
Abstract
Acquired disorders of platelet function are an underdiagnosed cause of bleeding tendency. A 14-year-old girl developed moderate mucocutaneous bleeding two weeks after a Mycoplasma pneumoniae infection successfully treated with clarithromycin. The patient was referred to us 7 months later for laboratory investigation of the persisting bleeding diathesis. The patient’s personal and family histories were negative for bleeding disorders. Complete blood count, von Willebrand Factor levels and coagulation tests were normal; platelet aggregation, ATP secretion, δ-granules content and serum thromboxane B2 levels were defective. At follow-up visits, laboratory parameters and the bleeding diathesis progressively normalized within 2 years. The patient’s condition is compatible with a diagnosis of acquired Storage Pool Deficiency (SPD), associated with defective thromboxane A2 production. To our knowledge, this is the first case of acquired, transient SPD with spontaneous remission. The pathogenic role of Mycoplasma pneumoniae infection or clarithromycin is possible, albeit uncertain.
Introduction
Disorders of platelet function – whether congenital or acquired – are associated with bleeding tendency and are underdiagnosed in clinical practice.Citation1 The most common congenital platelet function disorders are characterized by impairment of platelet secretion, caused by qualitative/quantitative defects in platelet granules or by abnormalities in signal transduction.Citation1 The term Storage Pool Deficiency (SPD) is referred to disorders caused by deficiency in platelet granules, which can be selective for alpha (α)- or delta (δ)-granules, or involve both types. Selective deficiency of α-granules has been identified in rare congenital disorders only, such as, for instance, the Gray Platelet Syndrome.Citation1 The most common congenital abnormality of δ-granules is δ-Storage Pool Deficiency (δ-SPD), characterized by a defect in the number and/or content of δ-granules, which can be syndromic or non-syndromic.Citation2 The lack of platelet δ-granules constituents (serotonin, adenine nucleotides, calcium and pyrophosphate) is associated with mucocutaneous bleeding of variable degree, ranging from mild to moderate. Mild thrombocytopenia can be observed in 20–40% of affected patients.Citation3 Abnormalities of platelet aggregation, characterized by lack of amplifying effects by secreted ADP, are of variable severity and may be absent in about 25% of affected patients.Citation4 The diagnosis of δ-SPD is based on the measurement of δ-granule constituents, or by inspection of platelets by transmission electron microscopy.Citation5 Cases of acquired SPD have been reported, which were most commonly associated with immune or hematological diseases.Citation6 Normalization or improvement of the acquired platelet defect never occurred spontaneously, but in association with successful treatment of the underlying disease. Here we report the case of a patient with acquired bleeding diathesis and SPD, associated with defective platelet production of thromboxane A2 (TxA2), temporally associated with Mycoplasma pneumoniae infection and treatment with clarithromycin, which normalized spontaneously within 2 years.
Case description
A 14-year old girl from Sicily was referred to our outpatient clinic in October 2014 for a moderate bleeding diathesis. She had experienced spontaneous bruising, bleeding from minor wounds, and frequent episodes of epistaxis and menorrhagia, which manifested for the first time in March 2014 in the absence of any clinically overt disorders (including immunologic and hematologic diseases), with the exception of a Mycoplasma pneumoniae infection which was diagnosed by means of a serological test 2 weeks before and was successfully treated with clarithromycin 500 mg twice daily for 21 days.
At the time of patient referral, the bleeding assessment tool score of the International Society on Thrombosis and Haemostasis was 6 (normal values for subjects under 18 years of age: 0–2Citation7); physical examination revealed a large bluish-purple ecchymosis on the left leg and few smaller resolving bruises on the lower limbs.
