
ABSTRACT
Okadaic acid (OA) and the analogs of dinophysistoxin (DTX) are important members of diarrhetic shellfish poisoning (DSP) toxins. In this study, five OA-specific mAbs (monoclonal antibodies) were developed from five stable cells of hybridoma. The anti-OA mAb-2D7 showed high sensitivity to OA, the IC50 of the antibody was 0.24 ng/mL, and its cross-reactivity was 91.6% with DTX-1 and 110.5% with DTX-2., In dcELISA, the IC50 was set at 0.182 ng/mL, and the detection limit was set at 0.023 ng/mL by using the anti-OA mAb-2D7. The level of OA recovered from spiked mussel samples was 2–50 ng/g, ranging from 97.6 ± 7.2% to 106.4 ± 9.8%. In contrast, the immunostrip assay with a limit of 5 ng/mL, conducted for detecting OA, was completed in 10 min. The mAb-based dcELISA and immunostrip assay developed were precise and sensitive enough to quickly assess OA, DTX-1, and DTX-2 in shellfish specimens.
Introduction
Diarrhetic shellfish poisoning (DSP) is a serious gastrointestinal condition that is caused by the consumption of seafood containing microalgal toxins of low molecular weight (Dominguez et al., Citation2010; Yasumoto et al., Citation1978). These toxins consist of several types of lipophilic polyethers, which accumulate in the shellfish digestive glands, known as the hepatopancreas. Most outbreaks of DSP are associated with Okadaic acid (OA) and DTX-1 (dinophysistoxin-1). Other OA-group toxins include dinophysistoxin-2 (DTX-2), which is an isomer of OA, dinophysistoxin-1 (DTX-1; the methylated OA derivative), and dinophysistoxin-3 (DTX-3; a group of toxins acylated on the C-7 hydroxyl group and containing long-chain fatty acids) (Gerssen et al., Citation2010; Van Egmond et al., Citation1993; Yasumoto et al., Citation1985).
OA-group toxins are acidic polyethers that can inhibit the serine/threonine protein phosphatase activity by binding to the receptor sites of the phosphatase, leading to a rapid increase in the phosphorylated protein levels (Bialojan & Takai, Citation1988; Fernández et al., Citation2014; Larsen et al., Citation2007; Munday, Citation2013). Additionally, animal studies have shown that these toxins can induce carcinogenic, mutagenic, and immunotoxic effects (Cembella, Citation1989). Despite the reported relationship of DSP toxins with immunoinhibitory, mutagenic, and carcinoma-promoting functions in animals, no conclusive evidence was found in humans6. However, chronic exposure to such toxins might increase the risk of gastrointestinal carcinomas (Cordier et al., Citation2000; Stamman et al., Citation1987).
Fast, reliable, and sensitive measures are needed for detecting OA and DTX to ensure the safety of seafood and minimize threats to public health. The maximum allowable level recommended by The European Union is 160 µg of OA equivalents/Kg according to Regulation (EC) No 853/2004. The EFSA (European Food Safety Authority) has recommended reducing the maximum limit of OA in bivalve mollusks from 160 µg to 45 µg OA equivalents/Kg (European Food Safety Authority, Citation2008). Murine bioassay is the initial screening strategy for identifying biotoxins (Campàs et al., Citation2007). Besides ethical problems, mouse bioassay is time-consuming and might provide false-positive results due to interferences from other marine toxins or fatty acids (Suzuki et al., Citation2005; Suzuki et al., Citation1996). This method was prohibited in 2014.
Other tests were suggested for replacing the rodent bioassays, including the OA molecular activity-based enzymatic protein phosphatase inhibition assays (Simon & Vemoux, Citation1994; Tubaro et al., Citation1996) and cytotoxicity assays (Croci et al., Citation1997). However, enzymatic assays cannot distinguish between toxins, and false positives arise due to certain interferences. The cell rounding response during a cytotoxicity assay is nonspecific against toxic substances. Such chromatographic approaches, such as HPLC (high-performance liquid chromatography) coupled with ADAM derivatization-based fluorescence, are universally recognized (Lee et al., Citation1987; Mouratidou et al., Citation2004; Prassopoulou et al., Citation2009; Vale et al., Citation1999). LC-MS/MS (liquid chromatography-tandem mass spectrometry) is the best technique for derivatization-free direct toxin determination (Ito & Tsukada, Citation2002; Shen et al., Citation1997; Suzuki et al., Citation2004; Suzuki & Yasumoto, Citation2000). The LC-FLD and LC-MS methods have high sensitivity, but these techniques are time-consuming, expensive, and require expertise, which is beyond the scope of many screening laboratories.
In contrast, immunoassays have been used to develop many sensitive and fast approaches for the surveillance and quantification of OA in contaminated shellfish (Campàs et al., Citation2007; Garthwaite et al., Citation2001; Matsuura et al., Citation1994; Sassolas et al., Citation2013; Stewart et al., Citation2009). Although immunoassays cannot evaluate toxicity, given that structural toxin identification is performed by monoclonal/polyclonal antibodies, they are a promising approach for detecting shellfish toxins routinely since neither expensive and sophisticated equipment nor skilled personnel is required. ELISA (enzyme-linked immunosorbent assay) and related immunoassays are unsuitable for in situ detection and rapid screening of specimens because the incubation, washing procedure, and application of instruments are time-consuming.
