
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
A clorprenaline (CLP) monoclonal antibody, 4G1, raised against the CLP-hapten was successfully prepared for the development of indirect competitive enzyme-linked immunosorbent assay (ic-ELISA) and immunochromatographic assays for the detection of CLP in pig urine samples. The half maximal inhibitory concentration of the antibody was 0.435 ng/mL, and the limit of detection was 0.104 ng/mL. The linear range was 0.104–1.818 ng/mL, and cross-reactivity with the CLP analogue was <5%. In the spiked sample and recovery test, the recovery ranged from 81.8% to 100.5%, indicating that the ic-ELISA was suitable for CLP analysis in pig urine. The critical value of the immunochromatographic strip in pig urine was 10 ng/mL and could be used for semi-quantitative analysis of CLP. Thus, this immunochromatographic strip was suitable for quickly and sensitively detecting CLP residues in pig urine samples.
Introduction
β2-agonists can significantly promote protein synthesis and fat decomposition in animals, and are often used as animal feed additives to raise the lean meat rate of keeping animals (Caloni, Montana, Pasqualucci, Brambilla, & Pompa, Citation1995; Zhou, Wang, Zhang, Ji, & Yang, Citation2017). However, metabolism of β2-agonists is slow and accumulation leads to toxicity (Bosak, Knežević, Smilović, Šinko, & Kovarik, Citation2017). When this meat is introduced to the human body, it is harmful to a person’s health (Liu, Ousmane, Gan, Wu, & Li, Citation2017). Clorprenaline (CLP) is a selective β2-agonist, also known as chlortane and often used as a substitute for clenbuterol (Yan, Liu, Gao, & Sun, Citation2013). Its chemical name is 1-(2-chlorophenyl)-2-isopropylamino-ethanol and its molecular formula is C11H16ClNO. It has a molecular weight of 213.7, and its sulfate is white or almost white crystalline powder (Shao, Zhang, Li, Yue, & Chen, Citation2016). CLP can be used to treat symptoms such as animal cold. When it reaches a certain dose in the animal body, it results in nutrient redistribution in animals similar to other β2-agonists (Lu, Luo, Huang, & Li, Citation2011; Wang, Zeng, Wang, Guo, & He, Citation2016). Based on this feature, it will be used as a new type of animal growth-promoting agent. Because of a lack of public attention, clenbuterol is often overlooked in routine testing, which brings a potential threat to animal food safety (Xiao et al., Citation2016). In 2013, it was discovered that in China clenbuterol was being illegally added to veterinary drugs, the culprit was also known as CLP. It was added in the “30% tilmicosin injection” and six other veterinary drugs that can treat animal cold, resulting in CLP residues in animals (Peng, Zhang, et al., Citation2014).
When consumers eat meat products with excess CLP residues, they will present with significant symptoms of poisoning, including the clinical manifestations of muscle tremor, limb weakness, fever, headache, nausea and so on (Zhou et al., Citation2014). At present, there are liquid chromatography-tandem mass spectrometry (LC-MS) (Wang et al., Citation2013; Xiong, Gao, Li, Yang, & Shimo, Citation2015), capillary electrophoresis (Yao, Song, et al., Citation2017; Zhou et al., Citation2008) and ion chromatography (Shen, Ouyang, Baeyens, Delanghe, & Yang, Citation2005) assays to detect these residues. However, the instrumental analysis methods are time-consuming, costly and complicated, and cannot meet the requirement of fast on-site inspection (Peilong, Ximeng, Xiaoou, & Ruohua, Citation2015; Wang, Zeng, Wang, & He, Citation2016). Moreover, the literature describing the rapid detection of CLP residues in animal foods is still non-existent. Therefore, to promote the healthy development of the animal husbandry industry in China, it is imperative to establish a quick and simple method for sensitive detection of CLP residues.
Immunoassays such as enzyme-linked immunosorbent assay (ELISA) (Bergmann et al., Citation2017), colloidal gold (CG) immunoassay technology (Yang et al., Citation2015) and other methods are well suited to meet the needs of the market because of their high sensitivity and fast detection speed. In 1971, Engvall & Perlmann (Citation1971) established an ELISA for the first time. As the name implies, the immunological antigen and antibody reaction and biochemical enzyme–catalytic substrate reaction were combined successfully (Bai et al., Citation2017). Thus, in this study, we prepared an anti-CLP monoclonal antibody (mAb) of high affinity and produced an immunochromatographic assay based on the mAb to detect CLP in pig urine samples.
