NLRP3 induces endothelial cell pyroptosis and releases inflammatory factors through activation of Caspase-1. In contrast, the classical pyroptosis pathway was inhibited by the blockade of NLRP3 in PMPs using MCC950, an NLRP3 inhibitor. These findings indicate that NLRP3 is a predominant PMPs-derived regulator of endothelial cell pyroptosis.
Considering that endothelial cells are in close contact with blood and are susceptible to PMPs, we investigated the efficacy of PMPs on endothelial cells. First, we demonstrated that IC-PMPs were internalized by HUVECs using in vitro co-incubation. Next, we explored other pathways affecting endothelial cell pyroptosis induced by PMPs. PMPs modulate signaling pathways in target cells at the transcription and translation levels depending on their composition. Ceroi et al showed that PMPs stimulated the activation of hepatic X receptors in dendritic cells [Citation49]. Laffont et al. demonstrated that PMPs regulate the gene expression of F-box/WD repeat protein 7 and epinephrine A1 in HUVECs [Citation14]. There is mounting support that NF-κB plays a pivotal role in pyroptosis. In the development of ulcerative colitis, PH domain and leucine rich repeat protein phosphatase 2(PHLPP2) deficiency activates the NF-κB signaling pathway, leading to intestinal epithelial pyroptosis [Citation50]. In osteoarthritis, P2× purinoceptor 7 activation induces chondrocyte pyroptosis and promotes inflammation through the NF-κB signaling pathway, thereby exacerbating the symptoms of osteoarthritis [Citation51]. In addition, many drugs reduce inflammatory damage in disease by downregulating NF-κB pathway-induced pyroptosis, including rheumatoid arthritis [Citation52], myocardial infarction [Citation53], and acute kidney injury [Citation54]. In the present study, we demonstrate that IC-PMPs increase the production of the pyroptosis protein GSDMD by inducing the NF-κB signaling pathway in endothelial cells. In contrast, untreated PMPs did not have this effect. Thus, the NF-κB pathway regulates endothelial cell pyroptosis induced by IC-PMPs.
Because collecting PMPs is challenging, we did not investigate their specific mechanism in raising NLRP3, which warrants exploring whether PMPs raise NLRP3 levels or prevent its rapid degradation under different stimuli. In addition, the detection limit of the PSS nanoparticle size is 0.1 µm, which is much smaller than the 0.4–0.5 µm limit of standard instruments. Therefore, a large population of previously undetectable PMPs may have been isolated in this study, yielding more detailed results. Further research is necessary to dissect the factors mediating PMP internalization by HUVECs and the potential impact of proteins on the membranes of PMPs in the mechanism of internalization. Our findings indicate that IC-PMPs can increase endothelial cell pyroptosis and enhance the inflammatory response.
In conclusion, NLRP3 released from PMPs stimulated by anti-β2GPI/β2GPI complexes induced endothelial cell pyroptosis. The underlying mechanism is related to NLRP3-induced activation of NF-κB as well as the caspase-1 pathway in endothelial cells. The sustained release of PMPs eventually leads to endothelial cell pyroptosis and an inflammatory response. Our findings may provide a potential pharmacological target for preventing the development of thrombosis in APS.
Authors’ contributions
Longjiang Di and Yanhong Liu designed the experiments; Longjiang Di and Yanhong Liu performed the experiments; Longjiang Di, Yanhong Liu and Caijun Zha wrote the paper.
Supplemental Material
Download TIFF Image (1.1 MB)Supplemental Material
Download TIFF Image (11.7 MB)Supplemental Material
Download PDF (70.3 KB)Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2022.2156492
Additional information
Funding
References
- Rand JH. Molecular pathogenesis of the antiphospholipid syndrome. Circ Res. 2002;90(1):29–10. doi: 10.1161/hh0102.102795.
- Petri M. Epidemiology of the antiphospholipid antibody syndrome. J Autoimmun. 2000;15(2):145–151. doi: 10.1006/jaut.2000.0409.
- Giannakopoulos B, Mirarabshahi P, Krilis SA. New insights into the biology and pathobiology of beta2-glycoprotein I. Curr Rheumatol Rep. 2011;13(1):90–95. doi: 10.1007/s11926-010-0151-9.
- McDonnell T, Wincup C, Buchholz I, Pericleous C, Giles I, Ripoll V, Cohen H, Delcea M, Rahman A. The role of beta-2-glycoprotein I in health and disease associating structure with function: more than just APS. Blood Rev. 2020;39:100610. doi: 10.1016/j.blre.2019.100610.