Colloidal gold granules have been applied to biosensors, immunocytochemistry, and immunostrips (Faulk & Taylor, Citation1971; Llamas et al., Citation2007; Lu et al., Citation2012; Paek et al., Citation2000; Zhou et al., Citation2010). The performance of immunostrip depends on the migration of test specimens and antibody-colloidal gold conjugates along the membrane strips, the sites of binding interactions. A few advantages of this assay over the routine screening approaches include high throughput, convenient implementation, and fast results (within 5–10 min).
While these immunoassays are sensitive and robust for detecting OA at the concentrations required by current legislation, these antibodies failed to detect DTX-1 and/or DTX-2 in the matrix in competitive indirect ELISA (CI-ELISA) and SPR (surface plasmon resonance) with low cross-reaction (Liu et al., Citation2014; Llamas et al., Citation2007). According to some studies, multitoxin SPR has 68% and 65% cross-reactivity for DTX-1 and DTX-2 compared to 78% and 2.6% for these two analogs by ELISA (Dubois et al., Citation2010; Mcnamee & Christopher, Citation2013). Based on toxicological experiments, the TEFs for the analogs OA, DTX-1, and DTX-2 were determined to be 1, 1, and 0.6, respectively (Aune et al., Citation2007). Toxicology experts believe that DTX-1 and DTX-2 should be measured more precisely. False positive and false negative results caused by low antibody cross-reactivity should be avoided when detecting OA and its derivatives. Therefore, this study describes the development of a mAb used in dcELISA (direct ELISA) and gold nanoparticle immunochromatographic strip that is most specific for in situ detection of OA but also has sensitivity and cross-reactivity useful for detecting DTX-1 and DTX-2 at relevant levels in shellfish.
Materials and methods
Chemicals
OA, DTX-1, DTX-2, and other phycotoxin standards and certified reference materials (CRM-Zero-Mus) were purchased from the National Research Council of the Canadian Institute of Marine Biosciences (Halifax, Canada). CDI (1,1'-carbonyldiimidazole), BSA (bovine serum albumin), KLH (keyhole limpet hemocyanin), Freund’s complete/incomplete adjuvants, HAT (hypoxanthine aminopterin thymidine), HT (hypoxanthine thymidine), PEG 1500 (polyethylene glycol solution), and Tween 20 were purchased from Sigma Aldrich (St. Louis, USA). The IgG-HRP (peroxidase-conjugated goat-anti-mouse IgG) was procured from Pierce Chemical Co. (Rockford, IL, USA). The virus-free female BALB/c mice (six to eight weeks old) and murine myeloma Sp2/0-Ag14 cells were procured from Dalian Medical University. The nitrocellulose membrane, absorbance pad, and glass fiber membrane were purchased from Whatman International (Middlesex, UK). The remaining chemicals and organic solvents, which were of reagent/analytical grade, were purchased from the Beijing Reagent Corp. (Beijing, China).
The buffers used in this study were as follows: (1) Carbonate buffer (0.05 M; pH 9.6) was used for coating; (2) the blocking buffer comprised phosphate-buffered saline (PBS; 0.01 M; pH 7.4), 1% polyvinyl pyrrolidone (PVP) (w/v); (3) PBST (0.05% Tween 20 mixed with 0.01 M PBS) was used as the washing buffer; (4) standard dilution and sample buffers were 10% methanol added to 0.01 M PBS; (5) the antibody buffer consisted of 1% BSA (w/v) in PBST; (6) the substrate buffer was a solution for HRP, which was prepared by adding 12 mL of citrate buffer (0.21 M, pH 3.95) to 600 µL of 1:9 acetone–methanol solution of 3,3’,5,5'-tetramethylbenzidine (TMB) (1 nM), and the subsequent instant addition of 30% H2O2 (4 µL) before use; (7) H2SO4 (2 M) served as the stopping solution.
Extraction and isolation of OA
OA was extracted and isolated from a marine dinoflagellate Prorocentrum lima following the method reported by Murakami (Murakami et al., Citation1982). The purified OA was stored at –80 °C.
Preparation of OA conjugates
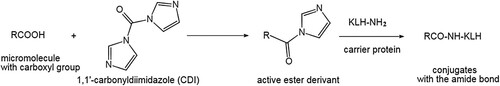
The OA protein conjugates were synthesized following the method reported by Tsubokawa (Tsubokawa & Hosoya, Citation1991) (). Briefly, 1 mg of OA was reacted for 0.5 h with 0.5 mg of CDI in 150 µL of dry acetone at ambient temperature. Water (0.02 mL) was slowly added, followed immediately by the addition of 3 mg of KLH in 1 mL of sodium carbonate buffer (0.1 M; pH 8.3). The resultant mixture was incubated for 12 h at 4 °C and then extensively dialyzed against 0.01 M PBS (pH 7.4). In the same way, OA was conjugated to BSA to generate the solid phase test antigen OA-BSA and to HRP to generate OA-HRP for competitive assays.
Figure 1. Chemical structures of okadaic acid and its derivatives.
Figure 2. Synthesis of OA-protein conjugates.
Immunization and monoclonal antibody preparation
Freund’s complete and incomplete adjuvants were used to emulsify OA-KLH. Emulsified OA-KLH was subcutaneously administered to five BALB/c female mice (Tochi et al., Citation2016). Fusion and screening of cells were conducted by methods described in a study by Peng et al. (Citation2017), and the antibody titer was monitored by performing indirect ELISA (icELISA). Preparation and purification of the antibodies were performed following the methods described in a study by Isanga et al. (Citation2017). The purified anti-OA mAbs were stored at –80 °C.
