
ABSTRACT
Artemisia is one of the largest genera of the family Asteraceae or Compositae, consisting of 500 species. Some Artemisia species, such as Artemisia afra, A. sacrorum, and A. annua, have been widely used as traditional medicine to treat inflammatory and malarial diseases. However, the biological activity of A. montana has not been broadly studied. Therefore, in this study, we investigated the anti-inflammatory activity of A. montana leaf extract (ALE) and its molecular mechanisms in lipopolysaccharide-activated RAW 264.7 cells. Non-cytotoxic concentrations of ALE significantly reduced the expression of inducible nitric oxide (NO) synthase and cyclooxygenase-2, resulting in the decrease in NO and prostaglandin E2. Moreover, ALE inhibited the production of tumour necrosis factor-α and interleukin-6. We also observed that ALE treatment repressed mitogen-activated kinase pathways by inhibiting the phosphorylation of extracellular signal-regulated kinase, c-Jun N-terminal kinase, and p38, suggesting that ALE is a therapeutic candidate to treat inflammatory diseases.
Introduction
Artemisia is one of the largest genera of the family Asteraceae or Compositae, consisting of 500 species; it mainly grows in the temperate regions of Asia, Europe, and North America (Abad, Bedoya, Apaza, & Bermejo, Citation2012; Pandey & Singh, Citation2017). As a folk medicine, Artemisia species have shown a wide range of pharmacological activities, such as antitussive, analgesic, antihypertensive, antitoxic, antiviral, antimalarial, and anti-inflammatory activities (Bora & Sharma, Citation2011; Tan, Zheng, & Tang, Citation1998).
Artemisia afra, one of the most popular species in South Africa, contains rich terpenes and has valuable biological activities (Afolayan & Sunmonu, Citation2011; Ntutela et al., Citation2009). The methanol extract of A. afra aerial parts possesses suppressive activity against microorganisms and inflammation (Bora & Sharma, Citation2011; van Wyk, Citation2008). A. asiatica Nakai, a common species in East Asia, has been used as a food ingredient and traditional medicine. Studies have shown that ethanol extracts of A. asiatica have antioxidative, anti-inflammatory, and antiviral activities (Bora & Sharma, Citation2011; Lim, Chung, Lee, Lee, & Suk, Citation2008). A. annua, one of the most famous medicinal plants in China, has been used for treating malaria (Daddy et al., Citation2017). The Chinese pharmaceutical chemist, Tu Youyou, a Nobel laureate, extracted artemisinin from A. annua leaves, and found that it possessed antimalarial activity (Miller & Su, Citation2011; Tu, Citation2011). Artemisinin has a lactone structure with an unusual peroxide bridge, and artemisinin and its endoperoxide derivatives have been widely used for the treatment of malaria (Klonis et al., Citation2011; Miller & Su, Citation2011; Tu, Citation2011). In addition, ethanol extracts of A. annua exhibited antiulcerogenic activity in ethanol- and indomethacin-induced rat ulcer models (Bora & Sharma, Citation2011; Foglio et al., Citation2002; He et al., Citation2009).
A. montana is a perennial plant, growing in woods and grassy fields in Japan, China, and Korea. A. montana has been used in cuisine and as a moxibustion, a traditional heat therapy in China and Korea, which involves the burning of mugwort on the body to facilitate healing. In addition, aerial parts of A. montana have been used in folk medicine as antihypertensive, anti-hepatotoxic, and antihaemorrhoid agents (Kim, Citation1996). Previous studies have shown that A. montana has antioxidant and anti-obesity activities (Jung et al., Citation2011; Kim, Kim, Chung, & Choi, Citation2000). However, the anti-inflammatory activity of A. montana has only been tested by measuring nitric oxide (NO) production following A. montana extract treatment (Choi, Park, Lee, & Kim, Citation2013). Therefore, we have more comprehensively investigated the anti-inflammatory effects of A. montana leaf extract (ALE) and its underlying molecular mechanisms in murine macrophage cells.
Materials and methods
Cell culture
RAW 264.7 cells, derived from murine macrophages, were cultured in Dulbecco’s Modified Eagle’s Medium (Welgene, Gyeongsan-si, Gyeongsangbuk-do, Korea) supplemented with 10% foetal bovine serum (Welgene), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, Grand Island, NY, USA) at 37°C in a humidified atmosphere containing 5% CO2.