- Martínez-Flores JA, Serrano M, Pérez D, Cámara a GDL, Lora D, Morillas L, Ayala R, Paz-Artal E, Morales JM, Serrano A. Circulating immune complexes of IgA bound to beta 2 glycoprotein are strongly associated with the occurrence of acute thrombotic events. J Atheroscler Thromb. 2016;23(10):1242–1253. doi: 10.5551/jat.34488.
- Zhang W, Gao F, Lu D, Sun N, Yin X, Jin M, Liu Y. Anti-β2 glycoprotein I antibodies in complex with β2 glycoprotein I induce platelet activation via two receptors: apolipoprotein E receptor 2′ and glycoprotein I bα. Front Med. 2016;10(1):76–84. doi: 10.1007/s11684-015-0426-7.
- Ando M, Iwata A, Ozeki Y, Tsuchiya K, Akiba T, Nihei H. Circulating platelet-derived microparticles with procoagulant activity may be a potential cause of thrombosis in uremic patients. Kidney Int. 2002;62(5):1757–1763. doi: 10.1046/j.1523-1755.2002.00627.x.
- Kohli S, Ranjan S, Hoffmann J, Kashif M, Daniel EA, Al-Dabet MM, Bock F, Nazir S, Huebner H, Mertens PR, et al. Maternal extracellular vesicles and platelets promote preeclampsia via inflammasome activation in trophoblasts. Blood. 2016;128(17):2153–2164. doi: 10.1182/blood-2016-03-705434.
- Nomura S, Tandon NN, Nakamura T, Cone J, Fukuhara S, Kambayashi J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis. 2001;158(2):277–287. doi: 10.1016/s0021-9150(01)00433-6.
- Olausson N, Mobarrez F, Wallen H, Westerlund E, Hovatta O, Henriksson P. Microparticles reveal cell activation during IVF - a possible early marker of a prothrombotic state during the first trimester. Thromb Haemost. 2016;116(3):517–523. doi: 10.1160/TH15-12-0970.
- Shomer E, Katzenell S, Zipori Y, Sammour RN, Isermann B, Brenner B, Aharon A. Microvesicles of women with gestational hypertension and preeclampsia affect human trophoblast fate and endothelial function. Hypertension. 2013;62(5):893–898. doi: 10.1161/HYPERTENSIONAHA.113.01494.
- Helal O, Defoort C, Robert S, Marin C, Lesavre N, Lopez-Miranda J, Risérus U, Basu S, Lovegrove J, McMonagle J, et al. Increased levels of microparticles originating from endothelial cells, platelets and erythrocytes in subjects with metabolic syndrome: relationship with oxidative stress. Nutr Metab Cardiovasc Dis. 2011;21(9):665–671. doi: 10.1016/j.numecd.2010.01.004.
- Brisson AR, Tan S, Linares R, Gounou C, Arraud N. Extracellular vesicles from activated platelets: a semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 2017;28(3):263–271. doi: 10.1080/09537104.2016.1268255.
- Laffont B, Corduan A, Plé H, Duchez A-C, Cloutier N, Boilard E, Provost P. Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood. 2013;122(2):253–261. doi: 10.1182/blood-2013-03-492801.
- Milbank E, Soleti R, Martinez E, Lahouel B, Hilairet G, Martinez MC, Andriantsitohaina R, Noireaud J. Microparticles from apoptotic RAW 264.7 macrophage cells carry tumour necrosis factor-α functionally active on cardiomyocytes from adult mice. J Extracell Vesicles. 2015;4(1):28621. doi: 10.3402/jev.v4.28621.
- Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124(14):2173–2183. doi: 10.1182/blood-2014-05-573543.
- Mobarrez F, He S, Bröijersen A, Wiklund B, Antovic A, Antovic J, Egberg N, Jörneskog G, Wallén H. Atorvastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb Haemost. 2011;106(2):344–352. doi: 10.1160/TH10-12-0810.
- Zhang W, Zha C, Lu X, Jia R, Gao F, Sun Q, Jin M, Liu Y. Anti-β2GPI/β2GPI complexes induce platelet activation and promote thrombosis via p38mapk: a pathway to targeted therapies. Front Med. 2019;13(6):680–689. doi: 10.1007/s11684-018-0673-5.
- Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214. doi: 10.1111/j.1600-065X.2011.01044.x.
- Walle VL, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568–572. doi: 10.1016/j.cub.2016.02.019.
- Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. doi: 10.1038/s41577-019-0228-2.
- Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, Qiu J, Rong X, Shi Z, Xiao J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–318. doi: 10.1016/j.intimp.2018.12.028.