Materials and methods
Chemicals and apparatus
CLP, salbutamol, clenbuterol, ractopamine, cimaterol, brombuterol hydrochloride, formoterol, mabuterol and terbutaline were bought from J&K Scientific (Shanghai, China). Bovine serum albumin (BSA; MW 67,000 Da), ovalbumin (OVA; MW 43,000 Da) and keyhole limpet haemocyanin (MW 5,000,000 Da) were purchased from Sigma Chemical Company (St. Louis, MO, USA). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), Freund’s complete adjuvant, Freund’s incomplete adjuvant and 3,3′,5,5′-tetramethylbenzidine (TMB) were obtained from Sigma–Aldrich (St. Louis, MO, USA). Goat anti-mouse immunoglobulin antibody was purchased from Jackson ImmunoResearch Laboratories. RPMI-1640 cell culture medium, hypoxanthine–aminopterin–thymidine supplement (HAT), hypoxanthine–thymidine supplement (HT) and foetal calf serum were purchased from Gibco (Shanghai, China). All the reagents used in this study were analytical or HPLC grade. Nitrocellulose (NC) high-flow-plus membrane (Pura-bindRP), including sample pad (CB-SB08), polyvinylchloride (PVC) backing card and absorption pad (SX18) was obtained from GoldBio Tech Co. (Shanghai, China).
Buffers and solutions
The following buffers were used in the ELISA: (1) standard dilution buffer was phosphate-buffered saline (PBS, 0.01M, pH 7.4); (2) washing buffer was PBS containing 0.05% Tween 20 (v/v); (3) coating solution was 0.05M carbonate–bicarbonate buffer (CB, pH 9.6); (4) blocking buffer was prepared by adding CB and 0.2% gelatin (w/v); (5) antibody dilution buffer consisted of PBS and 0.1% gelatin (w/v); (6) substrate buffer was composed of 0.01% TMB (w/v), 0.3% H2O2 (v/v) and phosphate buffer; and (7) stop reagent was 2M sulfuric acid.
Hapten (CLP) synthesis
In , 20 g of compound 1 was diluted into 150 mL ethyl alcohol and added to 4 mL H2SO4. The reaction mixture was stirred at 80°C overnight and then poured into ice water and extracted with ethyl acetate. The organic layer was collected and washed with brine, dried over Na2SO4 and evaporated to give compound 2. Compound 2 was diluted into 230 mL dichloromethane, then 19 mL acetyl chloride and 76 g of AlCl3 (in 750 mL dichloromethane) were added and the mixture was stirred at room temperature for 4 h. The reaction mixture was poured into ice water and extracted with dichloromethane. The organic layer was collected, washed with brine, dried over Na2SO4 and evaporated to give compound 3 as a yellow oil. The obtained compound 3 was diluted into 150 mL MeOH and 12 g N-bromosuccinimide was added and reacted at 60°C with stirring for 1 h.
Figure 1. The synthesis of CLP-hapten.
The mixture was added to water (200 mL) and extracted with 100 mL ethyl acetate three times. The organic layer was concentrated to the crude product. The residue was purified over a column (ethyl acetate/petroleum ether = 1/5) to yield product 4 as a yellow oil. Product 4 (5 g) was diluted in 50 mL dichloromethane and 2 g isopropylamine was added and stirred at room temperature for 2 h. The reaction mixture was poured into ice water and extracted with dichloromethane. The organic layer was washed with brine, dried over Na2SO4 and evaporated to give the crude compound 5. Compound 5 (2 g) in 20 mL methyl alcohol was added to 0.4 g NaBH4 at 0°C and stirred at room temperature for 2 h. The reaction mixture was added to water and extracted with ethyl acetate. The organic layer was concentrated to the crude product. The residue was purified over a column (ethyl acetate/petroleum ether = 1/2) to generate product 6 as a yellow oil. Compound 6 (1 g) in 10 mL Tetrahydrofuran and 1 mL water was added to 12 g LiOH·H2O and stirred at room temperature overnight. The mixture was concentrated under vacuum to generate the crude product, which was subjected to preparative HPLC to give the product JNDX-3-LR (0.7 g) as a colourless oil with a yield of 73.7%.