The patient’s personal and family histories of bleeding were negative. All parameters of blood count, including the platelet count (208×109/L) and blood cells morphology, were normal. Prothrombin time, activated partial thromboplastin time, von Willebrand factor (vWF) antigen and ristocetin cofactor activity were within their normal ranges. The closure time of the collagen/epinephrine cartridge of Platelet Function Analyzer-100 (PFA-100) was prolonged (>300 seconds; normal range: 76–178 seconds), while that of the collagen/ADP cartridge was normal (93 seconds; normal range: 60–116 seconds), compatible with the presence of a platelet function abnormality and normal vWF activity.Citation8 The following parameters of platelet function were investigated: I) platelet aggregation and ATP secretion induced by several agonists, measured simultaneously by lumi-aggregometry (Chrono-log series 560 Havertown, PA, USA) in citrated platelet-rich plasma prepared as previously described,Citation9,Citation10 according to SCC/ISTH recommendationsCitation11; II) platelet content of serotonin, measured by spectrofluorometry,Citation12 adenosine triphosphate (ATP) and adenosine diphosphate (ADP), measured by luminometryCitation13; III) phosphorylation of vasodilator-stimulated phosphoprotein (VASP) induced by prostaglandin E1 (flow cytometry); IV) serum levels of thromboxane B2 (TxB2, stable metabolite of the platelet agonist TxA2), measured by a selective, competitive enzyme immunoassay (Thromboxane B2 EIA kit, Cayman Chemicals, Ann Arbor, MI, USA).
Platelet aggregation and ATP secretion induced by ADP (2, 4 and 20 µM), epinephrine (10 µM), thrombin receptor activating peptide (TRAP, 10 and 20 µM), collagen (2, 4 and 10 µg/mL) and a thromboxane A2 analogue (U46619, 1 µM) were partially or severely impaired (, ). Only platelet aggregation induced by arachidonic acid (1 mM) was within the normal range. The platelet levels of serotonin and ADP were low and the ATP/ADP ratio was high, indicative of defective δ-granules content (). Serum T×B2 levels were lower than normal (45 pmol/108 platelets, normal values 63–472 pmol/108 platelets) (). Phosphorylation of VASP induced by prostaglandin E1 was normal (PRI: 75%), thus ruling out defects of the P2Y12 receptor for ADP.Citation14 Platelet aggregation, ATP secretion, ADP and serotonin contents were normal in the patient’s parents. In a follow-up visit 4 months later (February 2015), most platelet parameters normalized or improved, although some of them remained pathological (, ). Clinically, the hemorrhagic manifestations had improved, and only spontaneous ecchymoses were present. At the next follow-up visit in March 2016 the bleeding manifestations had disappeared and did not relapse thereafter. Subsequent testing in March 2016 showed normalization of platelet function, with δ-granule contents and serum T×B2 levels within their normal ranges, which persisted in two subsequent follow-up visits (, ).
Figure 1. Platelet aggregation and ATP secretion of citrated PRP from the patient performed during the first visit (October 2014), and subsequent follow-up visits in February 2015 and June 2021, compared to control. Platelets were stimulated with ADP (A), collagen (B) and TRAP (C). Platelet serotonin and adenine nucleotides contents (nmol/108 platelets) at each visit are reported in the table.
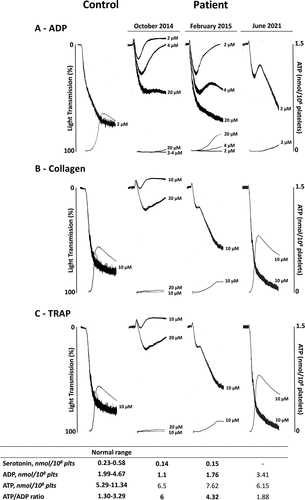
Table I. Results of the platelet function tests in the studied patient over time.