A. montana (Nakai) Pamp leaf ethanol extract (ALE)
An 80% ethanol extract of A. montana (Nakai) Pamp leaves was obtained from Jeju Biodiversity Research Institute (JBRI) (Code No: JBRI-10666). The extract was dissolved in dimethyl sulfoxide (DMSO; Duchefa Biochemie, Haarlem, The Netherlands) to prepare an 80 mg/mL stock solution. The stock solution was diluted to produce final concentrations ≤200 μg/mL. The final concentration of DMSO in the culture medium did not exceed 0.5%.
Cell viability assay
RAW 264.7 cells were seeded overnight at a density of 5 × 104 cells/well in a 96-well plate in 100 µL cell culture medium. The cells were treated with 0–200 μg/mL of ALE for 4 h, washed with cold phosphate-buffered saline (PBS), and stimulated with 1 μg/mL LPS (Sigma-Aldrich, St. Louis, MO, USA) for 20 h. Ten microliters of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) was added to each well, followed by 100 μL of 0.04 N HCl in isopropanol, 5 h later. The optical density was measured at 540 nm to detect viable cells using an EMax® microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
RAW 264.7 cells were treated with ALE and LPS, and lysed using RNAiso (TAKARA BIO INC., Otsu, Shiga, Japan). Total RNA was then extracted and reverse transcribed into complementary DNA (cDNA) using M-MLV Reverse Transcriptase (Promega, Madison, WI, USA). TOPreal™ qPCR 2X Premix (Enzynomics, Daejeon, Korea) and a CFX Connect™ Real-Time PCR Detection System (Bio-Rad Laboratories, San Francisco, CA, USA) were used to perform qPCR as directed by the manufacturer’s instructions. The sequences of primers used for qPCR are listed in .
Table 1. The list of primers used for quantitative PCR.
Measurement of NO production
RAW 264.7 cells were treated with ALE for 4 h, washed with cold PBS, and stimulated with LPS for 20 h. Fifty microliters of culture supernatant was then collected and mixed with 50 μL of Griess reagent (Sigma-Aldrich). The optical density was measured at 540 nm, and NO production was quantified using sodium nitrite (NaNO₂; Junsei Chemical CO., Chuo-ko, Tokyo, Japan) to generate a nitrite standard reference curve.
Enzyme-linked immunosorbent assay (ELISA)
A 96-well immunoplate was coated with purified rat anti-mouse interleukin-6 (IL-6) antibody (Ab; eBioscience, San Diego, CA, USA) or purified Armenian hamster anti-mouse/rat tumour necrosis factor-α (TNF-α) Ab (eBioscience) at 4°C overnight. The wells were rinsed with PBST (PBS with 0.05% Tween® 20), blocked with PBS containing 3% bovine serum albumin (BSA; MP Biomedicals, Solon OH, USA) at 25°C for 2 h, and washed with PBST. After washing, cytokine standards and samples were loaded into each well, incubated at 4°C overnight, and washed with PBST. Biotinylated anti-mouse IL-6 Ab (BD Biosciences) or biotinylated anti-mouse TNF-α Ab (eBioscience) were added to each well, incubated for 45 min, and washed with PBST. Streptavidin-conjugated AKP (alkaline phosphatase; BD Biosciences, San Diego, USA) was added for 30 min, followed by washing and incubation with phosphatase substrate, p-nitrophenyl phosphate (Sigma-Aldrich). The optical density was measured at 405 nm. The level of each cytokine was quantified using a standard curve generated with recombinant murine TNF-α (Peprotech, Rocky Hill, NJ, USA) or IL-6 (Peprotech).
The production of prostaglandin E2 (PGE2) was measured using a PGE2 ELISA kit (Cayman Chemical, Ann Arbor, MI, USA), following the manufacturer’s instructions.