Characterization of monoclonal antibodies
Sensitivity of different mAbs. The five anti-OA mAbs were examined, and their toxin sensitivities were compared by performing icELISA, based on indirect labeling. For fitting the calibration graphs, the sigmoidal logistic four-variable equation was used in OriginPro 9, and the procedure followed was the same as that described in a previous study (Sassolas et al., Citation2013).
In the equation, A1 and A2 represent the minimum and maximum asymptomatic values, x0 refers to the inflection point value of x, and p denotes the inflection point (IC50) slope. The detection limits corresponded to 85% of antibody binding, IC50, and correlation factors (R). The linear range for detecting OA was set at the IC20–IC80 OA levels.
Specificity of antibodies. The ability of a method to distinguish the analyte under examination from other substances is assessed by specificity. The specificity of the five antibodies was assessed by determining the degree of cross-reaction of each antibody by performing the icELISA, where analytes with a similar chemical structure were used to produce the antibodies. The icELISA was performed following previously reported procedures (Mukunzi et al., Citation2016) to predict the degree of antibody specificity within the diarrhetic poison families. The computational equation for cross-reactivity is shown below.
Cross-reactivity% = (IC50 of OA)/(IC50 of other analytes) 100%.
Determination of Isotypes. for the mAb isotype assessment, a mouse mAb isotyping kit was used, and the specific immunoglobulin was recognized following the manufacturer's guidelines.
Development and optimization of dcELISA
The dcELISA was modified for measuring OA in mussel samples. Briefly, the captured antibodies were coated by adding a PBS solution (100 µL) containing anti-OA antibodies (monoclonal) to each microplate well, followed by overnight incubation at 4 °C. Then, PBST was used to wash the microplate four times, and blocking was performed by adding 300 µL of the blocking buffer for 2 h at 37 °C. After washing with PBST, the OA standard or extracted samples (50 µL) were incubated in the wells with 50 µL of OA-HRP conjugate in PBST under ambient temperature for 10 min. After washing four times again, the peroxidase activity was measured at 450 nm. A standard curve was plotted based on a sigmoidal logistic four-variable dose–response (OriginPro 8.5, OriginLab, Northampton, USA).
Preparation of antibody-colloidal gold (CG) probe
After dialyzation against the boric acid/borax buffer at 4 °C for 24 h, the purified anti-OA mAb was centrifuged at 13,000 g for 10 min to yield a clear supernatant for conjugation. The CG-mAb probe was prepared following a procedure described in a previous study (Peng et al., Citation2017). Briefly, 1% (w/v) hydrogen tetrachloroaurate trihydrate (0.5 mL) and distilled water (50 mL) were mixed in a flask and subsequently heated to the boiling point. Then, freshly-prepared 1% sodium citrate solution (0.5 mL) was added to the flask while stirring constantly. The mixture was boiled till it turned red, and no further alteration of color was observed. The pH of the CG solution was adjusted to 8.0 by adding 0.15 M K2CO3. Then, 1 mL CG solution was added with 10 µL of anti-OA mAb and slowly stirred at ambient temperature for 2 h. Next, 1% (w/v) PEG-20000 solution (100 µL) was added, and the mixture was stirred for 30 min. Then, 10% (w/v) BSA solution (100 µL) was added, and the mixture was centrifuged at 13,000 g for 30 min after being stirred for an additional 30 min. Following centrifugation, the supernatant was removed. Finally, the resultant pellets were dissolved by adding a PBS resuspension buffer (200 µL) containing 5% sucrose, 2% trehalose, 1% BSA, 1% PEG 20000, and 0.25% Tween 20. The mixture was stored at 4 °C until further use. The IR spectrum were used to scan the anti-OA mAb-gold nanoparticle probes.
Preparation of immunostrips
The immunochromatographic strip consists of a sample pad, an absorbent pad, a PVC (polyvinylchloride) backing card, and nitrocellulose (NC) film containing the test and control zones. The NC membrane was coated with OA-BSA (1 mg/mL) as the test line and with anti-mouse rabbit IgG antibody (0.8 mg/mL) as the control line (T/C) using a dispenser. The membranes were dried at ambient temperature for 2 h. The anti-OA mAb-gold nanoparticle probes were added to the microplate wells. After the microplates were air-dried for 50 min, a borate buffer (50 mM; containing 1% BSA, 0.05% sodium azide, and 0.5% Tween 20; pH 7.4) was used to treat the sample pad, which was then dried at 60 °C for 2 h. The NC film, the absorbent pad, and the pretreated sample pad were assembled as a test strip.
OA extraction from certified reference mussels
The samples were prepared using a modified version of previously reported methods (Jawaid et al., Citation2015). We acquired the certified negative mussel samples (CRM-Zero-Mus) from the NRC. After adding 80:20 (v/v) methanol/deionized water (5 mL) to the mussel tissue (1 g), the mixture was centrifuged at 3,000 g for 10 min at ambient temperature after vortex-mixing for 5 min. Following aspiration of the supernatant (50 µL) and dilution in 0.01 M PBS (0.95 mL), dcELISA was performed. During the in situ immunochromatographic strip assay, 10-min precipitation of the sample pellet extract was achieved initially at ambient temperature, and then, clear supernatant (0.1 mL) was aspirated and diluted in water (0.2 mL) for conducting further assays.