The structure of the hapten was verified with LC-MS.
Preparation and identification of the antigen
In general, a method for coupling a hapten to a protein is selected based on the small molecule or the active group of the derivative. For example, a small molecule containing a benzene ring directly attached to an amino group is directly coupled using a diazo method; a carboxyl group may be coupled using the 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide method (EDC) (Kong, Liu, Song, Kuang, & Xu, Citation2017) or a mixed acid anhydride method (MA). The hapten is coupled to the carrier protein to obtain complete antigen, and the antigen is characterized by gel electrophoresis or UV spectrophotometry.
CLP-hapten contains a carboxyl group as an active group, so the carbodiimide reaction was chosen to couple with the carrier protein. CLP-hapten (5 mg) was dissolved in 1 mL of 0.1M MES buffer (pH 7.4), and 11.3 mg EDC and 6.8 mg NHS were added to the hapten solution. The reaction solution was stirred overnight at room temperature. The activating solution went from turbid to clean to obtain the active solution we needed. Then 5 mg OVA and 5 mg BSA were dissolved in 2 mL of 0.05M CB buffer (pH 9.6) each. The activated solution was divided between the two proteins; the protein solutions were slowly added dropwise. The pH of the two reaction solutions was adjusted with 1M NaOH to pH 9.0. The reaction was stirred overnight in the dark. Finally, the dialysis bag and dialysis folder were boiled in pure water for 15 min. The reaction solution was put in the dialysis bag and dialysed against PBS for 3 days. After dialysis, the immunogen and coating antigen were stored at −20°C until use. The final conjugate was characterized with SDS-PAGE.
Monoclonal antibody preparation
BALB/c mice (6–8 weeks of age) were immunized via subcutaneous injection. After three immunizations, the mouse sera were tested using the indirect ELISA method. The mice with a high-serum antibody titre and low 50% inhibitory concentration (IC50) were selected for cell fusion. The experiment used the PEG fusion method. On the third day after fusion, the cells were transferred to HAT selection medium. Five days after fusion, the cells were treated with HT medium. The supernatants from the hybridomas were analysed with an indirect competitive (ic)-ELISA. Cells that can produce an antibody with high OD value, good affinity and specificity were selected for subcloning (Peng, Meng, et al., Citation2014).
Subcloning generally uses a limiting dilution method. Three subclones were tested to obtain a clone that produced the antibody that best inhibited CLP. The clone was expanded, cultured and injected intraperitoneally into BALB/c mice (6–8 weeks of age). The antibody from the mouse ascites was purified by octanoic acid–saturated ammonium sulfate method and dialysed against 0.01M PBS at 4°C for 48 h (El-Rm, Citation2006).
The sensitivity of the mAb was assessed for its 50% inhibition (IC50) and the limit of detection (LOD, IC10). IC50 values and LOD were measured using standard curves. The specificity of the antibody was evaluated by measuring the cross-reactivity (CR) (Hua et al., Citation2013).
CR was tested by the addition of a series of related analogues, including salbutamol, clenbuterol, ractopamine, cimaterol, brombuterol hydrochloride, formoterol, mabuterol and terbutaline.
Synthesis of gold nanoparticles
The glass instruments required for the experiment were soaked in aquaregia and washed several times with ultra-pure water. According to a previously reported method, gold nanoparticles were prepared with an average diameter of 30 nm (Chen, Liu, Kuang, Song, & Xu, Citation2013). Firstly, 100 mL of chloroauric acid (0.01 g/L) was heated and allowed to boil for 30 min. Next, 2 mL of trisodium citrate solution (1%) was added quickly and stirred continuously. Within 1 min, the colour of the reaction solution became wine red and was heated with stirring for 15 min until the reaction was complete. Finally, ultra-pure water was added to a final volume of 100 mL, cooled to room temperature and stored at 4°C for usage.
The gold nanoparticles were characterized by transmission electron microscopy scanning (TEM) and UV/Vis spectrometry. TEM was used to observe the uniformity, dispersion and appearance of gold nanoparticles. UV/Vis spectrometry was used to determine the average particle size. The gold nanoparticle solution was scanned at 300–800 nm, and the average size of the particles was judged by the wavelength where the maximum absorption peak was seen (Xu, Kong, Song, Liu, & Kuang, Citation2016).