Western blotting
RAW 264.7 cells were seeded at a density of 1 × 107 cells/mL per well in a 6-well plate, pretreated with ALE, and stimulated with LPS for the indicated length of time. The cells were then lysed and protein was extracted using a RIPA lysis and extraction buffer (Thermo Fisher Scientific, Rockford, IL, USA). Protein concentration was determined by using a Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific), and the western blotting protocol was continued as previously described (Kim, Jeong, Lee, & Min, Citation2017). Each membrane was incubated with appropriate primary antibodies. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (Bio-Rad) was used as a secondary antibody, and West-Q Femto Clean ECL Solution (Gen Depot, Barker, TX, USA) was used as a substrate. To detect and quantify protein bands on the membranes, a ChemiDoc™ XRS densitometer and Quantity One software (Bio-Rad) were used. The primary antibodies that were used detected: inducible NO synthase (iNOS; Santa Cruz Biotechnology, Dallas, TX, USA), cyclooxygenase-2 (COX-2; Bioss Antibodies, Woburn, MA, USA), β-tubulin (Cell Signalling, Danvers, MA, USA), extracellular signal–regulated kinase (ERK, Cell Signalling), phosphorylated ERK (p-ERK, Cell Signalling), c-Jun N-terminal kinase (JNK, Cell Signalling), phosphorylated JNK (p-JNK, Cell Signalling), p38 (Cell Signalling), phosphorylated p38 (p-p38, Cell Signalling), inhibitor of κBα (IκBα, Cell Signalling), phosphorylated IκBα (p-IκBα; Thermo Fisher Scientific), signal transducer and activator of transcription 3 (STAT-3, Cell Signalling), and phosphorylated STAT-3 (p-STAT-3, Cell Signalling).
Statistical analysis
All data are shown as means ± standard deviations (S.D). Experiments were repeated four to six times, and representative images were chosen. Difference between treatments was tested using a one-way analysis of variance (ANOVA) followed by Tukey’s post-hock test. p < .05 was considered statistically significant.
Results and discussion
Cytotoxicity assay of ALE
An MTT assay was performed to determine the cytotoxicity of ALE in RAW 264.7 cells. As shown in , ALE did not show cytotoxic effects at concentrations up to 200 μg/mL, compared to that of DMSO controls. Thus, the anti-inflammatory effects of ALE were subsequently evaluated at concentrations ≤200 μg/mL.
Figure 1. Cytotoxicity of ALE. RAW 264.7 cells were treated with 25, 50, 100, or 200 μg/mL of ALE for 4 h and stimulated with LPS (1 μg/mL) for 20 h. Cell viability was measured by MTT assay and plotted as fold changes relative to LPS-treated cells (white bar), which was arbitrarily set to a value of 1.0. The data are expressed as means ± S.D. (n = 4).
Inhibition of iNOS expression and NO production
Upon microbial infection or inflammation, activated macrophages produce iNOS leading to a massive generation of NO, a major inflammatory mediator (Bogdan, Röllinghoff, & Diefenbach, Citation2000; Suleria, Addepalli, Masci, Gobe, & Osborne, Citation2017). To test the anti-inflammatory effects of ALE, iNOS expression was measured using RT-qPCR and western blot. The results show that ALE significantly reduced the expression of iNOS both at the mRNA and protein levels in RAW 264.7 cells upon LPS stimulation (A and B). In addition, ALE treatment decreased the production of NO, a major mediator and indicator of inflammation, in LPS-stimulated cells in a concentration-dependent manner (C).
Figure 2. Decrease in iNOS expression and NO production. RAW 264.7 cells were treated with 25, 50, 100, or 200 μg/mL of ALE for 4 h and stimulated with LPS (1 μg/mL) for 20 h. Total RNA and protein were extracted from the cells and analysed for iNOS mRNA (A) and protein (B) expression, respectively. The cell culture media was also collected for measuring NO using the Griess reaction (C). Representative western blots are shown in part (B). The data are expressed as means ± S.D. (n = 4). *p < .05, **p < .01, ***p < .001 compared to LPS-treated cells.
A previous study indicated that methanol extracts of various Artemisia species, such as A. stolonifera, A. selengensis, A. capillaris, and A. keiskeana, decreased NO production in LPS-stimulated RAW 264.7 cells (Choi et al., Citation2013). However, a methanol extract of A. montana did not affect NO production, while ALE, an ethanol extract of A. montana leaves, exerted a significant inhibitory effect on NO production, even at 25 μg/mL, suggesting that differences in extraction solvent and plant parts result in distinct biological activities.