Performing dcELISA to determine OA in mussels
The analytical recovery experiment was conducted to determine the validity of dcELISA for assessing OA in mussel specimens. CRM-Zero-Mus was spiked using OA (gradient of 2–50 ng/g concentrations). After diluting the clear extract (50 µL) in 0.01 M PBS (0.95 mL), dcELISA was performed.
Assessing OA in mussels using immunostrips
To the microplate wells, 120 µL of the extracted samples or OA solutions (after serial dilution; 2–50 ng/mL) were added using an anti-OA mAb-gold nanoparticle probe and incubated at ambient temperature for 5 min. Then, the immunostrips were vertically dipped into the wells for 5 min, following which the test outcomes were assessed through visual observations.
Results and discussion
Similar to other marine toxins, OA is a small-molecular non-immunogenic polyether toxin comprising 38 carbons (MW: 805). For triggering an immune response, the conjugation of OA to carrier proteins, such as OVA (ovalbumin), BSA, and KLH, is necessary. The design of the immunogen is the key to the successful generation of antibodies. Proper immunogens (with an optimal cross-linker, carrier protein, and conjugation strategy) and competent immunization schemes are required for producing high-quality mAbs. OA has a carboxyl group. In previous studies, for obtaining the most desirable designs of haptens, the carboxyl group of OA was cross-linked to carrier proteins with EDC or DCC and NHS, where OA acted as the immunogen (Hayat et al., Citation2011; Llamas et al., Citation2007; Sassolas et al., Citation2013). However, excessive EDC or DCC and NHS may react with the carboxylate (-COOH) and primary amine (-NH2) groups of the carrier proteins, resulting in cross-linking of the proteins. In this study, protein conjugates of OA were synthesized through the CDI and quench reactions by adding water; the excess CDI was decomposed. A study showed that the affinity of hapten antibodies varies with different protein carriers used for conjugation (Tsao et al., Citation2007). Therefore, in this study, we conjugated OA to KLH for immunization. Animal immunization and ELISA tests further indicated that the artificial antigen of OA-KLH had a good immune effect.
Hybridoma production and antibody characterization
In this study, the immunogen OA-KLH was applied to generate five hybridomas (2D7, 1C3, 9C7, 11H8, and 4B7). The icELISA graphs of the 5 mAbs obtained are presented in and and . Using an isotyping kit for mouse monoclonal antibodies, the five mAbs were determined as isotypes ().
Figure 3. The anti-OA mAbs produced by the five different stable hybridoma cell lines were compared by performing icELISA.
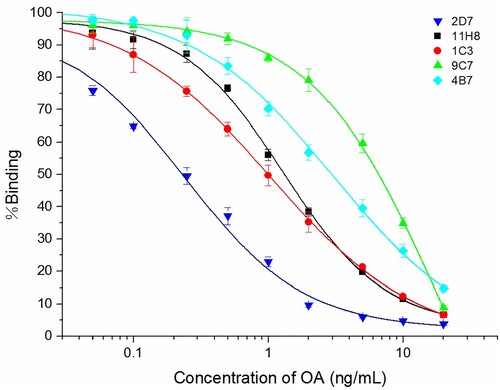
Table 1. The parameters of the anti-OA mAbs secreted by the hybridomas.
Table 2. The cross-reactivities of mAbs with OA analogs.
An ELISA targeted for DSP detection, which can allow equal detection of OA and DTX, has yet to be developed. For detecting DTX-3, an additional step of alkaline hydrolysis is necessary for preparing the extract to eliminate the acyl group and convert the toxin to DTX-1. Relevant commercial ELISAs include the DSP-Check®ELISA test kit (UBE Industries, Japan), which is a globally used kit for OA and DTX-1 screening with a 20 ng/g detection limit, and the Rougier Bio-Tech® ELISA test kit, where an anti-idiotypic antibody and a monoclonal anti-OA antibody compete for the binding sites on anti-OA antibody against OA. The sensitivity of the anti-OA antibody to OA is considerably higher than its sensitivity to DTX-1 or DTX-2 (10–20 times).
To determine the specificity of the five mAbs, analogs of OA were tested by performing icELISA. The anti-OA mAb-2D7 showed a high affinity to OA. A standard curve for detecting OA was plotted (), and the equation obtained was y = 2.054 + (94.082)/(1 + (x/0.240)0.981), with an R2 value of 0.9871. Based on the equation, the 2D7 antibody had an IC50 of 0.24 ng/mL to OA. On the other hand, the cross-reactivities of the anti-OA mAb-2D7 were high with both DTX-1 (91.6%) and DTX-2 (110.5%). No reaction of the 2D7 antibody was found with neosaxitoxin, STX, or domoic acid, even at a very high concentration (1 µg/mL) (). Thus, we used the anti-OA mAb-2D7 for the subsequent experiments.
Table 3. Comparisons of optimized ELISA based on direct vs. indirect labeling.
Development of dcELISA
The factors that improved the performance of dcELISA for assessing OA were evaluated. The optimal concentrations were determined for OA-HRP (1:10,000) and anti-OA mAb-2D7 (1:7,000) by performing ELISA checkerboard assays.