- Wang Y, Zhu X, Yuan S, Wen S, Liu X, Wang C, Qu Z, Li J, Liu H, Sun L, et al. TLR4/NF-κB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front Endocrinol (Lausanne). 2019;10:603. 10.3389/fendo.2019.00603.
- Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza FA, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122(20):3405–3414. doi: 10.1182/blood-2013-05-504449.
- Mitra S, Exline M, Habyarimana F, Gavrilin MA, Baker PJ, Masters SL, Wewers MD, Sarkar A. Microparticulate caspase 1 regulates gasdermin D and pulmonary vascular endothelial cell injury. Am J Respir Cell Mol Biol. 2018;59(1):56–64. doi: 10.1165/rcmb.2017-0393OC.
- Yan J, Bao H, Fan YJ, Jiang ZL, Qi YX, Han Y. Platelet-derived microvesicles promote endothelial progenitor cell proliferation in intimal injury by delivering TGF-β1. FEBS J. 2020;287(23):5196–5217. doi: 10.1111/febs.15293.
- Zhou Q, Lian Y, Zhang Y, Li L, Li H, Shen D, Zhou Y, Zhang M, Lu Y, Liu J, et al. Platelet-derived microparticles from recurrent miscarriage associated with antiphospholipid antibody syndrome influence behaviours of trophoblast and endothelial cells. Mol Hum Reprod. 2019;25(8):483–494. doi: 10.1093/molehr/gaz019.
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x.
- Orrenius S, Nicotera P, Zhivotovsky B. Cell death mechanisms and their implications in toxicology. Toxicol Sci. 2011;119(1):3–19. doi: 10.1093/toxsci/kfq268.
- Erkan D, Willis R, Murthy VL, Basra G, Vega J, Ruiz-Limón P, Carrera AL, Papalardo E, Martínez-Martínez LA, González EB, et al. A prospective open-label pilot study of fluvastatin on proinflammatory and prothrombotic biomarkers in antiphospholipid antibody positive patients. Ann Rheum Dis. 2014;73(6):1176–1180. doi: 10.1136/annrheumdis-2013-203622.
- Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, Salmon JE, Shoenfeld Y, Shovman O, Hunt BJ. Antiphospholipid syndrome [published correction appears in Nat Rev Dis Primers. 2018 Jan 25;4:18005]. Nat Rev Dis Primers. 2018;4:17103. doi: 10.1038/nrdp.2017.103.
- Zhou H, Ling S, Yu Y, Wang T, Hu H. Involvement of annexin A2 in anti-beta2gpi/beta2gpi-induced tissue factor expression on monocytes. Cell Res. 2007;17(8):737–739. doi: 10.1038/cr.2007.33.
- Xia L, Zhou H, Hu L, Xie H, Wang T, Xu Y, Liu J, Zhang X, Yan J. Both NF-κB and c-Jun/AP-1 involved in anti-β2GPI/β2GPI-induced tissue factor expression in monocytes. Thromb Haemost. 2013;109(4):643–651. doi: 10.1160/TH12-09-0655.
- Zha C, Zhang W, Gao F, Xu J, Jia R, Cai J, Liu Y. Anti-β2GPI/β2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology. 2018;138:140–150. doi: 10.1016/j.neuropharm.2018.06.001.
- Milioli M, Ibáñez-Vea M, Sidoli S, Palmisano G, Careri M, Larsen MR. Quantitative proteomics analysis of platelet-derived microparticles reveals distinct protein signatures when stimulated by different physiological agonists. J Proteomics. 2015;121:56–66. doi: 10.1016/j.jprot.2015.03.013.
- Shai E, Rosa I, Parguiña AF, Motahedeh S, Varon D, García Á. Comparative analysis of platelet-derived microparticles reveals differences in their amount and proteome depending on the platelet stimulus. J Proteomics. 2012;76:287–296. doi: 10.1016/j.jprot.2012.02.030.
- Zhang Y, Zhang W, Zha C, Liu Y. Platelets activated by the anti-β2GPI/β2GPI complex release microRnas to inhibit migration and tube formation of human umbilical vein endothelial cells. Cell Mol Biol Lett. 2018;23(1):24. doi: 10.1186/s11658-018-0091-3.
- Kaptan K, Beyan C, Ifran A, Pekel A. Platelet-derived microparticle levels in women with recurrent spontaneous abortion. Int J Gynaecol Obstet. 2008;102(3):271–274. doi: 10.1016/j.ijgo.2008.04.007.