Labelling the mAb with CG particles
The coupling between the gold nanoparticles and antibodies is based on electrostatic absorption. After adjusting 10 mL of CG solution to pH 8.8 with 0.1M K2CO3, the mAb was added to 1 mL of CG solution with continuous stirring.
The mixture was stirred continuously for 50 min at room temperature. Then, 1 mL of blocking buffer (0.5% casein, w/v) was added and the solution was stirred for 2 h. Next, the mixture was centrifuged at 8000 g for 25 min to remove the blocking agent and excess antibody. The soft sediment was collected and washed by gold-labelled resuspension buffer three times (20 mM Tris [pH 8.2], 0.1% PEG, 0.1% Tween, 5% sucrose, 5% trehalose, 0.2% BSA, 5% Brij). Finally, the mAb was reconstituted to a volume of 1 mL with gold-labelled resuspension buffer including 0.02% NaN3 and stored at 4°C until use.
Preparation of the immunochromatographic strip
Under the action of electrostatic absorption, the antibody with a positive charge was coupled with the CG nanoparticles of negative charge. The test strip is composed of an NC membrane that was fixed to the PVC backing pad, the bottom of the sample pad and the top absorption pad. Both of these were pressured in the NC membrane. To make the control line (C-line) and test line (T-line), 30 μL of goat anti-mouse antibody (1 mg/mL) and coating antigen (0.5 mg/mL) were sprayed on the NC film and dried at 37°C for 2 h. The distance between them was 0.5 cm to ensure that the reactants fully reacted. Test strips were cut 3 mm wide with a cutting machine for use (Chen et al., Citation2017). The gold-labelled antibody was immobilized on the solid support as previously described (Xing et al., Citation2015). Specifically, the gold-labelled antibody was dried into a powder, to be dissolved by the application of liquid samples. The sample was reacted at room temperature for about 3 min. The mixture was added to the test strip, migrating to the coating antigen within 5 min. The coating antigen can bind with the excess gold-labelled antibody.
The principle of immunochromatographic strip test
An immunochromatographic strip test is based on a labelling technique (Yao, Liu, Song, Kuang, & Xu, Citation2017). When the liquid samples were added to the sample pad, the result can be identified by the naked eye in 3–5 min. The T-line was combined with the coating antigen, and C-line was the goat anti-mouse IgG antibody. Through capillary action, the sample solution reacted with gold-labelled antibody after it was added to the sample pad and migrated towards the test line. If there is no small molecule to be tested in the sample, then the gold-labelled antibody will bind to the coating antigen at T-line. The colour of the test line became red and the sample was negative. If the sample to be tested contained the small molecule of interest, it competed with the coating antigen for the gold-labelled antibody. The colour of the test line became weaker, until it ultimately disappeared.
Test procedure of the immunochromatographic strip assay
The sample solution was added to the sample pad, the solutions reacted with the gold-labelled antibody and migrated upwards by capillary action. After 3–5 min, if both the C- and T-lines appeared dark red, the sample was negative. If the C-line was darker, the T-line appeared lighter or even completely free of colour, then the sample being tested was positive (Peng, Liu, Kuang, Cui, & Xu, Citation2016).
Sample analysis
Samples were prepared in order to identify the monoclonal antibody which was resistant to urine matrix interference and demonstrated high affinity for the antigen (Liu, Liu, et al., Citation2017). In this experiment, pig urine samples were used as a negative sample during mouse serum detection and cell detection operation. The experiment was divided into two steps to identify whether the pig urine samples were negative. First, kits (such as R-Biopharm AG, Germany) were purchased to do a simple screening. Second, samples were sent to the Jiangsu Provincial Entry-Exit Inspection and Quarantine Bureau to be confirmed.
Results and discussion
Identification of the hapten
From the hapten identification chromatogram shown in (a), the main product was retained for 1.449 min and the main UV peak also appeared at 1.449 min. Additionally, the signal was strong, indicating that the purity of the sample was high. The hapten contained a carboxyl reactive group, so the detection was carried out in the negative ion state. From the mass spectrum shown in (b), the corresponding molecular ion peak at 1.449 min was 265.9, and the major fragment ion added was 267.0. Element composition was consistent with CLP-hapten, which indicated that the synthesis of CLP-hapten was successful with high purity.