According to the equation y = −5.517 + (105.740)/(1 + (x/0.182)0.850), the IC20, IC50, and IC80 values of mAb 2D7 to OA were 0.036, 0.182, and 0.932 ng/mL, respectively, with an R2 value of 0.9972. A detection limit of 0.023 ng/mL in PBS and a linear range of 0.036–0.932 ng/mL were recorded. The average intraplate CV for the replicates was 1.36% and 12.84%, while the average interplate CV was 6.23%.
Analysis of OA in mussels based on mAb
CRM-Zero-Mus is a Mytilus edulis (mussel) tissue matrix CRM that is used as a negative control for assessing PST (paralytic shellfish toxins), DA (domoic acid), and its isomers, GYM (gymnodimine), AZA (azaspiracids), PTX (pentenotoxins), YTX (yessotoxins), as well as, OA and DTX. Therefore, the sample can be used to perform relevant experiments for determining the recovery rate.
To evaluate the underestimation and overestimation of the recovery rate, the standard analytes of OA were spiked into the extract and the buffer solution to obtain the standard curves (). No noticeable matrix extract intersection appeared in the standard curves of the buffer and the mussel matrix extract. The recovery rates for tissue homogenate extracts for six doses of spiked OA are shown in . The recovery rate for spiked OA in mussel extracts was 97.6 ± 7.2%∼106.4 ± 9.8%.
Figure 4. Standard curves of OA in the assay buffer and the matrix (extracted mussel sample, diluted 50 times).
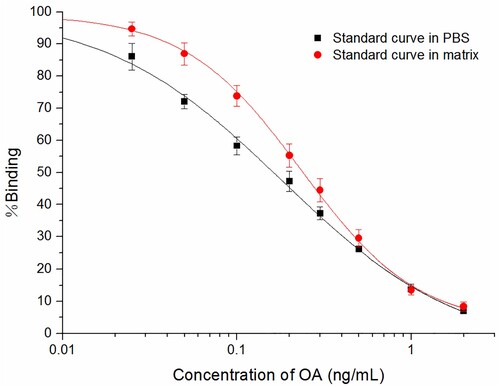
Table 4. Recovery rate for spiked OA in mussel extracts for the OA dcELISA (n = 5).
Characterization of the immunostrip for OA
The IR spectrum image () revealed no discernible changes in characteristic absorption peaks following antibody conjugation. The sensitivity and visibility of the immunostrip are affected by the mAb concentration. For the synthesis of the anti-OA mAb-gold nanoparticle probes, a series of mAb concentrations (5, 10, 15, and 20 µg/mL) were optimized. Therefore, we discovered that the optimal mAb concentration was 15 µg/mL, and the optimal immunostrip conditions were: coating antigen at 1 mg/mL, pH value at 8.0, and mAb at 15 µg/mL.
Figure 5. IR spectrum of colloidal gold (a) and gold nanoparticle probes labeled mAb (b).
Detection limit of the OA test strip
The immunostrip assay was performed using different concentrations (0–10 ng/mL) of the OA standard solutions. The duration of the assay was less than 10 min, while the detection limit for the OA test was about 5 ng/mL (a). At a concentration of 10 ng/mL, OA can occupy the entire antibody-CG conjugates and avoid OA-BSA binding on the test line; this leads to the formation of only one control line.
Figure 6. Detection of OA with immunochromatographic strips in (a) standard solutions and (b) mussel samples.
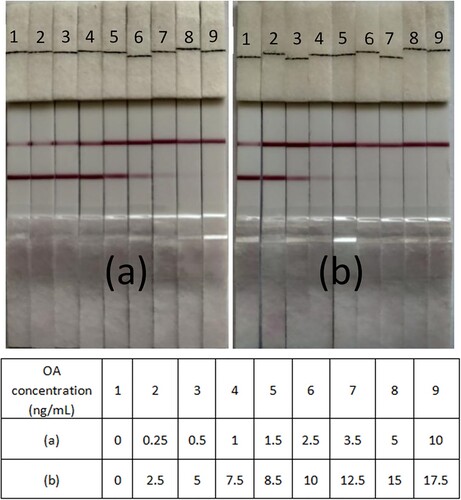
Analysis of OA in mussels by the immunostrip assay
Matrix effects are traits of mussel specimens that distinguish them from PBS. The immunostrip assay was performed to detect OA in the mussel specimens; the relevant outcomes are shown in (b). At OA doses below 8.5 ng/mL, the result was negative. A positive result was found at OA doses exceeding 10 ng/mL. This implies that the detection technique can be used to determine OA residues in mussel samples.