Inhibition of COX-2 expression and PGE2 production
In addition to NO, PGE2 is another inflammatory mediator that induces fever and vasodilation. PGE2 is synthesized by sequential oxygenation of arachidonic acid by cyclooxygenases (COX-1 and COX-2) and prostaglandin E synthase during inflammation (Ahn et al., Citation2015; Echizen, Hirose, Maeda, & Oshima, Citation2016; Yu, Lao, & Zheng, Citation2016). As shown in , ALE remarkably reduced the expression of COX-2 in LPS-stimulated macrophages at both mRNA and protein levels in a concentration-dependent manner. In accordance with the decreased COX-2 expression, the level of PGE2 (PGE2) produced by COX-2 decreased from 631.0 ± 70.53 pg/mL to 248.33 ± 47.65 pg/mL after treatment with 200 μg/mL ALE.
Figure 3. Decrease in COX-2 expression and PGE2 production. ALE suppressed the expression of COX-2 at the mRNA (A) and protein (B) levels, and the production of PGE2 (C). PGE2 present in the cell culture media was measured by ELISA. Representative western blots are shown in part (B). The data are expressed as means ± S.D. (n = 4). *p < .05, **p < .01, ***p < .001 compared to LPS-treated cells.
The decrease in PGE2 production was also observed with a methanol extract of a leaf mixture of A. argyi and A. princeps, an ethanol extract of A. asiatica Nakai, and a supercritical carbon dioxide extract of A. annua. These findings support the prevailing anti-inflammatory activity of Artemisia species, including A. montana, through the suppression of PGE2 production (Hunt, Yoshida, Davis, Greenhill, & Davis, Citation2015; Jeong et al., Citation2014; Yun et al., Citation2016).
Inhibition of proinflammatory cytokine production
One of the hallmarks of an inflammatory response is the production of proinflammatory cytokines, such as TNF-α and IL-6, by macrophages (Im et al., Citation2016). Similarly, LPS treatment was shown previously to stimulate the production of TNF-α and IL-6 in RAW 264.7 cells (Wu & Schauss, Citation2012). In order to confirm the anti-inflammatory effects of ALE, the secretion of TNF-α and IL-6 was measured by ELISA. As shown in , LPS treatment induced the secretion of TNF-α and IL-6. However, the induction of these pro-inflammatory cytokines was repressed by ALE treatment in a concentration-dependent manner, which has been detected in other Artemisia species, such as A. argyi, A. princeps, A. absinthium, and A. asiatica (Jeong et al., Citation2014; Krebs, Omer, & Omer, Citation2010; Yun et al., Citation2016).
Figure 4. The inhibitory effect of ALE on cytokine production. RAW 264.7 cells were treated with 25, 50, 100, or 200 μg/mL of ALE for 4 h followed by LPS stimulation. ELISAs were used to measure the production of the proinflammatory cytokines, TNF-α (A) and IL-6 (B). The data are expressed as means ± S.D. (n = 4). **p < .01, ***p < .001 compared to LPS-treated cells.
Effects of ALE on mitogen-activated kinase (MAPK) signalling pathways
To investigate the effects of ALE on intracellular signalling pathways, the phosphorylation of MAPKs, including ERK, JNK, and p38 was assessed in ALE-treated RAW 264.7 cells. In addition, a downstream transcription factor, STAT-3, and IκBα, which mediates the nuclear factor-κB (NF-κB) signalling pathway, were evaluated in ALE-treated RAW 264.7 cells. As shown in A–C, the phosphorylation of ERK, p38, and JNK was decreased by ALE treatment in response to LPS stimulation. However, ALE treatment had no effect on the activation of IκBα and STAT-3, indicating that ALE controls inflammatory responses by inhibiting the activation of MAPKs, such as ERK, p38, and JNK.
Figure 5. The inhibitory effects of ALE on the activation of MAPK. ALE inhibited the phosphorylation of MAPK including ERK, p38, and JNK. Cells were treated with ALE for 4 h, followed by LPS stimulation (1 μg/mL) for 15 min. For STAT3 analysis, cells were treated with 10 ng/mL of IL-6 for 30 min. Total cell lysates were used, and the expression levels of ERK, p-ERK (A), p38, p-p38 (B) JNK, p-JNK (C), IκB, p-IκB (D), and STAT-3, p-STAT-3 (E) were determined by western blot analysis. p-MAPK expression was normalized to the corresponding total MAPK level. IκBα, p-IκBα, STAT-3, and p-STAT-3 expression levels were normalized to the corresponding β-tubulin level. The images shown are representative of three independent experiments with similar results. The intensity of protein bands from the western blots was quantified and expressed as bar graphs. Data are expressed as the means ± S.D. (n = 3). *p < .05, **p < .01 versus LPS-treated cells.
