
Abstract
Background
Most systemic agents used for moderate-to-severe atopic dermatitis (AD) may lead to adverse events requiring routine laboratory monitoring, increasing patient burden and possibly decreasing treatment adherence.
Objective
To evaluate clinical laboratory findings in adults with moderate-to-severe AD treated with dupilumab up to 3 years.
Methods
LIBERTY AD OLE (NCT01949311) was a phase 3, multicenter, open-label extension study in adults with moderate-to-severe AD receiving dupilumab 300 mg weekly.
Results
2,677 patients were treated up to 3 years. No clinically meaningful changes in mean hematology/serum chemistry parameters from baseline were observed. Few laboratory abnormalities were reported as treatment-emergent adverse events. Serious events included one event each of thrombocytopenia, hematuria, and hemolytic anemia, all unrelated to treatment. Abnormalities leading to treatment withdrawal included thrombocytopenia (one patient), increased hepatic enzymes (two patients), and blood creatine phosphokinase increased (one patient). No patients had Grade 3 anemia or Grade 3/4 thrombocytopenia; one patient had Grade 3 neutropenia (Week 100); two patients had Grade 3 eosinophilia (baseline visit); no eosinophil abnormalities were associated with clinically symptomatic events/permanent treatment discontinuation.
Conclusion
Dupilumab treatment of adults with moderate-to-severe AD up to 3 years showed no clinically meaningful changes in mean laboratory parameters, supporting continuous long-term use without laboratory monitoring.
ClinicalTrials.gov identifier
NCT01949311
Introduction
Patients with moderate-to-severe atopic dermatitis (AD) often require treatment with systemic immunosuppressants (Citation1), most of which are used off label with poorly characterized long-term efficacy and safety (Citation1–5). Furthermore, most systemic treatments available for moderate-to-severe AD, including methotrexate, cyclosporine A, mycophenolate, and azathioprine (Citation5), require routine laboratory monitoring. Such monitoring increases patient burden and may decrease treatment adherence.
Dupilumab, a fully human VelocImmune®-derived (Citation6,Citation7) monoclonal antibody, blocks the shared receptor subunit for interleukin (IL)-4 and IL-13, thus inhibiting signaling of these key and central inflammatory cytokines in AD pathophysiology. Dupilumab is approved for patients with type 2 inflammatory diseases, including AD, asthma, and chronic rhinosinusitis with nasal polyps (Citation8,Citation9). In multiple phase 3 trials, dupilumab significantly improved clinical signs, symptoms, and quality of life, and it had an acceptable safety profile in adults and adolescents with moderate-to-severe AD (Citation10–13). Dupilumab also provided acceptable safety and sustained efficacy up to 3 years in adults with moderate-to-severe AD enrolled in an open-label extension study (Citation14). Adults with moderate-to-severe AD treated with dupilumab for up to 52 weeks had no clinically meaningful changes in mean laboratory parameters attributable to dupilumab, thereby supporting its use without laboratory monitoring (Citation15). Similar findings were reported in adolescents and children aged 6–11 years old treated with dupilumab for up to 16 weeks (Citation16,Citation17).
Here we further characterize the safety of dupilumab by evaluating clinical laboratory findings from up to 3 years in adults with moderate-to-severe AD enrolled in LIBERTY AD OLE, an open-label extension (OLE) study.
Materials and methods
Study design
LIBERTY AD OLE is an ongoing phase 3, multicenter, OLE clinical trial (NCT01949311) in adults with moderate-to-severe AD. The detailed study design and the safety and efficacy of dupilumab up to 3 years have been previously reported (Citation14). Patients were eligible for inclusion if they had previously participated (including in the placebo groups) in phase 1–3 dupilumab studies in moderate-to-severe AD and if they adequately completed the required assessments of the parent studies. Patients were excluded if they had an adverse event (AE) deemed related to dupilumab in the parent study that led to treatment discontinuation or if they had a serious AE deemed related to dupilumab in the parent study. Treatment consisted of subcutaneous dupilumab 300 mg weekly. The approved dose for treatment of AD in adults is 300 mg every other week. However, the 300 mg weekly dose was chosen for this study to increase the likelihood of identifying safety signals and to generate safety data that could adequately support both dose regimens. Rescue medications included systemic corticosteroids and nonsteroidal systemic immunosuppressive medications (including phototherapy); patients were allowed to receive other concomitant treatments for AD, including topical corticosteroids and topical calcineurin inhibitors (Citation14).