Discussion
Based on these findings and on the absence of pathological bleeding in the remote patient’s history, we hypothesized that the patient was affected by an acquired, transient form of SPD associated with defective T×A2 production. The platelet aggregation abnormalities of our patient, similarly to those of congenital δ-SPD, are a consequence of defective amplification by secreted ADP.Citation9 They are characterized by normal first-wave aggregation in response to adenosine diphosphate (ADP) and epinephrine, diminished or absent second-wave aggregation, and impaired aggregation induced by secretion-inducing agonists, such as collagen and TRAP. Moreover, defective T×A2 production, which contributes with secreted ADP to the amplification of platelet aggregation in normal platelets, could have played an additional role in impairing the aggregation response of our patient’s platelets. Combined defects of δ-granules and T×A2 production have not been described previously in either congenitalCitation15 or acquired forms of SPD, suggesting that the pathogenic mechanism of the acquired defect observed in our patient was not selective for a particular platelet target. The observation that platelet aggregation induced by arachidonic acid (the precursor of TxA2) was normal in our patient, despite partially defective T×A2 production, suggests that either the patient's platelet phospholipase A2 activity was defective or that the amount of T×A2 produced by platelets upon their exposure to high concentrations of exogenous arachidonic acid (1 mM) was sufficient to support normal platelet aggregation. In the absence of alternative potential causes of acquired SPD, we postulate a causal relationship with Mycoplasma pneumoniae infection and/or with the exposure of the patient to clarithromycin. Mycoplasma pneumoniae causes upper and lower respiratory tract infections especially in children and young adults, which may associate with extrapulmonary clinical manifestations, including thrombocytopenia and autoimmune hemolytic anemia.Citation16 Given that acquired SPD may have an autoimmune genesis in some cases, one plausible hypothesis could be that immunoglobulins generated during Mycoplasma pneumoniae infection or its pharmacological treatment could have caused the platelet defect. Alternative pathogenic mechanisms can be hypothesized, such as direct or indirect toxic effects on the megakaryocytic lineage. Although there have been reports of thrombocytopenia secondary to Mycoplasma pneumoniaeCitation17 or macrolide treatment,Citation18 no cases of acquired platelet dysfunction have been described so far.
In summary, we report the case of a young girl with acquired bleeding diathesis, normal platelet count and transient severe and complex platelet function defect characterized by acquired SPD and impaired T×A2 production, with spontaneous complete remission. The temporal association with Mycoplasma pneumoniae infection and treatment with clarithromycin could suggest, but not prove, a cause-effect relationship with the acquired platelet defect.
Author contributions statement
M.S., E.A.F., C.G., A.F. and E.B. performed the laboratory analyses. M.S. and B.C. wrote the first draft of the manuscript. M.C. and G.M.P. designed the study, contributed to data interpretation and critically reviewed and edited the manuscript. All authors read and approved the final manuscript.
Acknowledgments
The authors are thankful to the patient and her family and to the nursing staff of the Hematology Day Hospital of Presidio Ospedaliero San Paolo, ASST Santi Paolo e Carlo, whose cooperation was essential for the collection of samples for this study. The authors acknowledge the support of the APC central fund of the University of Milan.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The raw data supporting the results of this study will be made available upon reasonable request.
Additional information
Funding
References
- Cattaneo M. Inherited disorders of platelet function. In: Michelson A, Cattaneo M, Frelinger A Newman P. editors. Platelets. 4th ed. London, UK: Elsevier - Academic Press; 2019. p. 877–5.
- Flaumenhaft R, Sharda A. Platelet secretion. In: Michelson A, Cattaneo M, Frelinger A Newman P. editors. Platelets. 4th ed. London, UK: Elsevier - Academic Press; 2019. p. 349–70.
- Balduini A, Di Buduo CA, Malara A, Lecchi A, Rebuzzini P, Currao M, Pallotta I, Jakubowski JA, Cattaneo M. Constitutively released adenosine diphosphate regulates proplatelet formation by human megakaryocytes. Haematologica. 2012;97(11):1657–65. doi:10.3324/haematol.2011.059212.