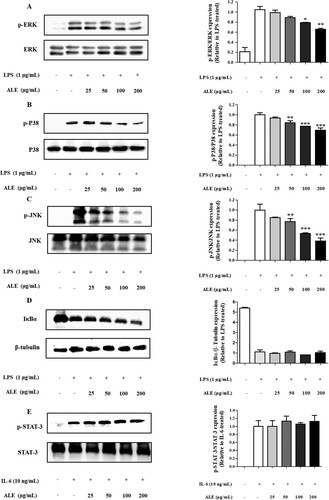
Although various Artemisia species have been shown to possess anti-inflammatory activities in common, the molecular mechanisms for these effects are varied. For example, a leaf extract of A. argyi and A. princeps inhibited phosphorylation of p38 but had no effect on the NF-κB pathway (Yun et al., Citation2016). However, A. asiatica ethanol extract suppressed the Src/Syk/NF-κB and TRAF6/JNK/AP-1 signalling pathways and JNK phosphorylation without affecting p38 or ERK. In addition, an ethyl acetate extract of A. anomala S. Moore inhibited the activation of MAPK signalling pathways, including JNK, ERK, and p38, along with the NF-κB signalling pathway (Tan, Wang, Yang, & Zhang, Citation2014). The disparity in underlying molecular mechanisms among diverse Artemisia species could be due to the differences in chemical constituents. For example, artemisinin, isolated from A. annua, exerts anti-inflammatory effects by repressing the NF-κB and MAPK pathways, accompanied by a decrease in phosphorylation of p38 and ERK without affecting JNK (Wang et al., Citation2017). Jaceosidin, which is present in A. argyi and leaves of A. montana, blocks the phosphorylation of ERK-1 and ERK-2 (Jeong, Lee, Yoon, & Lee, Citation2007; Kim, Lee, & Kim, Citation2013). Similarly, eupatilin, derived from A. asiatica and leaves of A. montana, was shown to inhibit the activation of Akt, as well as ERK-1 and ERK-2 (Kim et al., Citation2005; Kim et al., Citation2013). The combination of numerous compounds present in Artemisia extracts would result in anti-inflammatory activity through diverse molecular mechanisms.
In conclusion, this study show that ALE has anti-inflammatory activity in LPS-induced RAW 264.7 cells. ALE significantly reduced the production of NO, PGE2, and proinflammatory cytokines by repressing MAPK pathways without affecting IκBα and STAT-3, suggesting a potential therapeutic use of ALE for treating inflammatory diseases.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Abad, M. J., Bedoya, L. M., Apaza, L., & Bermejo, P. (2012). The artemisia L. genus: A review of bioactive essential oils. Molecules, 17(3), 2542–2566. doi: https://doi.org/10.3390/molecules17032542
- Afolayan, A. J., & Sunmonu, T. O. (2011). Artemisia Afra Jacq. Ameliorates oxidative stress in the pancreas of streptozotocin-induced diabetic Wistar rats. Bioscience, Biotechnology, and Biochemistry, 75(11), 2083–2086. doi: https://doi.org/10.1271/bbb.100792
- Ahn, S.-I., Lim, S. J., Gu, G.-J., Kim, J.-S., Paek, J. H., Kim, S., & Youn, H.-S. (2015). Eupartoium makinoi suppresses lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 expression. Food and Agricultural Immunology, 26(4), 496–503. doi: https://doi.org/10.1080/09540105.2014.968768
- Bogdan, C., Röllinghoff, M., & Diefenbach, A. (2000). The role of nitric oxide in innate immunity. Immunological Reviews, 173(1), 17–26. doi: https://doi.org/10.1034/j.1600-065X.2000.917307.x
- Bora, K. S., & Sharma, A. (2011). The genus artemisia: A comprehensive review. Pharmaceutical Biology, 49(1), 101–109. doi: https://doi.org/10.3109/13880209.2010.497815
- Choi, E., Park, H., Lee, J., & Kim, G. (2013). Anticancer, antiobesity, and anti-inflammatory activity of Artemisia species in vitro. Journal of Traditional Chinese Medicine, 33(1), 92–97. doi: https://doi.org/10.1016/S0254-6272(13)60107-7
- Daddy, N. B., Kalisya, L. M., Bagire, P. G., Watt, R. L., Towler, M. J., & Weathers, P. J. (2017). Artemisia annua dried leaf tablets treated malaria resistant to ACT and i.v. Artesunate: Case reports. Phytomedicine, 32(Supplement C), 37–40. doi: https://doi.org/10.1016/j.phymed.2017.04.006
- Echizen, K., Hirose, O., Maeda, Y., & Oshima, M. (2016). Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and toll-like receptor/MyD88 pathways. Cancer Science, 107(4), 391–397. doi: https://doi.org/10.1111/cas.12901
- Foglio, M. A., Dias, P. C., Antônio, M. A., Possenti, A., Rodrigues, R. A. F., da Silva, ÉF, & de Carvalho, J. E. (2002). Antiulcerogenic activity of some sesquiterpene lactones isolated from Artemisia annua. Planta Medica, 68(06), 515–518. doi: https://doi.org/10.1055/s-2002-32570
- He, S.-P., Tan, G.-Y., Li, G., Tan, W.-M., Nan, T.-G., Wang, B.-M., & Li, Q. X. (2009). Development of a sensitive monoclonalantibody-based enzyme-linked immunosorbent assay for the antimalaria active ingredient artemisinin in the Chinese herb Artemisia annua L. Analytical and Bioanalytical Chemistry, 393(4), 1297–1303. doi: https://doi.org/10.1007/s00216-008-2527-5
- Hunt, S., Yoshida, M., Davis, C. E. J., Greenhill, N. S., & Davis, P. F. (2015). An extract of the medicinal plant Artemisia annua modulates production of inflammatory markers in activated neutrophils. Journal of Inflammation Research, 8, 9–14. doi: https://doi.org/10.2147/JIR.S75484
- Im, K., Lee, J. Y., Byeon, H., Hwang, K. W., Kang, W., Whang, W. K., & Min, H. (2016). In vitro antioxidative and anti-inflammatory activities of the ethanol extract of eggplant (Solanum melongena) stalks in macrophage RAW 264.7 cells. Food and Agricultural Immunology, 27(6), 758–771. doi: https://doi.org/10.1080/09540105.2016.1150427
- Jeong, M. A., Lee, K. W., Yoon, D.-Y., & Lee, H. J. (2007). Jaceosidin, a pharmacologically active flavone derived from artemisia argyi, inhibits phorbol-ester-induced upregulation of COX-2 and MMP-9 by blocking phosphorylation of ERK-1 and -2 in cultured human mammary epithelial cells. Annals of the New York Academy of Sciences, 1095(1), 458–466. doi: https://doi.org/10.1196/annals.1397.049
- Jeong, D., Yi, Y. S., Sung, G. H., Yang, W. S., Park, J. G., Yoon, K., & Cho, J. Y. (2014). Anti-inflammatory activities and mechanisms of Artemisia asiatica ethanol extract. Journal of Ethnopharmacology, 152(3), 487–496. doi: https://doi.org/10.1016/j.jep.2014.01.030
- Jung, H. A., Islam, M. D. N., Kwon, Y. S., Jin, S. E., Son, Y. K., Park, J. J., & Choi, J. S. (2011). Extraction and identification of three major aldose reductase inhibitors from Artemisia montana. Food and Chemical Toxicology, 49(2), 376–384. doi: https://doi.org/10.1016/j.fct.2010.11.012
- Kim, T. (1996). Korean resources plants. Seoul: Seoul National University, pp. 169–169.
- Kim, J., Jeong, S. H., Lee, W., & Min, H. (2017). In vitro anti-inflammatory activity of Pothos scandens extract in RAW 264.7 cells. Food Science and Biotechnology, 26(3), 791–799. doi: https://doi.org/10.1007/s10068-017-0093-3
- Kim, N. M., Kim, J., Chung, H. Y., & Choi, J. S. (2000). Isolation of luteolin 7-O-rutinoside and esculetin with potential antioxidant activity from the aerial parts of Artemisia montana. Archives of Pharmacal Research, 23(3), 237–239. doi: https://doi.org/10.1007/BF02976451
- Kim, M. J., Kim, D. H., Na, H. K., Oh, T. Y., Shin, C. Y., & Surh Ph, D. P. Y. J. (2005). Eupatilin, a pharmacologically active flavone derived from Artemisia plants, induces apoptosis in human gastric cancer (AGS) cells. Journal of Environmental Pathology, Toxicology and Oncology, 24(4), 261–270. doi: https://doi.org/10.1615/JEnvironPatholToxicolOncol.v24.i4.30
- Kim, Y. J., Lee, J.-H., & Kim, S.-J. (2013). Cultivation characteristics and flavonoid contents of wormwood (Artemisia montana Pamp.). Journal of Agricultural Chemistry and Environment, 2(4), 6.