The study was conducted following ethical principles derived from the Declaration of Helsinki, the International Conference on Harmonization guideline, Good Clinical Practice, and local applicable regulatory requirements. Written informed consent was obtained from all patients prior to commencement of any study procedure.
Outcomes
Blood and urine samples were collected for laboratory monitoring and reported at baseline and weeks 4, 12, 24, 48, 100, and 148. Hematology, serum chemistry, and urinalysis were analyzed by a central laboratory (Covance). Laboratory outcomes of interest included hematocrit (%), hemoglobin (g/L), mean corpuscular hemoglobin (pg), mean corpuscular hemoglobin concentration (g/dL), mean corpuscular volume (fl), erythrocytes (×1012/L), platelets (×109/L), basophils (×109/L), leukocytes (×109/L), lymphocytes (×109/L), eosinophils (×109/L), neutrophils (×109/L), alanine aminotransferase (ALT; U/L), aspartate aminotransferase (AST; U/L), total bilirubin (μmol/L), creatine phosphokinase (CPK; U/L), lactate dehydrogenase (LDH; U/L), cholesterol (mmol/L), triglycerides (mmol/L), and glucose (mmol/L).
The number and proportion of patients reporting treatment‐emergent AEs (TEAEs) were encoded using Medical Dictionary for Regulatory Activities system Preferred Terms.
Investigators were instructed to report laboratory outcomes as TEAEs if the test result was associated with accompanying symptoms; and/or the test result required additional diagnostic testing or medical/surgical intervention; and/or the test result led to a change in dosing (outside of protocol-stipulated dose adjustments); discontinuation from the study; significant additional concomitant drug treatment; or other therapy. A patient was permanently withdrawn from the study in case of severe laboratory abnormalities deemed related to dupilumab, including neutrophil count ≤0.5 × 103/μL; platelet count ≤0.5 × 103/μL; confirmed (by 2 separate tests at least 2 weeks apart) ALT and/or AST values >3× upper limit of normal (ULN) and total bilirubin >2× ULN, excluding confirmed Gilbert’s syndrome; and confirmed AST and/or ALT values >5× ULN (for >2 weeks). In case of severe laboratory abnormalities where a causal relationship to dupilumab could be reasonably excluded (i.e. an alternative cause is evident), the drug could be temporarily interrupted (≥1 dose). The study drug could be resumed when the laboratory parameters normalize sufficiently. Laboratory abnormalities that could lead to temporary interruptions included neutrophil count ≤1.0 × 103/μL but >0.5 × 103/μL; platelet count ≤100 × 103/μL but >50 × 103/μL; and CPK >10× ULN, unless the increase could be clearly attributed to physical exertion and considered clinically inconsequential.
Analysis
All analyses were carried out in the safety analysis set, which included all patients who received ≥1 dose of the study drug. All analyses are descriptive. The counts and proportions of patients with laboratory values classified as AEs are provided. Some parameters are shown as change from baseline by visit or absolute counts by visit (weeks 4, 12, 24, 48, 100, and 148); only the values available at that particular time point were used in calculations, with no imputation for missing values. Data of patients with Grades 1, 2, 3, or 4 changes in platelets, eosinophils, and neutrophils are provided as counts with proportions, with grades defined as per Common Terminology Criteria for Adverse Events version 5.0 (Citation18) as follows: Grade 1 is defined as mild, Grade 2 as moderate, Grade 3 as severe or medically significant but not immediately life-threatening, and Grade 4 as potentially life threatening. For eosinophilia, grades were defined as per the Nordic MPN Study group recommendation as: Grade 1, mild (≥0.5 to ≤1.5 × 109/L), Grade 2, moderate (>1.5 to ≤5.0 × 109/L) and Grade 3, severe (>5.0 × 109/L) (Citation19). Statistical Analysis Software version 9.4 (SAS Institute, Inc., Cary, NC, USA) was used for all analyses.