- Nieuwenhuis HK, Akkerman JW, Sixma JJ. Patients with a prolonged bleeding time and normal aggregation tests may have storage pool deficiency: studies on one hundred six patients. Blood. 1987;70(3):620–3. doi:10.1182/blood.V70.3.620.620.
- Cattaneo M. Inherited platelet-based bleeding disorders. J Thromb Haemost. 2003;1(7):1628–36. doi:10.1046/j.1538-7836.2003.00266.x.
- Bacci M, Ferretti A, Marchetti M, Alberelli MA, Falanga A, Lodigiani C, De Candia E. Autoimmune disorders of platelet function: systematic review of cases of acquired glanzmann thrombasthenia and acquired delta storage pool disease. Blood Transfus. 2022;20(5):420–32. doi:10.2450/2021.0119-21.
- Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, Hopman W, Clark DS, Mauer AC, Bowman M. et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20(6):831–5. doi:10.1111/hae.12503.
- Podda GM, Bucciarelli P, Lussana F, Lecchi A, Cattaneo M. Usefulness of PFA-100 testing in the diagnostic screening of patients with suspected abnormalities of hemostasis: comparison with the bleeding time. J Thromb Haemost. 2007;5(12):2393–8. doi:10.1111/j.1538-7836.2007.02752.x.
- Cattaneo M. Light transmission aggregometry and ATP release for the diagnostic assessment of platelet function. Semin Thromb Hemost. 2009;35(2):158–67. doi:10.1055/s-0029-1220324.
- Cattaneo M, Hayward CP, Moffat KA, Pugliano MT, Liu Y, Michelson AD. Results of a worldwide survey on the assessment of platelet function by light transmission aggregometry: a report from the platelet physiology subcommittee of the SSC of the ISTH. J Thromb Haemost. 2009;7(6):1029. doi:10.1111/j.1538-7836.2009.03458.x.
- Cattaneo M, Cerletti C, Harrison P, Hayward CPM, Kenny D, Nugent D, Nurden P, Rao AK, Schmaier AH, Watson SP. et al. Recommendations for the standardization of light transmission aggregometry: a consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J Thromb Haemost. 2013 Apr 10; 11(6):1183–9. doi:10.1111/jth.12231.
- Drummond AH, Gordon JL. Letter: rapid, sensitive microassay for platelet 5HT. Thromb Diath Haemorrh. 1974;31(2):366–7. doi:10.1055/s-0038-1649172.
- Harker LA, Zimmerman TS. Platelet dense granule and lysosome content. Measurement of Platelet Function: Methods in Haematology. Churchill Livingstone; 1983. p. 92–114.
- Zighetti ML, Carpani G, Sinigaglia E, Cattaneo M. Usefulness of a flow cytometric analysis of intraplatelet vasodilator-stimulated phosphoprotein phosphorylation for the detection of patients with genetic defects of the platelet P2Y(12) receptor for ADP. J Thromb Haemost. 2010;8(10):2332–4. doi:10.1111/j.1538-7836.2010.04002.x.
- Podda G, Femia EA, Pugliano M, Cattaneo M. Congenital defects of platelet function. Platelets. 2012;23(7):552–63. doi:10.3109/09537104.2012.724737.
- Narita M. Classification of extrapulmonary manifestations due to mycoplasma pneumoniae infection on the basis of possible pathogenesis. Front Microbiol. 2016;28(7):23. doi:10.3389/fmicb.2016.00023.
- Aviner S, Miskin H, London D, Horowitz S, Schlesinger M. Mycoplasma pneumonia infection: a possible trigger for immune thrombocytopenia. Indian J Hematol Blood Transfus. 2011;27(1):46–50. doi:10.1007/s12288-011-0054-6.
- Rossi M, Capecchi M, Lazzerini PE. Roxithromycin-associated acute thrombocytopenia. Am J Case Rep. 2021;30(22):e932039. doi:10.12659/AJCR.932039.