Conclusions
In this study, we prepared five specific mAbs (2D7, 1C3, 9C7, 11H8, and 4B7) against OA through immunization with protein conjugates of the synthesized haptens rather than using rare natural toxins. The production of mAbs with high affinity to the OA-group toxins highly depends on the synthesis of hapten and the protein conjugate synthetic scheme. Furthermore, we used them to perform the immunochromatographic strip (with gold nanoparticles) assay and dcELISA for elucidating OA in mussel specimens. The dcELISA showed an IC50 of 0.182 ng/mL in the specimens and a detection limit of 0.023 ng/g. Additionally, the visual detection limit for the gold nanoparticle immunochromatographic strip was 5 ng/mL. Moreover, visual evidence was provided by using red-colored gold nanoparticles as tracers, and it required 10 min to obtain the results of the immunostrip assay. Hence, the one-step immunostrip assay developed in this study was suitable for in situ OA detection in uncleaned mussel specimens and might be used to evaluate other compounds and matrices that have a low molecular weight.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No 41106078, No 21405026), and foundation of State Environmental Protection Key Laboratory of Coastal Ecosystem (No. 201606, No.201806).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Aune, T., Larsen, S., Aasen, J. A. B., Rehmann, N., Satake, M., & Hess, P. (2007). Relative toxicity of dinophysistoxin-2 (dtx-2) compared with okadaic acid, based on acute intraperitoneal toxicity in mice. Toxicon, 49(1), 1–7. https://doi.org/10.1016/j.toxicon.2006.07.033
- Bialojan, C., & Takai, A. (1988). Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochemical Journal, 256(1), 283–290. https://doi.org/10.1042/bj2560283
- Campàs, M., Prieto-Simón, B., & Marty, J. L. (2007). Biosensors to detect marine toxins: Assessing seafood safety. Talanta, 72(3), 884–895. https://doi.org/10.1016/j.talanta.2006.12.036
- Cembella, A. (1989). Occurrence of okadaic acid, a major diarrheic shellfish toxin, in natural populations ofDinophysis spp. From the eastern coast of North America. Journal of Applied Phycology, 1(4), 307–310. https://doi.org/10.1007/BF00003466
- Cordier, S., Monfort, C., Miossec, L., Richardson, S., & Belin, C. (2000). Ecological analysis of digestive cancer mortality related to contamination by diarrhetic shellfish poisoning toxins along the coasts of France. Environmental Research, 84(2), 145–150. https://doi.org/10.1006/enrs.2000.4103
- Croci, L., Cozzi, L., Stacchini, A., De Medici, D., & Toti, L. (1997). A rapid tissue culture assay for the detection of okadaic acid and related compounds in mussels. Toxicon, 35(2), 223–230. https://doi.org/10.1016/S0041-0101(96)00124-9
- Dominguez, H. J., Paz, B., Daranas, A. H., Norte, M., Franco, J. M., & Fernández, J. J. (2010). Dinoflagellate polyether within the yessotoxin, pectenotoxin and okadaic acid toxin groups: Characterization, analysis and human health implications. Toxicon, 56(2), 191–217. https://doi.org/10.1016/j.toxicon.2009.11.005
- Dubois, M., Demoulin, L., Charlier, C., Singh, G., Godefroy, S.B., Campbell, K., Elliott, C.T., Delahaut, Ph.. ( 2010). Development of elisas for detecting domoic acid, okadaic acid, and saxitoxin and their applicability for the detection of marine toxins in samples collected in Belgium. Food Additives & Contaminants: Part A, 27(6), 859–868. https://doi.org/10.1080/19440041003662881
- European Food Safety Authority. (2008). Marine biotoxins in shellfish--okadaic acid and analogues-scientific opinion of the panel on contaminants in the food chain. EFSA Journal, 6(1), 589, 1–64. https://doi.org/10.2903/j.efsa.2008.589
- Faulk, W. P., & Taylor, G. M. (1971). Communication to the editors: An immunocolloid method for the electron microscope. Immunochemistry, 8(11), 1081–1083. https://doi.org/10.1016/0019-2791(71)90496-4
- Fernández, D. A., Louzao, M. C., Fraga, M., Vilariño, N., Vieytes, M. R., & Botana, L. M. (2014). Experimental basis for the high oral toxicity of dinophysistoxin 1: A comparative study of DSP. Toxins, 6(1), 211–228. https://doi.org/10.3390/toxins6010211
- Garthwaite, I., Ross, K. M., Miles, C. O., Briggs, L. R., Towers, N. R., Borrell, T., & Busby, P. (2001). Integrated enzyme-linked immunosorbent assay screening system for amnesic, neurotoxic, diarrhetic, and paralytic shellfish poisoning toxins found in New Zealand. Journal of AOAC International, 84(5), 1643–1648. https://doi.org/10.1093/jaoac/84.5.1643
- Gerssen, A., Pol-Hofstad, I. E., Poelman, M., Mulder, P. P., Top, V. d., J, H., & De Boer, J. (2010). Marine toxins: Chemistry, toxicity, occurrence and detection, with special reference to the Dutch situation. Toxins, 2(4), 878–904. https://doi.org/10.3390/toxins2040878
- Hayat, A., Barthelmebs, L., & Marty, J. L. (2011). Enzyme-linked immunosensor based on super paramagnetic nanobeads for easy and rapid detection of okadaic acid. Analytica Chimica Acta, 690(2), 248–252. https://doi.org/10.1016/j.aca.2011.02.031
- Isanga, J., Tochi, B. N., Mukunzi, D., Chen, Y., Liu, L., Kuang, H., & Xu, C. (2017). Development of a specific monoclonal antibody assay and a rapid testing strip for the detection of apramycin residues in food samples. Food and Agricultural Immunology, 28(1), 49–66. https://doi.org/10.1080/09540105.2016.1202211
- Ito, S., & Tsukada, K. (2002). Matrix effect and correction by standard addition in quantitative liquid chromatographic–mass spectrometric analysis of diarrhetic shellfish poisoning toxins. Journal of Chromatography A, 943(1), 39–46. https://doi.org/10.1016/S0021-9673(01)01429-7