Results
Patients
A total of 2,677 patients were included in the OLE and received study treatment. At the time of database lock, 82.4% of the patients (2,207/2,677) had completed up to week 52, 1,028 (38.4%) up to week 100, and 347 (13.0%) up to week 148 (Citation14). Of the 1,325 (49.5%) patients who withdrew from the study, most (807 [30.1%]) did so due to study termination by the sponsor upon regulatory approval/commercial availability of the drug. Baseline demographics, disease characteristics, efficacy, and overall safety for this patient population have been reported previously (Citation14).
Red blood cells and platelets
No clinically meaningful changes were observed in mean values from baseline in hematocrit, hemoglobin, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, mean corpuscular volume, or erythrocytes over time in the safety analysis set (not shown). Twenty-eight (1.0%) patients reported 29 events of anemia (including the Preferred Terms of anemia, blood iron decreased, iron deficiency, iron deficiency anemia, microcytic anemia, and hemolytic anemia; ). The single event of hemolytic anemia was classified as serious (not related), but no other anemia event was serious or severe. Of these events, 18 were recovered/resolved or recovering/resolving at the time of the analysis, and none resulted in treatment discontinuation. Grade 2 (moderate) anemia was reported in 10 (0.4%) patients at the baseline of the study and in 8 (0.3%), 1 (0.1%), and 1 (0.3%) patients at weeks 48, 100, and 148, respectively ().
Table 1. Laboratory abnormalities reported as TEAEs.a
Table 2. Proportion of patients with anemia by grade, n/N1 (%).
Mean platelet levels decreased over time but remained within the normal range for the duration of this analysis (). No patients had Grade 2 (moderate) or 3 (severe) decreases in platelets during the study (). Thrombocytopenia/platelet count decreased was reported as an AE in 8 (0.3%) patients. One serious event in a woman aged 61 years with a prior medical history of thrombocytopenia resulted in permanent withdrawal of drug at study week 12; the event was considered not related to dupilumab. The study drug was temporarily interrupted in 5 (0.2%) patients with AEs of thrombocytopenia/platelet count decreased. None of these events were considered related to study drug by the investigator, and 4 events were recovered/resolved. One event occurred in a patient with a history of alcohol abuse for whom study drug was subsequently permanently withdrawn.
Figure 1. (A) Mean change from baseline over time in platelets. (B) Mean change from baseline over time in lymphocytes. (C) Mean change from baseline over time in eosinophils. (D) Absolute eosinophil count over time. (E) Mean change from baseline over time in neutrophils. Upper and lower error bars in the figures show minimum and maximum values, respectively. SD: standard deviation. Footnote for D: Interrupted line shows the upper limit of normal; X show means; upper and lower edges of the boxes show first and third quartile, respectively; upper and lower error bars show minimum and maximum, respectively. SD: standard deviation.
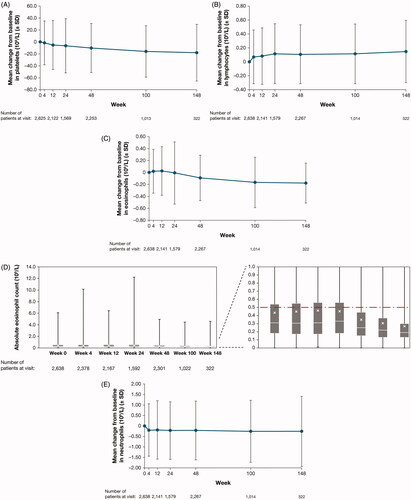
Table 3. Proportion of patients with thrombocytopenia by grade, n/N1 (%).
White blood cells
No clinically meaningful changes in mean values from baseline were observed in basophil or leukocyte counts. Furthermore, no meaningful mean changes were seen in lymphocytes (), mean change in eosinophils (), absolute eosinophils (), or mean change in neutrophil counts (); however, a trend was observed for increased eosinophil counts at early time points (weeks 4 and 12) followed by decreases at subsequent time points ().