Figure 2. LC-MS analysis of CLP-hapten.
Characterization of the immunogen and coating antigen
Since the CLP-hapten had no UV-absorbing group, it was impossible to use the migration of the UV absorption peak to determine whether the protein conjugation was successful, this could only be determined by denaturing gel electrophoresis. This method is mainly based on the molecular weight of the protein for its separation. A large amount of SDS binds to the denatured protein or the hydrophobic part of the polypeptide, eliminating the differences in the protein charge and forming a non-folded derivative with a negative charge. Under the action of the electric field, the protein migrates from negative to positive. The greater the molecular weight, the slower the migration speed. If the small molecule was successfully coupled with the protein, the molecular weight of the final conjugate will be heavier than the original carrier protein, resulting in the protein-hapten conjugate band migrating slower than the unconjugated carrier protein electrophoresis band. This can be used as a qualitative measure to determine the success of coupling.
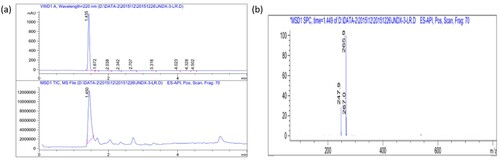
SDS-PAGE of BSA, immunogen, OVA and coating antigen are shown in . The electrophoresis direction was from top to bottom. It was apparent that the migration speed of the CLP-hapten and the carrier protein was slower than the protein, and the conjugate bands were behind the electrophoresis bands of the carrier bands. Due to the successful coupling between the small molecule and a carrier protein, the conjugate molecular weight increased. This indicated that the immunogen and the coating antigen were successfully coupled with the carrier proteins.
Figure 3. Polyacrylamide gel electrophoresis image of immunogen (a) (1 = BSA; 2/3 = MCLP-hapten : MBSA (80:1;120:1)) and coating antigens (b) of CLP (1 = MCLP-hapten: MOVA (30:1); 2 = OVA).
Sensitivity and specificity of mAb 4G1
The sensitivity and specificity of mAb 4G1 were determined by ic-ELISA. A standard curve () was constructed under optimized conditions. As shown in , the IC50 of 4G1 was 0.435 ng/mL, the LOD was calculated to be 0.052 ng/mL and the linear range of detection (IC20–IC80) was 0.104–1.818 ng/mL. The purified CLP-4G1 mAb was used for the immunochromatographic strip assay. The CR was used to determine whether the antibody can specifically recognize the analyte. To determine the specificity of the anti-CLP antibody, the CR with salbutamol, clenbuterol, ractopamine, cimaterol, brombuterol, formoterol, mabuterol and terbutaline was measured. The CR of 4G1 to all of the analogues tested was <1%, indicating that the mAb is highly specific for CLP and could be used for subsequent experiments ().
Figure 4. The standard curve of developed Ic-ELISA method. IC50 of 4G1 was 0.095 ng/mL, LOD was 0.013 ng/mL and the linear range of detection was from 0.0258 to 0.356 ng/mL.
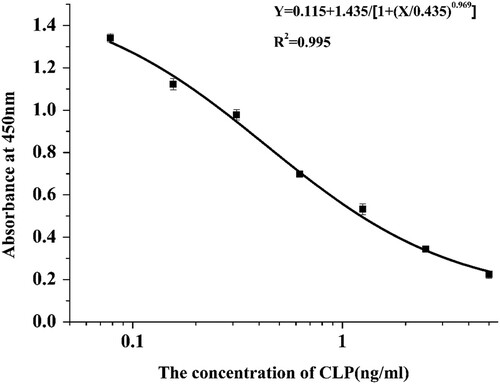
Table 1. Cross-reaction results of McAb 4G1.
Analytical characteristics of the immunochromatographic strip for CLP
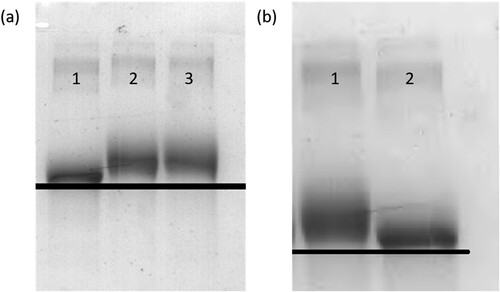
In this experiment, pig urine was selected as the matrix for the samples, and the results of the test were judged by the specific line value. A series of pig urine samples spiked with CLP (0, 0.1, 0.25, 0.5, 1, 2.5, 5 and 10 ng/mL) were prepared to test the cut-off values (). When the concentration of CLP was 0.1 ng/mL, the colour of the test line began to change. The test line became weaker when the concentration was 10 ng/mL. Therefore, the LOD was 0.1 ng/mL and the cut-off value was 10 ng/mL.