- Jawaid, W., Meneely, J. P., Campbell, K., Melville, K., Holmes, S. J., Rice, J., & Elliott, C. T. (2015). Development and validation of a lateral flow immunoassay for the rapid screening of okadaic acid and all dinophysis toxins from shellfish extracts. Journal of Agricultural and Food Chemistry, 63(38), 8574–8583. https://doi.org/10.1021/acs.jafc.5b01254
- Larsen, K., Petersen, D., Wilkins, A. L., Samdal, I. A., Sandvik, M., Rundberget, T., Goldstone, D., Arcus, V., Hovgaard, P., & Rise, F. (2007). Clarification of the C-35 stereochemistries of dinophysistoxin-1 and dinophysistoxin-2 and its consequences for binding to protein phosphatase. Chemical Research in Toxicology, 20(6), 868–875. https://doi.org/10.1021/tx700016m
- Lee, J. S., Yanagi, T., Kenma, R., & Yasumoto, T. (1987). Fluorometric determination of diarrhetic shellfish toxins by high-performance liquid chromatography. Agricultural and Biological Chemistry, 51(3), 877–881. https://doi.org/10.1271/bbb1961.51.877
- Liu, B. H., Hung, C. T., Lu, C. C., Chou, H. N., & Yu, F. Y. (2014). Production of monoclonal antibody for okadaic acid and its utilization in an ultrasensitive enzyme-linked immunosorbent assay and one-step immunochromatographic strip. Journal of Agricultural and Food Chemistry, 62(6), 1254–1260. https://doi.org/10.1021/jf404827s
- Llamas, N. M., Stewart, L., Fodey, T., Higgins, H. C., Velasco, M. L. R., Botana, L. M., & Elliott, C. T. (2007). Development of a novel immunobiosensor method for the rapid detection of okadaic acid contamination in shellfish extracts. Analytical and Bioanalytical Chemistry, 389(2), 581–587. https://doi.org/10.1007/s00216-007-1444-3
- Lu, S. Y., Lin, C., Li, Y. S., Zhou, Y., Meng, X. M., Yu, S. Y., Li, Z. H., Li, L., Ren, H. L., & Liu, Z. S. (2012). A screening lateral flow immunochromatographic assay for on-site detection of okadaic acid in shellfish products. Analytical Biochemistry, 422(2), 59–65. https://doi.org/10.1016/j.ab.2011.12.039
- Matsuura, S., Kita, H., & Takagaki, Y. (1994). Specificity of mouse monoclonal anti-okadaic acid antibodies to okadaic acid and its analogs among diarrhetic shellfish toxins. Bioscience, Biotechnology, and Biochemistry, 58(8), 1471–1475. https://doi.org/10.1271/bbb.58.1471
- Mcnamee, S. E., & Christopher, T. E. (2013). Multiplex biotoxin surface plasmon resonance method for marine biotoxins in algal and seawater samples. Environmental Science and Pollution Research, 20(10), 6794–6807. https://doi.org/10.1007/s11356-012-1329-7
- Mouratidou, T., Kaniou-Grigoriadou, I., Samara, C., & Koumtzis, T. (2004). Determination of okadaic acid and related toxins in Greek mussels by HPLC with fluorimetric detection. Journal of Liquid Chromatography & Related Technologies, 27(14), 2153–2166. https://doi.org/10.1081/JLC-200025690
- Mukunzi, D., Tochi, B. N., Isanga, J., Liu, L., Kuang, H., & Xu, C. (2016). Development of an immunochromatographic assay for hexestrol and diethylstilbestrol residues in milk. Food and Agricultural Immunology, 27(6), 855–869. https://doi.org/10.1080/09540105.2016.1183601
- Munday, R. (2013). Is protein phosphatase inhibition responsible for the toxic effects of okadaic acid in animals? Toxins, 5(2), 267–285. https://doi.org/10.3390/toxins5020267
- Murakami, Y., Oshima, Y., & Yasumoto, T. (1982). Identification of okadaic acid as a toxic component of a marine dinoflagellate prorocentrum Lima. NIPPON SUISAN GAKKAISHI, 48(1), 69–72. https://doi.org/10.2331/suisan.48.69
- Paek, S. H., Lee, S. H., Cho, J. H., & Kim, Y. S. (2000). Development of rapid one-step immunochromatographic assay. Methods, 22(1), 53–60. https://doi.org/10.1006/meth.2000.1036
- Peng, J., Liu, L., Kuang, H., Cui, G., & Xu, C. (2017). Development of an icELISA and immunochromatographic strip for detection of norfloxacin and its analogs in milk. Food and Agricultural Immunology, 28(2), 288–298. https://doi.org/10.1080/09540105.2016.1263987
- Prassopoulou, E., Katikou, P., Georgantelis, D., & Kyritsakis, A. (2009). Detection of okadaic acid and related esters in mussels during diarrhetic shellfish poisoning (DSP) episodes in Greece using the mouse bioassay, the PP2A inhibition assay and HPLC with fluorimetric detection. Toxicon, 53(2), 214–227. https://doi.org/10.1016/j.toxicon.2008.11.003
- Sassolas, A., Catanante, G., Hayat, A., Stewart, L. D., Elliott, C. T., & Marty, J. L. (2013). Improvement of the efficiency and simplification of ELISA tests for rapid and ultrasensitive detection of okadaic acid in shellfish. Food Control, 30(1), 144–149. https://doi.org/10.1016/j.foodcont.2012.05.028
- Shen, J. L., Hummert, C., & Luckas, B. (1997). Sensitive HPLC-fluorometric and HPLC-MS determination of diarrhetic shellfish poisoning (SP)-toxins as 4-bromomethyl-7-methoxycoumarin esters. Fresenius' Journal of Analytical Chemistry, 357(1), 101–104. https://doi.org/10.1007/s002160050120
- Simon, J. F., & Vemoux, J. P. (1994). Highly sensitive assay of okadaic acid using protein phosphatase and paranitrophenyl phosphate. Natural Toxins, 2(5), 293–301. https://doi.org/10.1002/nt.2620020508
- Stamman, E., Segar, A., & Davis, P. G. (1987). A preliminary epidemiological assessment of the potential for diarrhetic shellfish poisoning in the northeast United States. N. O. S. o. t. N. O. a. A. A. (NOAA). https://repository.library.noaa.gov/view/noaa/2847/noaa_2847_DS1.pdf.