A total of 29 events of eosinophilia/eosinophil counts increased were reported as AEs in 29 (1.1%) patients (). None of these events were serious, and 19 were resolved or resolving at the time of the analysis. Eosinophilia/eosinophil count increased led to temporary drug interruption in 5 patients. Grade 3 (severe) eosinophilia was reported in 2 (0.1%) patients at the baseline of the study, but in 0 patients at subsequent analyzed time points (). Grade 2 eosinophilia at the last reported assessment (either at the time of data cutoff or end of treatment) was observed in 4 patients, all of whom had ≥ Grade 1 eosinophilia (eosinophil values >0.5 × 109/L) at the parent study baseline. The highest reported value at last assessment/end of treatment was 2.46 × 109/L. No eosinophil abnormalities were associated with clinically symptomatic events or permanent discontinuation of treatment.
Table 4. Proportion of patients with eosinophilia by grade, n/N1 (%).
A total of 39 AEs of neutrophil count decreased/neutropenia occurred in 32 (1.2%) patients (). All events were mild or moderate, and all but 4 were recovered/resolved or recovering/resolving by the time of the analysis; 15 of these events were considered to be related to the study drug by the investigator. These events led to temporary treatment interruption in 3 cases. At week 100, there was one report of Grade 3 (severe) neutropenia (0.93 × 109/L) that was considered unrelated to treatment and did not lead to treatment withdrawal ().
Table 5. Proportion of patients with neutropenia by grade, n/N1 (%).
There were 26 events of leukopenia/white blood cell count decreased reported as an AE in 22 (0.8%) patients (), with none of the events reported as severe or serious. None of these events led to permanent treatment discontinuation; 2 events led to temporary drug interruption. Nine events were considered as related to treatment in 7 (0.3%) patients, and 21 events were recovered/resolved or recovering/resolving.
Serum chemistry
No clinically meaningful changes in mean values were observed from baseline in serum AST () or ALT (). Eighty AEs related to hepatic enzymes (ALT increased, AST increased, hepatic enzyme increased, transaminases increased, liver function test abnormal) were reported in 54 (2.0%) patients. None of the events were serious; 6 (0.2%) patients had temporary dose interruptions and 2 (0.1%) patients had a permanent drug withdrawal due to these AEs, including one in a patient with a known history of elevated transaminase levels and another in a patient with ALT/AST elevations <2 × ULN. A total of 60 events were recovered or recovering and 1 mild event recovered with sequalae by the time of the analysis.
Figure 2. (A) Mean change from baseline over time in AST. (B) Mean change from baseline over time in ALT. (C) Mean change from baseline over time in LDH. ALT: alanine aminotransferase; AST: aspartate aminotransferase; LDH: lactate dehydrogenase; SD: standard deviation.
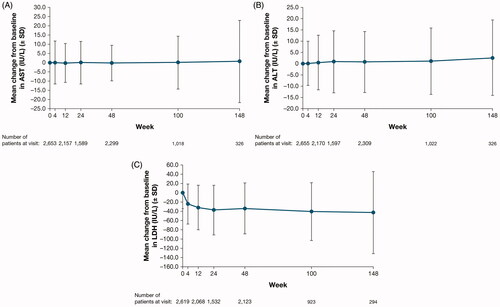
Except for LDH (), no clinically meaningful changes in mean values were observed from baseline in serum chemistry parameters of interest, including total bilirubin, CPK, cholesterol, triglycerides, and glucose levels. An increase in blood LDH was reported as an AE in 12 (0.4%) patients. None of these events were serious or severe or led to permanent drug withdrawal, and all recovered/resolved or were recovering/resolving at the time of analysis.
A total of 101 events of increase in blood CPK was reported as an AE in 89 (3.3%) patients, including 8 patients with 2 separate events each, 2 patients with 3 events, and 79 patients with 1 event. None of these events were serious. Two events led to permanent study drug withdrawal: one in a patient with an AE of ‘myopathy of both thighs’ that was moderate in intensity, with a CPK increase to >10 × ULN after 48 weeks of treatment that returned to normal after withdrawal of study intervention. A second patient with an AE of ‘generalized muscle pain’ that was moderate in intensity and considered to be related to study intervention had a mild CPK elevation at baseline of the current study, peaking at 305 U/L at study week 24 and declining to 244 U/L at study week 48. The study drug was temporarily interrupted in 13 (0.5%) patients with an AE of blood CPK increased, all of which were mild or moderate in severity. Recovery/resolution of these AEs was seen in 79 patients.