Figure 5. Image of detection by clorprenaline immunochromatographic strip in urine. A series of urine samples spiked clorprenaline were subjected to the immunochromatographic strip test. 1 = 0 ng/mL, 2 = 0.1 ng/mL, 3= 0.25 ng/mL, 4 = 0.5 ng/mL, 5= 1 ng/mL, 6 = 2.5 ng/mL, 7 = 0.5 ng/mL, 8 = 10 ng/mL. Cut-off value was 10 ng/mL.

Recovery test in pig urine samples
Based on the linear range of the CLP antibody, the concentration of CLP in the negative pig urine samples was 0.3, 0.6 and 1.2 ng/mL. The ic-ELISA was used to measure the recovery (). The recovery rate of pig urine samples ranged from 81.8% to 100.5%, indicating that the kit based on the preparation of CLP-4G1 antibody has good stability and accuracy, and can be applied to the detection of CLP in samples.
Table 2. Recovery of clorprenaline in urine by ic-ELISA and strip assay (n = 3).
Conclusion
In the present study, a novel hapten of CLP was synthesized. An mAb, 4G1, of high specificity and great sensitivity was prepared successfully using the CLP-hapten. Both ic-ELISA and immunochromatographic assays showed high sensitivity and stability in the analysis of spiked urine samples. In addition, the results of the immunochromatographic assays appeared in 5–10 min. Thus, the antibody can be used to detect CLP in pig urine samples, and immunochromatographic assays are more sensitive and faster than instrumental analysis methods.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Bai, Y., Hu, J., Liu, S., Zhang, W., Zhang, J., He, J., & Li, P. (2017). Production of antibodies and development of an enzyme-linked immunosorbent assay for 17β-estradiol in milk. Food & Agricultural Immunology, 28(6), 1519–1529.
- Bergmann, S. M., Wang, Q., Zeng, W., Li, Y., Wang, Y., Matras, M. … Morin, T. (2017). Validation of a KHV antibody enzyme-linked immunosorbent assay (ELISA). Journal of Fish Diseases, 40, 1511–1527. doi: https://doi.org/10.1111/jfd.12621
- Bosak, A., Knežević, A., Smilović, G., Šinko, G., & Kovarik, Z. (2017). Resorcinol-, catechol- and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. Journal of Enzyme Inhibition & Medicinal Chemistry, 32(1), 789–797. doi: https://doi.org/10.1080/14756366.2017.1326109
- Caloni, F., Montana, M., Pasqualucci, C., Brambilla, G., & Pompa, G. (1995). Detection of β2-agonists in milk replacer. Veterinary Research Communications, 19(4), 285–293. doi: https://doi.org/10.1007/BF01839311
- Chen, Y., Guo, L., Liu, L., Song, S., Kuang, H., & Xu, C. (2017). Ultrasensitive immunochromatographic strip for fast screening of 27 sulfonamides in honey and pork liver samples based on a monoclonal antibody. Journal of Agricultural & Food Chemistry, 65, 8248–8255. doi: https://doi.org/10.1021/acs.jafc.7b03190
- Chen, X., Liu, L., Kuang, H., Song, S., & Xu, C. (2013). A strip-based immunoassay for rapid determination of fenpropathrin. Analytical Methods, 5(21), 6234–6239. doi: https://doi.org/10.1039/c3ay41030g
- El-Rm, R. (2006). Comparison between therapeutic antitoxin F(ab)2 fractionated with ammonium sulfate and caprylic acid. Journal of Immunoassay & Immunochemistry, 27(4), 319. doi: https://doi.org/10.1080/15321810600861993
- Engvall, E., & Perlmann, P. (1971). Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry, 8(9), 871–874. doi: https://doi.org/10.1016/0019-2791(71)90454-X
- Hua, K., Liu, L., Xu, L., Wei, M., Guo, L., Wang, L., & Xu, C. (2013). Development of an enzyme-linked immunosorbent assay for dibutyl phthalate in liquor. Sensors, 13(7), 8331–8339. doi: https://doi.org/10.3390/s130708331