- Stewart, L. D., Hess, P., Connolly, L., & Elliott, C. T. (2009). Development and single-laboratory validation of a pseudofunctional biosensor immunoassay for the detection of the okadaic acid group of toxins. Analytical Chemistry, 81(24), 10208–10214. https://doi.org/10.1021/ac902084a
- Suzuki, T., Beuzenberg, V., Mackenzie, L., & Quilliam, M. (2004). Discovery of okadaic acid esters in the toxic dinoflagellateDinophysis acuta from New Zealand using liquid chromatography/tandem mass spectrometry. Rapid Communications in Mass Spectrometry, 18(10), 1131–1138. https://doi.org/10.1002/rcm.1455
- Suzuki, T., Jin, T., Shirota, Y., Mitsuya, T., Okumura, Y., & Kamiyama, T. (2005). Quantification of lipophilic toxins associated with diarrhetic shellfish poisoning in Japanese bivalves by liquid chromatography-mass spectrometry and comparison with mouse bioassay. Fisheries Science, 71(6), 1370–1378. https://doi.org/10.1111/j.1444-2906.2005.01104.x
- Suzuki, T., & Yasumoto, T. (2000). Liquid chromatography–electrospray ionization mass spectrometry of the diarrhetic shellfish-poisoning toxins okadaic acid, dinophysistoxin-1 and pectenotoxin-6 in bivalves. Journal of Chromatography A, 874(2), 199–206. https://doi.org/10.1016/S0021-9673(00)00094-7
- Suzuki, T., Yoshizawa, R., Kawamura, T., & Yamasaki, M. (1996). Interference of free fatty acids from the hepatopancreas of mussels with the mouse bioassay for shellfish toxins. Lipids, 31(6), 641–645. https://doi.org/10.1007/BF02523835
- Tochi, B., Peng, J., Song, S., Liu, L., Kuang, H., & Xu, C. (2016). Determination of sarafloxacin and its analogues in milk using an enzyme-linked immunosorbent assay based on a monoclonal antibody. Analytical Methods, 8(7), 1626–1636. https://doi.org/10.1039/C5AY02702K
- Tsao, Z. J., Liao, Y. C., Liu, B. H., Su, C. C., & Yu, F. Y. (2007). Development of a monoclonal antibody against domoic acid and its application in enzyme-linked immunosorbent assay and colloidal gold immunostrip. Journal of Agricultural and Food Chemistry, 55(13), 4921–4927. https://doi.org/10.1021/jf0708140
- Tsubokawa, N., & Hosoya, M. (1991). Reactive carbon black having acyl imidazole or acid anhydride groups: Preparation and reaction with functional polymers having hydroxyl or amino groups. Reactive Polymers, 14(1), 33–40. https://doi.org/10.1016/0923-1137(91)90245-J
- Tubaro, A., Florio, C., Luxich, E., Sosa, S., Della Loggia, R., & Yasumoto, T. (1996). A protein phosphatase 2A inhibition assay for a fast and sensitive assessment of okadaic acid contamination in mussels. Toxicon, 34(7), 743–752. https://doi.org/10.1016/0041-0101(96)00027-X
- Vale, P., Antónia, M., & Sampayo, M. (1999). Esters of okadaic acid and dinophysistoxin-2 in Portuguese bivalves related to human poisonings. Toxicon, 37(8), 1109–1121. https://doi.org/10.1016/S0041-0101(98)00247-5
- Van Egmond, H., Aune, T., Lassus, P., Speijers, G., & Waldock, M. (1993). Paralytic and diarrhetic shellfish poisons: Occurrences in Europe, toxicity, analysis and regulation. Journal of Natural Toxins, 2, 41–83.
- Yasumoto, T., Murata, M., Oshima, Y., Sano, M., Matsumoto, G., & Clardy, J. (1985). Diarrhetic shellfish toxins. Tetrahedron, 41(6), 1019–1025. https://doi.org/10.1016/S0040-4020(01)96469-5
- Yasumoto, T., Oshima, Y., & Yamaguchi, M. (1978). Occurrence of a new type of shellfish poisoning in the tohoku district. Nippon Suisan Gakkaishi, 44(11), 1249–1255. https://doi.org/10.2331/suisan.44.1249
- Zhou, Y., Li, Y., Lu, S., Ren, H., Li, Z., Zhang, Y., Pan, F., Liu, W., Zhang, J., & Liu, Z. (2010). Gold nanoparticle probe-based immunoassay as a new tool for tetrodotoxin detection in puffer fish tissues. Sensors and Actuators B: Chemical, 146(1), 368–372. https://doi.org/10.1016/j.snb.2010.02.049