Cholesterol- and lipid-related AEs were reported among the following Preferred Terms: blood triglycerides increased, blood cholesterol increased, low density lipoprotein increased, high density lipoprotein decreased, hypertriglyceridemia, hypercholesterolemia, hyperlipidemia, and dyslipidemia. There were 68 of these events reported among 58 (2.2%) patients in total; none were serious. Temporary dose interruption occurred in 1 patient, and there were no permanent dose withdrawals; 39 events were resolved or were resolving at the time of this analysis.
Blood glucose-related AEs (including blood glucose increased, hyperglycemia, and hypoglycemia) were reported in 19 (0.7%) patients (totaling 21 events). None of these events were serious or severe, and none led to dose interruptions or permanent discontinuations; 13 were resolved or were resolving by the time of the analysis.
Laboratory AEs associated with renal function were reported in 3 (0.1%) patients (a total of 5 events). None of the events were serious, severe, or considered to be related to the investigational drug, and none led to treatment discontinuation or withdrawal. All events resolved.
Urinalysis
There were no trends in changes in mean values for urinalysis parameters during the course of the OLE. Hematuria was reported as an AE in 11 (0.4%) patients. All 11 of these events were mild or moderate; one event was serious in a patient with a prior medical history of hematuria. None of the events were considered related to treatment by the investigator; none led to treatment discontinuation; and all but one event resolved/were resolving.
Discussion
In this long-term OLE study in adults with moderate-to-severe AD treated with dupilumab for up to 3 years, no clinically meaningful changes were observed in mean laboratory parameters. Mean values over time were either similar to baseline values or had small deviations from baseline. Few laboratory abnormalities were reported as TEAEs, of which only a small number was serious or led to treatment withdrawal. None of the reports of neutropenia and eosinophilia were serious, and only 1 event of thrombocytopenia was serious. One patient with thrombocytopenia, 2 patients with increased hepatic enzymes, and 1 patient with blood CPK increased withdrew from treatment. Only 1 patient had ≥ Grade 3 neutropenia, and no ≥ Grade 3 anemia, thrombocytopenia, or eosinophilia were reported during the study. Furthermore, no eosinophil abnormalities were associated with clinical symptoms. The mean overall eosinophil count was within the normal range at each visit. Mean eosinophil levels tended to decrease over time with dupilumab treatment, a result also previously observed in adults and adolescents with moderate-to-severe AD (Citation15,Citation16). In the pivotal phase 3 SOLO studies (Citation15), a modest increase in eosinophil levels was noted in the first 4–8 weeks of treatment that diminished by week 16. This initial increase followed by a decrease at later time points was also seen in these analyses from OLE safety data.
LDH, a marker of tissue damage, is strongly correlated to disease severity in AD (Citation20–23). In previous reports of dupilumab treatment in adults and adolescents with moderate-to-severe AD (Citation15,Citation16), baseline LDH levels were elevated but tended to normalize following treatment with dupilumab. A tendency for LDH levels to decrease over time with dupilumab treatment was also observed in the current analysis.
The laboratory safety of dupilumab reported in the present study adds further supportive evidence to the overall long-term safety profile of dupilumab. Overall, these laboratory findings in adults who received up to 3 years treatment with dupilumab are consistent with those reported in adults up to 1 year (Citation15) and in adolescents up to 16 weeks (Citation16). The lack of significant treatment-related laboratory toxicities differentiates dupilumab from other existing long-term systemic treatments for moderate-to-severe AD, including commonly used off-label systemic steroid-sparing agents, which require close laboratory surveillance to ensure safe use (Citation5). Moreover, emerging immunosuppressants in clinical development, such as Janus kinase inhibitors (Citation24–26), may similarly mandate regular laboratory monitoring for drug-induced metabolic derangements and clinically significant myelosuppression.