- Wei D, Wu Q, Shi H. Apoptosis and p53 expression in the placental villi of females with unexplained recurrent spontaneous abortion. Exp Ther Med. 2014;7(1):191–194. doi: 10.3892/etm.2013.1399.
- Vikerfors A, Mobarrez F, Bremme K, Holmström M, Ågren A, Eelde A, Bruzelius M, Antovic A, Wallén H, Svenungsson E. Studies of microparticles in patients with the antiphospholipid syndrome (APS). Lupus. 2012;21(7):802–805. doi: 10.1177/0961203312437809.
- Willemze R, Bradford RL, Mooberry MJ, Roubey RA, Key NS. Plasma microparticle tissue factor activity in patients with antiphospholipid antibodies with and without clinical complications. Thromb Res. 2014;133(2):187–189. doi: 10.1016/j.thromres.2013.11.027.
- Jiang J, Kao CY, Papoutsakis ET. How do megakaryocytic microparticles target and deliver cargo to alter the fate of hematopoietic stem cells? J Control Release. 2017;247:1–18. doi: 10.1016/j.jconrel.2016.12.021.
- Michael JV, Wurtzel JGT, Mao GF, Rao AK, Kolpakov MA, Sabri A, Hoffman NE, Rajan S, Tomar D, Madesh M, et al. Platelet microparticles infiltrating solid tumors transfer miRnas that suppress tumor growth. Blood. 2017;130(5):567–580. doi: 10.1182/blood-2016-11-751099.
- Zhang Y, Ma KL, Gong YX, GH Wang, ZB Hu, Liu L, Lu J, PP Chen, CC Lu, XZ Ruan, BC Liuet al. Platelet microparticles mediate glomerular endothelial injury in early diabetic nephropathy [published correction appears in J Am Soc Nephrol. 2019 Jan;30(1):182]. J Am Soc Nephrol. 2018;29(11):2671–2695. doi: 10.1681/ASN.2018040368.
- Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat Immunol. 2017;18(8):861–869. doi: 10.1038/ni.3772.
- Lu F, Lan Z, Xin Z, He C, Guo Z, Xia X, Hu T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J Cell Physiol. 2020;235(4):3207–3221. doi: 10.1002/jcp.29268.
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. Cutting edge: nF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–791. doi: 10.4049/jimmunol.0901363.
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–617. doi: 10.4049/jimmunol.1100613.
- Ceroi A, Delettre FA, Marotel C, Gauthier T, Asgarova A, Biichle S, Duperrier A, Mourey G, Perruche S, Lagrost L, et al. The anti-inflammatory effects of platelet-derived microparticles in human plasmacytoid dendritic cells involve liver X receptor activation. Haematologica. 2016;101(3):e72–76. doi: 10.3324/haematol.2015.135459.
- Li DF, Chang X, Zhao JL, Chen X-M, Xu Z-L, Zhang D-G, Wu B-H, Wang L-S, Bai Y, Yao J. Colonic epithelial PHLPP2 deficiency promotes colonic epithelial pyroptosis by activating the NF-κB signaling pathway.Oxid Med Cell Longev. 2021;2021:5570731. doi: 10.1155/2021/5570731.
- Li Z, Huang Z, Zhang H, Lu J, Tian Y, Wei Y, Yang Y, Bai L. et al. P2X7 receptor induces pyroptotic inflammation and cartilage degradation in osteoarthritis via NF-κB/NLRP3 crosstalk. Oxid Med Cell Longev. 2021;2021:8868361. doi: 10.1155/2021/8868361.
- Ge G, Bai J, Wang Q, Liang X, Tao H, Chen H, Wei M, Niu J, Yang H, Xu Y, et al. Punicalagin ameliorates collagen-induced arthritis by downregulating M1 macrophage and pyroptosis via NF-κB signaling pathway. Sci China Life Sci. 2022;65(3):588–603. doi: 10.1007/s11427-020-1939-1.
- Chen F, Chen ZQ, Zhong GL, Zhu JJ. Nicorandil inhibits TLR4/MyD88/NF-κB/NLRP3 signaling pathway to reduce pyroptosis in rats with myocardial infarction. Exp Biol Med (Maywood). 2021;246(17):1938–1947. doi: 10.1177/15353702211013444.
- Li X, Zou Y, Fu YY, Xing J, Wang K-Y, Wan P-Z, Wang M, Zhai X-Y. Ibudilast attenuates folic acid–induced acute kidney injury by blocking pyroptosis through TLR4-mediated NF-κB and MAPK signaling pathways. Front Pharmacol. 2021;12:650283. doi: 10.3389/fphar.2021.650283.