- Kong, D., Liu, L., Song, S., Kuang, H., & Xu, C. (2017). Development of sensitive, rapid, and effective immunoassays for the detection of vitamin B 12 in fortified food and nutritional supplements. Food Analytical Methods, 10(1), 1–9. doi: https://doi.org/10.1007/s12161-016-0543-1
- Liu, R., Liu, L., Song, S., Cui, G., Zheng, Q., Kuang, H., & Xu, C. (2017). Development of an immunochromatographic strip for the rapid detection of 10 β-agonists based on an ultrasensitive monoclonal antibody. Food & Agricultural Immunology, 28(4), 625–638. doi: https://doi.org/10.1080/09540105.2017.1309358
- Liu, H., Ousmane, D., Gan, N., Wu, D., & Li, T. (2017). Novel stir bar array sorptive extraction coupled with gas chromatography–mass spectrometry for simultaneous determination of three β2-agonist residues in pork. Chromatographia, 80(3), 473–482. doi: https://doi.org/10.1007/s10337-017-3242-1
- Lu, C., Luo, Z., Huang, L., & Li, X. (2011). Cheminform abstract: The Ru-catalyzed enantioselective preparation of chiral halohydrins and their application in the synthesis of (R)-clorprenaline and (S)-sotalol. Cheminform, 42(48), 722–727.
- Peilong, W., Ximeng, L., Xiaoou, S., & Ruohua, Z. (2015). Sensitive detection of β-agonists in pork tissue with novel molecularly imprinted polymer extraction followed liquid chromatography coupled tandem mass spectrometry detection. Food Chemistry, 184, 72–79. doi: https://doi.org/10.1016/j.foodchem.2015.03.073
- Peng, J., Liu, L., Kuang, H., Cui, G., & Xu, C. (2016). Development of an icELISA and immunochromatographic strip for detection of norfloxacin and its analogs in milk. Food & Agricultural Immunology, 28(2), 1–11.
- Peng, J., Meng, X., Deng, X., Zhu, J., Kuang, H., & Xu, C. (2014). Development of a monoclonal antibody-based sandwich ELISA for the detection of ovalbumin in foods. Food & Agricultural Immunology, 25(1), 1–8. doi: https://doi.org/10.1080/09540105.2012.716398
- Peng, T., Zhang, F. S., Yang, W. C., Li, D. X., Chen, Y., Xiong, Y. H., … Lai, W. H. (2014). Lateral-flow assay for rapid quantitative detection of clorprenaline residue in swine urine. Journal of Food Protection, 77(10), 1824–1829. doi: https://doi.org/10.4315/0362-028X.JFP-14-103
- Shao, X., Zhang, J., Li, D., Yue, J., & Chen, Z. (2016). Preparation of Fe2O3-clorprenaline/tetraphenylborate nanospheres and their application as ion selective electrode for determination of clorprenaline in pork. Nanoscale Research Letters, 11(1), 178. doi: https://doi.org/10.1186/s11671-016-1388-7
- Shen, S., Ouyang, J., Baeyens, W. R. G., Delanghe, J. R., & Yang, Y. (2005). Determination of β2-agonists by ion chromatography with direct conductivity detection. Journal of Pharmaceutical and Biomedical Analysis, 38(1), 166–172. doi: https://doi.org/10.1016/j.jpba.2004.11.060
- Wang, L. Q., Zeng, Z. L., Wang, Z., Guo, J. Y., & He, L. M. (2016). Influence of water in samples on residues analysis of beta-agonists in porcine tissues and urine using liquid chromatography tandem mass spectrometry. Food Analytical Methods, 9(7), 1904–1911. doi: https://doi.org/10.1007/s12161-015-0359-4
- Wang, L. Q., Zeng, Z. L., Wang, Z., & He, L. M. (2016). Correction function for biased results due to matrix effects in residue analysis of beta-agonists in porcine tissues and urine with LC-MS/MS. Rsc Advances, 6(32), 26967–26976. doi: https://doi.org/10.1039/C5RA28050H
- Wang, L., Zeng, Z., Wang, X., Yang, J., Chen, Z., & He, L. (2013). Multiresidue analysis of nine β-agonists in animal muscles by LC-MS/MS based on a new polymer cartridge for sample cleanup. Journal of Separation Science, 36(11), 1843–1852. doi: https://doi.org/10.1002/jssc.201201088
- Xiao, Y. P., Hai-Yan, L. U., Shao-Jun, LÜ, Xie, S. X., Wang, Z. Z., & Chen, H. W. (2016). Rapid analysis of trace salbutamol and clenbuterol in pork samples by mass spectrometry. Chinese Journal of Analytical Chemistry, 44(11), 1633–1638. doi: https://doi.org/10.1016/S1872-2040(16)60968-4
- Xing, C., Liu, L., Song, S., Feng, M., Kuang, H., & Xu, C. (2015). Ultrasensitive immunochromatographic assay for the simultaneous detection of five chemicals in drinking water. Biosensors & Bioelectronics, 66(66C), 445–453. doi: https://doi.org/10.1016/j.bios.2014.12.004
- Xiong, L., Gao, Y.-Q., Li, W.-H., Yang, X.-L., & Shimo, S. P. (2015). Simple and sensitive monitoring of β2-agonist residues in meat by liquid chromatography–tandem mass spectrometry using a QuEChERS with preconcentration as the sample treatment. Meat Science, 105(Suppl. C), 96–107. doi: https://doi.org/10.1016/j.meatsci.2015.03.013
- Xu, C., Kong, D., Song, S., Liu, L., & Kuang, H. (2016). Development of an immunochromatographic strip for the semi-quantitative and quantitative detection of biotin in milk and milk products. Analytical Methods, 8(7), 1595–1601. doi: https://doi.org/10.1039/C5AY02659H
- Yan, H., Liu, S., Gao, M., & Sun, N. (2013). Ionic liquids modified dummy molecularly imprinted microspheres as solid phase extraction materials for the determination of clenbuterol and clorprenaline in urine. Journal of Chromatography A, 1294(11), 10. doi: https://doi.org/10.1016/j.chroma.2013.04.024
- Yang, X. D., Wang, F. Y., Song, C. M., Wu, S. Y., Zhang, G. P., & Zeng, X. Y. (2015). Establishment of a lateral flow colloidal gold immunoassay strip for the rapid detection of estradiol in milk samples. LWT – Food Science and Technology, 64(1), 88–94. doi: https://doi.org/10.1016/j.lwt.2015.04.022
- Yao, L., Liu, L., Song, S., Kuang, H., & Xu, C. (2017). Development of indirect competitive enzyme-linked immunosorbent and immunochromatographic strip assays for carbofuran detection in fruits and vegetables. Food & Agricultural Immunology, 28(4), 639–651. doi: https://doi.org/10.1080/09540105.2017.1309359
- Yao, Y., Song, P., Wen, X., Deng, M., Wang, J., & Guo, X. (2017). Chiral separation of 12 pairs of enantiomers by capillary electrophoresis using heptakis-(2,3-diacetyl-6-sulfato)-β-cyclodextrin as the chiral selector and the elucidation of the chiral recognition mechanism by computational methods. Journal of Separation Science, 40, 2999–3007. doi: https://doi.org/10.1002/jssc.201700137
- Zhou, C., Xu, P., Tang, K.-W., Jiang, X.-Y., Yang, T., Zhang, P.-L., & Zhu, Z. (2014). Enantioselective extraction of clorprenaline enantiomers with hydrophilic selector of sulfobutylether-β-cyclodextrin by experiment and modeling. Journal of Central South University, 21(3), 891–899. doi: https://doi.org/10.1007/s11771-014-2015-3
- Zhou, L., Wang, Q., Zhang, Y., Ji, Y., & Yang, X. (2017). Aquatic photolysis of β2-agonist salbutamol: Kinetics and mechanism studies. Environmental Science & Pollution Research International, 24(6), 5544. doi: https://doi.org/10.1007/s11356-016-8207-7
- Zhou, S., Wang, Y., De, B. T., Baeyens, W. R., Fei, G. T., Dilinuer, M., & Ouyang, J. (2008). Simultaneous separation of eight beta-adrenergic drugs using titanium dioxide nanoparticles as additive in capillary electrophoresis. Electrophoresis, 29(11), 2321–2329. doi: https://doi.org/10.1002/elps.200700589