- Klonis, N., Crespo-Ortiz, M. P., Bottova, I., Abu-Bakar, N., Kenny, S., Rosenthal, P. J., & Tilley, L. (2011). Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proceedings of the National Academy of Sciences, 108(28), 11405–11410. doi: https://doi.org/10.1073/pnas.1104063108
- Krebs, S., Omer, T. N., & Omer, B. (2010). Wormwood (Artemisia absinthium) suppresses tumour necrosis factor alpha and accelerates healing in patients with Crohn’s disease – A controlled clinical trial. Phytomedicine, 17(5), 305–309. doi: https://doi.org/10.1016/j.phymed.2009.10.013
- Lim, B. O., Chung, H. G., Lee, W.-H., Lee, H. W., & Suk, K. (2008). Inhibition of microglial neurotoxicity by ethanol extract of Artemisia asiatica Nakai. Phytotherapy Research, 22(2), 279–282. doi: https://doi.org/10.1002/ptr.2304
- Miller, L. H., & Su, X. (2011). Artemisinin: Discovery from the Chinese herbal garden. Cell, 146(6), 855–858. doi: https://doi.org/10.1016/j.cell.2011.08.024
- Ntutela, S., Smith, P., Matika, L., Mukinda, J., Arendse, H., Allie, N., & Jacobs, M. (2009). Efficacy of Artemisia Afra phytotherapy in experimental tuberculosis. Tuberculosis (Edinb), 89(Suppl 1), S33–S40. doi: https://doi.org/10.1016/S1472-9792(09)70009-5
- Pandey, A. K., & Singh, P. (2017). The genus artemisia: A 2012–2017 literature review on chemical composition, antimicrobial, insecticidal and antioxidant activities of essential oils. Medicines (Basel), 4(3), 68. doi: https://doi.org/10.3390/medicines4030068
- Suleria, H. A. R., Addepalli, R., Masci, P., Gobe, G., & Osborne, S. A. (2017). In vitro anti-inflammatory activities of blacklip abalone (Haliotis rubra) in RAW 264.7 macrophages. Food and Agricultural Immunology, 28(4), 711–724. doi: https://doi.org/10.1080/09540105.2017.1310186
- Tan, X., Wang, Y.-L., Yang, X.-L., & Zhang, D.-D. (2014). Ethyl acetate extract of artemisia anomala S. Moore displays potent anti-inflammatory effect. Evidence-based Complementary and Alternative Medicine : eCAM, 2014, 681352.
- Tan, R. X., Zheng, W. F., & Tang, H. Q. (1998). Biologically active substances from the genus Artemisia. Planta Medica, 64(4), 295–302. doi: https://doi.org/10.1055/s-2006-957438
- Tu, Y. (2011). The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nature Medicine, 17(10), 1217–1220. doi: https://doi.org/10.1038/nm.2471
- van Wyk, B. E. (2008). A broad review of commercially important Southern African medicinal plants. Journal of Ethnopharmacology, 119(3), 342–355. doi: https://doi.org/10.1016/j.jep.2008.05.029
- Wang, K. S., Li, J., Wang, Z., Mi, C., Ma, J., Piao, L. X., & Jin, X. (2017). Artemisinin inhibits inflammatory response via regulating NF-κB and MAPK signaling pathways. Immunopharmacology and Immunotoxicology, 39(1), 28–36. doi: https://doi.org/10.1080/08923973.2016.1267744
- Wu, X., & Schauss, A. (2012). Mitigation of Inflammation with Foods.
- Yu, T., Lao, X., & Zheng, H. (2016). Influencing COX-2 activity by COX related pathways in inflammation and cancer. Mini-Reviews in Medicinal Chemistry, 16(15), 1230–1243. doi: https://doi.org/10.2174/1389557516666160505115743
- Yun, C., Jung, Y., Chun, W., Yang, B., Ryu, J., Lim, C., & Cho, S. I. (2016). Anti-inflammatory effects of artemisia leaf extract in mice with contact dermatitis in vitro and in vivo. Mediators of Inflammation, 2016, 8027537. doi: https://doi.org/10.1155/2016/8027537