Strengths of this analysis include the large study size and long-term study duration. Limitations include the open-label study design with no control arm, as well as fewer patients available at later time points because the study is ongoing, and the required withdrawal of patients upon regulatory approval of dupilumab in the country in which the patient enrolled (Citation14). Another limitation is that laboratory analyses were only included at fixed time points, not at time points when AEs occurred. Furthermore, patients enrolled in OLE originated from controlled trials which excluded patients with significant comorbidities such as active HIV, hepatitis B, and hepatitis C infections.
Conclusions
In this OLE study of adults with moderate-to-severe AD who had previously participated in randomized, controlled trials, dupilumab treatment for up to 3 years resulted in no clinically meaningful changes in mean laboratory parameters, supporting its continuous long-term use without routine laboratory monitoring.
Author contributions
All authors contributed to manuscript concept and design. LAB, DT, MD, MdB-W acquired data. ZC conducted the statistical analyses on the data. All authors interpreted the data, provided critical feedback on the manuscript, approved the final manuscript for submission, and are accountable for the accuracy and integrity of the manuscript.
Disclosure statement
Disclosure statement Beck LA: Abbvie, Allakos, AstraZeneca, Benevolent AIBio, Eli Lilly, Incyte, LEO Pharma, NAOS/Bioderma, Novartis, Pfizer, Principia Biopharma, Rapt Therapeutics, Regeneron Pharmaceuticals, Inc., Sanofi, UCB, Vimalan – consultant; Abbvie, LEO Pharma, Pfizer, Regeneron Pharmaceuticals, Inc., Sanofi – investigator. Gilead, Medtronics, Moderna, 3M – stock ownership. Thaçi D: AbbVie, Almirall, Amgen, Bristol Myers Squibb, Biogen-Idec, Boehringer Ingelheim, Dermira, DS Biopharma, Eli Lilly, Galapagos, Galderma, Janssen-Cilag, La Roche Posay, LEO Pharma, Novartis, Pfizer, Regeneron Pharmaceuticals, Inc., Samsung, Sandoz, Sanofi, Sun Pharma, UCB – consultant, advisory board member, and/or investigator. Deleuran M: AbbVie, Almirall, Eli Lilly, Galapagos, LEO Pharma, Meda Pharma, Pfizer, Pierre Fabre, Regeneron Pharmaceuticals, Inc., Sanofi Genzyme – research support, consulting/advisory board agreements, and/or honoraria for lectures. de Bruin-Weller M: Regeneron Pharmaceuticals, Inc., Sanofi Genzyme – Principal Investigator, advisory board member, consultant; AbbVie, Pfizer, LEO Pharma – Principal Investigator, advisory board member; Eli Lilly, Galderma, Janssen, UCB – advisory board member. Chen Z, Khokhar FA, Shumel B: Regeneron Pharmaceuticals, Inc. – employees and shareholders. Zhang M: Sanofi – employee, may hold stock and/or stock options in the company Ozturk ZE: Sanofi Genzyme – employee, may hold stock and/or stock options in the company.
Additional information
Funding
References
- Boguniewicz M, Alexis AF, Beck LA, et al. Expert perspectives on management of moderate-to-severe atopic dermatitis: a multidisciplinary consensus addressing current and emerging therapies. J Allergy Clin Immunol Pract. 2017;5(6):1519–1531.
- Katayama I, Aihara M, Ohya Y, Japanese Society of Allergology, et al. Japanese guidelines for atopic dermatitis 2017. Allergol Int. 2017;66(2):230–247.
- Wollenberg A, Barbarot S, Bieber T, European Dermatology Forum (EDF), the European Academy of Dermatology and Venereology (EADV), the European Academy of Allergy and Clinical Immunology (EAACI), the European Task Force on Atopic Dermatitis (ETFAD), European Federation of Allergy and Airways Diseases Patients’ Associations (EFA), the European Society for Dermatology and Psychiatry (ESDaP), the European Society of Pediatric Dermatology (ESPD), Global Allergy and Asthma European Network (GA2LEN) and the European Union of Medical Specialists (UEMS), et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol. 2018;32(6):850–878.
- Sidbury R, Davis DM, Cohen DE, American Academy of Dermatology, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327–349.
- Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? Recommendations from an expert panel of the International Eczema Council. J Am Acad Dermatol. 2017;77(4):623–633.
- Macdonald LE, Karow M, Stevens S, et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci USA. 2014;111(14):5147–5152.
- Murphy AJ, Macdonald LE, Stevens S, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014;111(14):5153–5158.
- European Medicines Agency. DUPIXENT® (dupilumab). Summary of product characteristics. [cited 2020 Dec 01]. Available from: https://ec.europa.eu/health/documents/community-register/2019/20190801145601/anx_145601_en.pdf.
- US Food and Drug Administration. DUPIXENT® (dupilumab). Highlights of prescribing information. [cited 2020 Dec 01]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761055s014lbl.pdf.
- Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–2348.
- Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–2303.
- de Bruin-Weller M, Gooderham M, Cather JC, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to cyclosporine A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018;178(5):1083–1101.
- Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(1):44–56.
- Beck LA, Thaçi D, Deleuran M, et al. Dupilumab provides favorable safety and sustained efficacy for up to 3 years in an open-label study of adults with moderate-to-severe atopic dermatitis. Am J Clin Dermatol. 2020;21(4):567–577.
- Wollenberg A, Beck LA, Blauvelt A, et al. Laboratory safety of dupilumab in moderate-to-severe atopic dermatitis: results from three phase III trials (LIBERTY AD SOLO 1, LIBERTY AD SOLO 2, LIBERTY AD CHRONOS). Br J Dermatol. 2020;182(5):1120–1135.
- Siegfried EC, Simpson EL, Paller AS, et al. Laboratory safety findings for dupilumab in adolescent patients with moderate-to-severe atopic dermatitis inadequately controlled by or ineligible for topical therapy. Am J Clin Dermatol. 2021; in press.
- Wollenberg A, Siegfried E, Thaçi D, et al. Laboratory safety of dupilumab in pediatric patients aged ≥6 to < 12 years with severe atopic dermatitis: results from a phase 3 trial (LIBERTY AD PEDS). Poster presented at the 29th Congress of the European Academy of Dermatology and Venereology; Virtual meeting; 29–31 October, 2020.
- U.S. Department of Health and Human Services. Common terminology criteria for adverse events (CTCAE); version 5.0. November 27, 2017. [cited 2020 Dec 01]. Available from: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf.
- Nordic MPN Study Group. Guidelines for the diagnosis and treatment of eosinophilia. 3rd version. May 2018. [cited 2020 Dec 01]. Available from: http://www.nmpn.org./index.php/guidelines/18-care-program-for-the-diagnosis-and-treatment-of-eosinophilia-3rd-version-may-2018/file.
- Thijs J, Krastev T, Weidinger S, et al. Biomarkers for atopic dermatitis: a systematic review and meta-analysis. Curr Opin Allergy Clin Immunol. 2015;15(5):453–460.
- Simpson EL, Villarreal M, Jepson B, et al. Patients with atopic dermatitis colonized with Staphylococcus aureus have a distinct phenotype and endotype. J Invest Dermatol. 2018;138(10):2224–2233.
- Mukai H, Noguchi T, Kamimura K, et al. Significance of elevated serum LDH (lactate dehydrogenase) activity in atopic dermatitis. J Dermatol. 1990;17(8):477–481.
- Kou K, Aihara M, Matsunaga T, et al. Association of serum interleukin-18 and other biomarkers with disease severity in adults with atopic dermatitis. Arch Dermatol Res. 2012;304(4):305–312.
- US Food and Drug Administration 2017. JAKAFI® (ruxolitinib). Highlights of prescribing information. [cited 2020 Dec 01]. Available from: www.accessdata.fda.gov/drugsatfda_docs/label/2017/202192s015lbl.pdf.
- US Food and Drug Administration 2018. XELJANZ® (tofacitinib). Highlights of prescribing information. [cited 2020 Dec 01]. Available from: www.accessdata.fda.gov/drugsatfda_docs/label/2018/203214s018lbl.pdf.
- US Food and Drug Administration 2017. OLUMIANT® (baricitinib). Highlights of prescribing information. [cited 2020 Dec 01]. Available from: www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf.