
Abstract
Purpose
Radiation-induced bystander effect (RIBE) frequently is seen as DNA damage in unirradiated bystander cells, but the repair processes initiated in response to that DNA damage are not well understood. RIBE-mediated formation of micronuclei (MN), a biomarker of persistent DNA damage, was previously observed in bystander normal fibroblast (AG01522) cells, but not in bystander human chondrosarcoma (HTB94) cells. The molecular mechanisms causing this disparity are not clear. Herein, we investigate the role of DNA repair in the bystander responses of the two cell lines.
Methods
Cells were irradiated with X-rays and immediately co-cultured with un-irradiated cells using a trans-well insert system in which they share the same medium. The activation of DNA damage response (DDR) proteins was detected by immunofluorescence staining or Western blotting. MN formation was examined by the cytokinesis-block MN assay, which is a robust method to detect persistent DNA damage.
Results
Immunofluorescent foci of γH2AX and 53BP1, biomarkers of DNA damage and repair, revealed a greater capacity for DNA repair in HTB94 cells than in AG01522 cells in both irradiated and bystander populations. Autophosphorylation of ATR at the threonine 1989 site was expressed at a greater level in HTB94 cells compared to AG01522 cells at the baseline and in response to hydroxyurea treatment or exposure to 1 Gy of X-rays. An inhibitor of ATR, but not of ATM, promoted MN formation in bystander HTB94 cells. In contrast, no effect of either inhibitor was observed in bystander AG01522 cells, indicating that ATR signaling might be a pivotal pathway to preventing the MN formation in bystander HTB94 cells. Supporting this idea, we found an ATR-dependent increase in the fractions of bystander HTB94 cells with pRPA2 S33 and RAD51 foci. A blocker of RAD51 facilitated MN formation in bystander HTB94 cells.
Conclusion
Our results indicate that HTB94 cells were likely more efficient in DNA repair than AG01522 cells, specifically via ATR signaling, which inhibited the bystander signal-induced MN formation. This study highlights the significance of DNA repair efficiency in bystander cell responses.
Introduction
The radiation-induced bystander effect (RIBE) is the responses of unirradiated cells to bystander signaling released from nearby irradiated cells. Although radiotherapy aims to give a sufficient dose to the target lesion to kill all tumor cells, there might be adjacent or distant tumor cells (e.g. metastases or micro-extensions from the tumor) that are not irradiated. These cells may die or exhibit genetic instability as a result of bystander signals emitted from irradiated cells. Furthermore, the bystander signals may cause toxicity to neighboring normal tissues. Therefore, RIBE could have significant impacts on radiotherapy efficacy. Understanding the molecular mechanisms of RIBE may provide insight into suitable strategies to optimize radiotherapy by maximizing cancer cell killing and sparing healthy cells (Prise and O’Sullivan Citation2009).
Multiple mechanisms mediate RIBE including diffusion of soluble factors or gap junction-intercellular communication (Mothersill and Seymour Citation1997; Azzam et al. Citation2001; Azzam et al. Citation2002). Central to RIBE is DNA damage induction caused by elevated bystander signaling factors such as reactive oxygen species (ROS), nitric oxide, or cytokines (Klammer et al. Citation2015; Mothersill et al. Citation2018). Endpoints of RIBE range from short-term responses like formation of DNA damage response (DDR)-associated protein foci to longer-term responses such as persistent DNA damage to form micronuclei (MN) or sister chromatid exchanges that in general contribute to mutation induction and chromosomal instability (Klammer et al. Citation2015).
DNA damage could activate cellular DDR signaling consisting of various mediator proteins to orchestrate an extensive network of cellular processes such as DNA repair, DNA replication, or cell cycle control. Ataxia-telangiectasia mutated (ATM) and Ataxia-telangiectasia and Rad3-related protein (ATR) are two primary kinases involved in regulating DDR signaling. ATM primarily responds to double-stranded DNA breaks (DSB). On the other hand, ATR is activated by single-stranded DNA that arises from replication stress, DSB resection, or the processing of bulky lesions (Shiotani and Zou Citation2009; Awasthi et al. Citation2015). Once activated, both kinases induce a broad spectrum of signal transduction pathways to initiate the cellular responses to DNA damage.
In contrast to irradiated cells, DNA repair in unirradiated bystander cells is still little understood. Prise’s group has proposed a model of DDR signaling whereby bystander cells recruit ATR-dependent signaling to repair bystander DNA damage, probably replication stress, while DNA damage in irradiated cells, mainly DSB, is regulated by ATM-dependent signaling (Burdak-Rothkamm et al. Citation2007; Prise et al. Citation2007; Burdak-Rothkamm and Prise Citation2009). This model was presented based on investigations performed with only a few cell types, including human glioma and astrocyte cells (Burdak-Rothkamm et al. Citation2007; Burdak-Rothkamm et al. Citation2008; Burdak-Rothkamm et al. Citation2015), and this pattern may be specific to these cell lines due to their genetic features. There is a lack of data on how other cell lines respond to DNA damage produced by bystander signaling.
In a previous study, we reported a significant difference in bystander responses of human normal fibroblast (AG01522) cells and human chondrosarcoma (HTB94) cells (Wakatsuki et al. Citation2012). While bystander AG01522 cells produced MN, no increase in MN was found in bystander HTB94 cells regardless of linear energy transfer of irradiation to the treated cells (Wakatsuki et al. Citation2012). The mechanism underlying this difference remains unclear. Furthermore, some evidence showed that DNA repair deficiency could enhance the sensitivity to bystander signals, and the repair phenotype of bystander cells could determine their overall responses (Nagasawa et al. Citation2003; Kashino et al. Citation2004; Mothersill et al. Citation2004; Tu et al. Citation2019). In this study, we aim to clarify the involvement of the DNA repair capacity of AG01522 and HTB94 cells in their bystander responses. We hypothesize that HTB94 cells repair DNA damage more efficiently than AG01522 cells, thereby suppressing the RIBE-induced formation of MN in the bystander cells. We also investigated the DDR signaling that might be involved in RIBE-mediated MN induction in bystander cells.
Materials and methods
Materials
ATR kinase inhibitor VE821 was purchased from APExBIO (Houston, TX, USA). ATM inhibitor KU55933 was obtained from Tocris Bioscience (Ellisville, MO, USA). RAD51 inhibitor RI-1 was from Cayman Chemical Company (MI, USA). Hydroxyurea (HU) was obtained from Tokyo Chemical Industry Co. (Tokyo, Japan). Falcon cell culture inserts and companion 6-well plates were purchased from Corning Incorporated (NY, USA). Phospho-ATR Thr1989 (D5K8W) antibody (#30632) and ATR (E1S3S) antibody (#2790) obtained from Cell Signaling Technology (Danvers, MA, USA) were diluted by Can Get Signal solution (TOYOBO Inc., Osaka, Japan). RAD51 (H-92) antibody (#sc-8349) was acquired from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Phospho-RPA32 S33 antibody (#A300-246A-M) was obtained from Bethyl Laboratories Inc. (Montgomery, TX, USA). 53BP1 antibody (#ab21083) and vinculin antibody (EPR8185) (#ab129002) were obtained from Abcam (Cambridge, UK). Phospho-histone H2AX (Ser139) clone JBW301 antibody (#05-636) was purchased from Millipore (Temecula, CA, USA). All other reagents were of the highest purity available.
Cell culture and co-culture system
HTB94 cells (ATCC, Manassas, VA, USA) were grown at 37 °C in a humidified incubator consisting of no CO2 and 100% room air atmosphere with Leibovitz’s L-15 medium (ATCC) supplemented with 10% fetal bovine serum (FBS, Sigma) and antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin). AG01522 cells (Genetic Cell Repository at the Coriell Institute for Medical Research, Camden, NJ) were cultured in an 95% air and 5% CO2 humidified incubator with alpha-modified MEM medium (Sigma) containing 20% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. The cells were maintained in T75 flasks (Thermo Fisher Scientific, Rockford, IL, USA) until reaching 80–100% confluence and passage into the appropriate dishes for each experiment. A trans-well insert co-culture system, as described previously (Yang et al. Citation2005), was used to study the effects of medium-mediated bystander signaling. Glass coverslips (Matsunami glass IND., Osaka, Japan) with a diameter of 18 mm were used to grow cells for the MN and immunofluorescence (IF) assays. One day before experiments, cells were seeded on an insert at a density of 0.6 × 105 cells for MN assay or 1 × 105 cells for IF assay, respectively. Simultaneously, cells were seeded on a well of a six-well companion plate (1.38 × 105 cells for MN assay or 2.3 × 105 cells for IF assay, respectively). The cells on an insert were X-irradiated, then immediately transferred to co-culture with the un-irradiated cells in a well of a six-well companion plate until the time at which the cells were fixed. The control samples were treated in the same way, except X-irradiation.
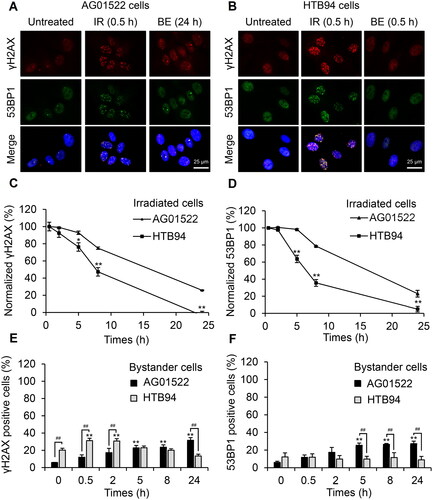
Figure 1. Different kinetics of DNA repair in normal fibroblast AG01522 and human chondrosarcoma HTB94 cells. Cells were irradiated with 1 Gy X-rays and co-cultured with un-irradiated cells using an insert trans-well system. The cells were fixed at the specified time points and immunofluorescently stained with γH2AX and 53BP1 antibodies. (A, B) Representative images of induced foci were shown at the time points when their maximum formation was observed, including post-30 min in irradiated (IR) AG01522 and HTB94 cells, post-24 h in bystander (BE) AG01522 cells, and post-30 min in bystander HTB94 cells. The repair kinetics of cells following 1 Gy X-rays were monitored by following γH2AX (C) and 53BP1 (D) foci. Cells with ≥ 5 foci were considered positive and data are shown as the percentage of total cells from which the background was subtracted and normalized by the data at 30 min after irradiation. (E, F) Quantification of fractions of γH2AX and 53BP1 positive cells in bystander cell populations, respectively. The experiments with AG01522 cells were done independently three times, while seven replicates with γH2AX staining and three repeats with 53BP1 staining were performed with HTB94 cells. **p < .01 vs 0 h in each group; #p < .05, ##p < .01.
X-irradiation
X-rays were generated by the MultiRad 225 system (Faxitron Bioptics, Tucson, AZ, USA) at Gunma University and the X-RAD 320 machine (Precision X-Ray Inc, North Brandford, CT, USA) at Emory University. The former was set at 200 kV and 14.6 mA with a dose rate of 1.12 Gy/min through a 0.5 mm-copper and aluminum filter, while the latter was set at 300 kV and 10 mA with a dose rate of 1.67 Gy/min through a 2 mm-aluminum filter.
Western blotting
Cells were seeded on a 35-mm plate with a density of 4.6 × 105 cells per plate one day before treatment with HU. After treatment, the cells were washed three times with cold phosphate buffered saline (PBS), then lysed using a lysis buffer containing 4% SDS, 125 mM Tris-HCl (pH 7.5), and 1% protease inhibitor cocktail (Takara Bio Inc., CA, USA), followed by heating at 95 °C for 20 min. The protein concentration of each sample was determined by the Pierce bicinchoninic acid protein assay (Thermo Fisher Scientific) and normalized before mixing with a quarter volume of SDS-PAGE sample loading buffer (Wako) containing 20% 2-mercaptoethanol, then heated for 5 min at 95 °C. The cell lysates (20 µg/sample) were separated by SDS-PAGE and then electro-transferred onto PVDF membranes (Bio-Rad Laboratories, CA, USA) at 2 mA/cm2 for 1 h. The membranes were blocked with 5% skim milk in TTBS [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% Tween 20] for 1 h at room temperature, incubated with primary antibodies overnight at 4 °C, and then reacted with HRP-conjugated IgG secondary antibodies at room temperature for 1 h. The immunoreactive proteins on the membranes were detected via enhanced chemiluminescence using Immobilon Western Chemiluminescent HRP substrate (Millipore, Burlington, MA, USA). The presented results are representative of four independent experiments. The band intensity was measured using Fiji software (Wayne Rasband, National Institutes of Health, USA).
Micronucleus assay
The cytokinesis-block MN assay was used to measure the frequency of MN formation as described previously with modification (Yang et al. Citation2005). Briefly, bystander cells seeded in wells of a six-well companion plate were pretreated with DMSO or the indicated inhibitors for 30 min prior to start of co-culture. Immediately after irradiation, irradiated cells on an insert were added into co-culture with the unirradiated bystander cells. Cytochalasin B (Wako, Osaka, Japan) was added to the cultures to a final concentration of 1.5 µg/mL. Media containing the inhibitors of ATM or ATR were changed to fresh media containing only cytochalasin B after 5 h, and cells were incubated up to a total of 72 h, while media containing the RAD51 inhibitor were kept on cells for the total 72 h. The cells were then fixed with methanol:acid acetic (3:1, v/v) and stained with 10 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) solution (Dojindo, Kumamoto, Japan) for 10 min at room temperature. A minimum of 400 binucleate cells in at least 30 view fields was counted for each sample in each experiment.
Immunofluorescence
Bystander cells were pretreated with DMSO or ATR inhibitor (2 µM VE821) for 30 min. To determine S-phase dependence, cells were pretreated with the inhibitor in the presence of 1 µM 5-ethynyl-2′-deoxyuridine (EdU) for 30 min. The cells were co-cultured with irradiated cells and subsequently fixed at the indicated time points with 4% paraformaldehyde in PBS (Wako) for 15 min at room temperature. Fixed cells were then permeabilized with 0.5% Triton X-100 in PBS for 15 min on ice, blocked with 10% bovine serum albumin (BSA, Sigma Aldrich, St. Louis, MO, USA) and 0.1% Triton X-100 in PBS for 45 min at room temperature, and incubated with primary antibodies at 4 °C overnight. The cells reacted with secondary antibodies Alexa Fluor 568 goat anti-mouse IgG or Alexa Fluor 488 goat anti-rabbit IgG (1:1000 dilution, Thermo Fisher Scientific) for 45 min at room temperature. Incorporated EdU was visualized by Click-iT EdU Alexa Fluor 647 imaging kit (Life Technologies Corporation, OR, USA). Cell nuclei were counterstained with 10 µg/mL DAPI for 10 min at room temperature. The coverslips were mounted with PermaFluor mountant (Thermo Fisher Scientific) on glass slides. Images were captured by an EVOS M5000 microscope (Invitrogen, Thermo Fisher Scientific). At least 400 cells in 30 fields were examined. Cells with at least five foci were considered positive cells.
Statistical analysis
Data were shown as the mean ± standard error from at least three independent experiments. Statistical significance was assessed using a one-way or two-way ANOVA test, followed by a Dunnett’s or Bonferroni’s multiple-comparison test using GraphPad Prism version 9.3.1 software (San Diego, CA, USA). P < .05 was considered significant.
Results
More proficient DNA repair capacity of HTB94 cells than AG01522 cells
To evaluate the kinetics of DNA repair in the two cell lines, we immunostained irradiated and bystander cells for two biomarkers of DNA damage and repair including phosphorylated histone H2AX at serine 139 (γH2AX) and p53-binding protein 1 (53BP1). Irradiation of both cell lines with 1 Gy X-rays triggered the greatest increase in the fraction of cells with γH2AX and 53BP1 foci in the irradiated cells by 30 min, with the fraction of cells with foci declining in subsequent times (). Notably, the fraction of HTB94 cells with γH2AX foci and 53BP1 decreased faster than in AG01522 cells (). Twenty-four hours after irradiation, the number of HTB94 cells with γH2AX and 53BP1 foci was reduced to nearly the baseline, while over 20% of AG01522 cells still retained foci in comparison to the untreated controls ().
On the other hand, the bystander populations of the two cell lines co-cultured with the 1 Gy-irradiated cells in an insert trans-well system exhibited a distinct kinetic response. The bystander HTB94 cells co-cultured with irradiated cells showed a significantly increased fraction of cells with γH2AX foci at 30 min and 2 h, compared to cells co-cultured with unirradiated cells, then the fraction of cells with γH2AX foci decreased to the basal level after 5 h, whereas no bystander HTB94 cells positive for 53BP1 were detected (). In the bystander AG01522 cells, the fraction of cells with γH2AX foci gradually increased throughout the incubation period, while the fraction of cells with 53BP1 foci increased to about 5 h then remained constant ().
Involvement of DDR signaling in the RIBE-induced MN formation
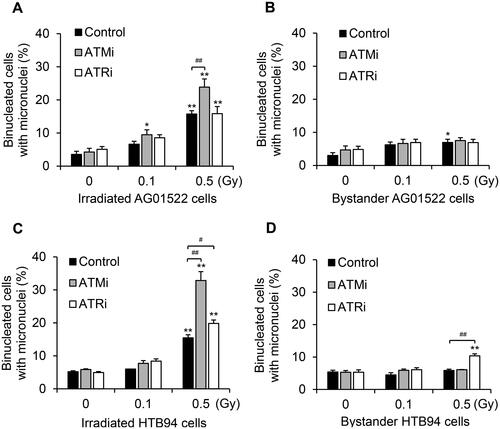
To explore the DDR signaling involved in RIBE-mediated MN induction, we used the cytokinesis-block MN assay following co-culture of irradiated and bystander cells with ATM or ATR inhibitors in the media. Treatment with an ATM inhibitor (KU55933), but not an ATR inhibitor (VE821), significantly increased the fraction of 0.5 Gy-irradiated AG01522 cells with MN (), yet neither inhibitor affected the RIBE-induced MN generation of these cells (). On the other hand, both inhibitors boosted MN formation in the irradiated HTB94 cells, with the ATM inhibitor causing significantly greater effect than the ATR inhibitor (). No increased MN formation was observed in the bystander HTB94 cells, without inhibitor treatment (), which is consistent with our previous study (Wakatsuki et al. Citation2012). However, an inhibitor of ATR, but not ATM, considerably promoted MN induction in the 0.5-Gy co-cultured bystander cells (). These data indicate that although both ATM and ATR signaling were activated to repair DNA in the irradiated HTB94 cells, only ATR signaling displayed a role in responding to damage in the bystander HTB94 cells. In comparison, AG01522 cells showed a weak capacity of ATR signaling to repair DNA in both irradiated and bystander populations.
Figure 2. Role of DNA damage response (DDR) pathways in micronuclei (MN) formation of two cell lines in response to radiation-induced bystander signals. Cells were co-cultured with irradiated cells (0–0.5 Gy) after pretreatment with inhibitors of ATM (ATMi, 10 µM KU55933) or ATR (ATRi, 2 µM VE821) for 30 min. The frequency of MN formation was measured by a cytokinesis-block assay. Results are the mean ± standard error of four independent experiments with AG01522 cells and three independent experiments with HTB94 cells. *p < .05; **p < .01 vs 0 Gy in each group; #p < .05; ##p < .01.
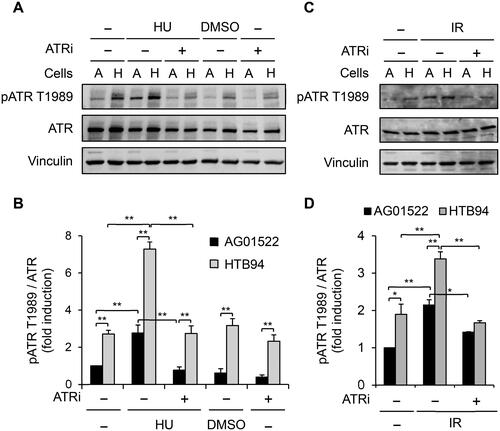
The higher level of autophosphorylation of ATR at threonine 1989 site (pATR T1989) in HTB94 cells than in AG01522 cells
pATR T1989 is a marker of active ATR signaling beca use the phosphorylation can stimulate ATR kinase activity and potentiate ATR substrate recognition (Liu SZ et al. Citation2011; Nam et al. Citation2011). Therefore, we employed Western blotting to examine pATR T1989 in the two cell lines to evaluate their ability to activate ATR signaling. shows that pATR T1989 in HTB94 cells was much higher at the baseline level than in AG01522 cells. Exposure of the cells to HU, a compound known to cause replication stress-dependent activation of ATR signaling (Cliby et al. Citation1998), or irradiation of the cells with 1 Gy X-rays significantly increased pATR T1989 in both cell lines, with a greater level in HTB94 cells than in AG01522 cells, whereas incubation with an ATR inhibitor abolished the activation (). These data suggest that HTB94 cells might have a strong capacity for DNA repair, specifically via ATR signaling, thereby limiting the RIBE-induced MN formation in the bystander cells. In subsequent experiments, we investigated ATR signaling in bystander HTB94 cells.
Figure 3. Phosphorylation of ATR at threonine 1989 (pATR T1989) in AG01522 and HTB94 cells. Cells were pretreated with 2 µM ATRi for 30 min and exposed to hydroxyurea (HU, 20 mM) (A, B) or irradiated with 1 Gy X-rays (C, D). The cells were collected after 5 h. (A, C) The cell lysate preparation was analyzed by Western blotting. A: AG01522 cells, H: HTB94 cells. (B, D) The relative intensity of pATR T1989 bands was measured by Fiji software, normalized to ATR, and shown as fold induction compared to untreated AG01522 cells. Each value is the mean ± standard error of four independent experiments for (A, B) and three times for (C, D). *p < .05; **p < .01.
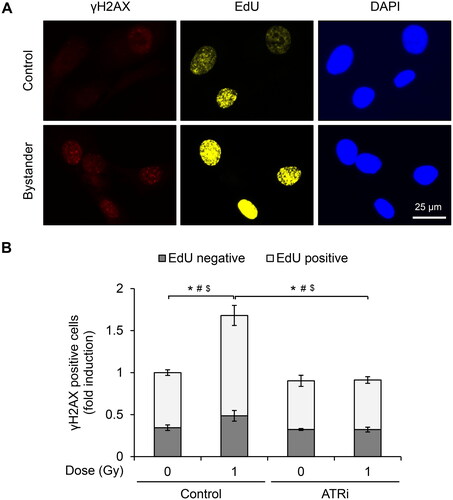
ATR-dependent increase of γH2AX foci mainly in the actively replicating HTB94 cells upon bystander signaling
Burdak-Rothkamm et al. previously showed that γH2AX formation is downstream of ATR signaling and restricted to the S-phase of bystander human glioma and astrocyte cells (Burdak-Rothkamm et al. Citation2007). We tested this finding in the bystander HTB94 cells. Before co-culture with irradiated cells, the bystander cells were pulse-labelled for 30 min with EdU (), a compound that can efficiently incorporate into replicating DNA and has been used frequently to mark S-phase cells (Salic and Mitchison Citation2008; Dhuppar et al. Citation2020). We found that γH2AX foci formation was predominantly localized to EdU-positive cells, while an ATR inhibitor prevented this increase ().
Figure 4. ATR-dependent induction of γH2AX foci in bystander HTB94 cells actively replicating their DNA. HTB94 cells were treated with 2 µM ATRi in the presence of 1 µM 5-ethynyl-2´-deoxyuridine (EdU) for 30 min before co-culturing with 1-Gy irradiated HTB94 cells. After 30 min, the cells were fixed and stained with γH2AX antibody. EdU was detected by click chemistry. (A) Representative images of γH2AX and EdU staining. (B) Quantification of γH2AX positive cells in EdU-positive and EdU-negative cells. The cells with ≥ 5 foci of γH2AX were considered positive and expressed as fold induction. Results are the mean ± standard error of five independent experiments. *, #, $, p < .05 compared in total cells, EdU-positive, and EdU-negative cells, respectively.
Activation of ATR signaling cascades in the bystander HTB94 cells
In response to DNA damage, DDR kinase regulators could phosphorylate RPA2 on multiple N-terminal residues as downstream targets (Maréchal and Zou Citation2015). Among the phosphorylated sites of RPA2, serine 33 (pRPA2 S33) is phosphorylated upon ATR activation in response to DSB or DNA replication stress (Vassin et al. Citation2009; Liu SQ et al. Citation2012; Shiotani et al. Citation2013; Maréchal and Zou Citation2015). Hence, to examine the activation of ATR signaling downstream, we co-cultured the irradiated and bystander cells for 0–24 h in the presence and absence of an ATR inhibitor and immunostained with pRPA2 S33 antibody. The fraction of irradiated cells with pRPA2 S33 foci peaked at 30 min and remained elevated at 8 h, but the fraction of bystander cell with elevated pRPA2 S33 foci levels had returned to baseline by 5 h (). The ATR inhibitor significantly abolished the fraction of bystander cells with pRPA2 S33 foci, which had increased after 30 min and 2 h (), but it negligibly reduced the number of irradiated cells with pRPA2 S33 foci ().
Figure 5. Activation of downstream ATR substrates in bystander HTB94 cells. HTB94 cells were pretreated with 2 µM ATRi for 30 min then co-cultured with 1-Gy irradiated HTB94 cells and fixed at the indicated times for immunostaining. Representative images of foci formation of (A) RPA2 phosphorylated at serine 33 (pRPA2 S33) and (D) RAD51. Foci images are shown at the time point when maximum induction of proteins was observed, including (A) 30 min after irradiation in irradiated (IR) and bystander (BE) cells stained for pRPA2 S33, (D) 2 h after irradiation for irradiated cells and 30 min after irradiation and start of co-culture for bystander cells stained for RAD51, respectively. (B, C, E, F) Quantification of positive cells. The cells with ≥ 5 foci were considered positive, and data are expressed as the percentage of total cells. Each value is the mean ± standard error of four independent experiments with pRPA2 S33 (B, C) and RAD51 (E, F). *p < .05, **p < .01 vs 0 h in each group; #p < .05, ##p < .01.
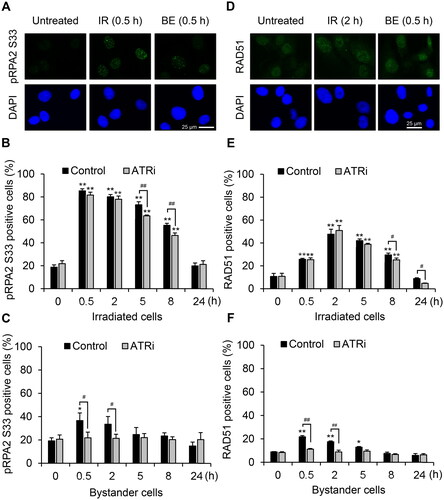
Furthermore, we detected foci formation of RAD51 that is a well-known downstream effector of ATR signaling involved in DNA repair by the homologous recombination pathway (Tutt and Ashworth Citation2002; Buisson et al. Citation2017) in both irradiated and bystander HTB94 cells (). The ATR inhibitor had a minor effect on the fraction of irradiated cells with RAD51 foci () but had more impact on the increased fraction of bystander HTB94 cells with RAD51 foci at 0.5 and 2 h (). The kinetics of RAD51 foci formation in the bystander HTB94 cells are similar to γH2AX and pRPA2 S33 responses ( and ). Our data suggest that ATR signaling was activated by the DNA damage in bystander cells and then signals to downstream cascades including γH2AX, pRPA2 S33, and RAD51.
Role of RAD51 in suppression of MN formation in bystander HTB94 cells
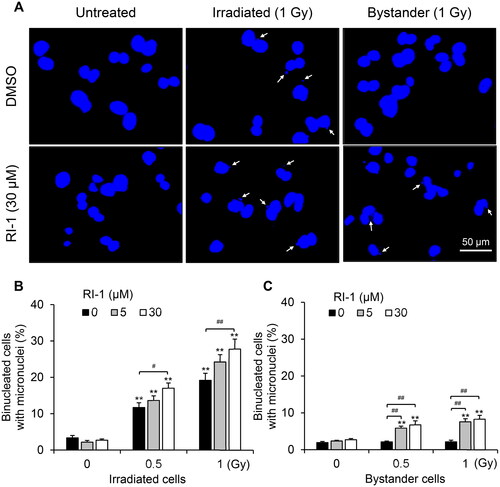
To evaluate the contribution of RAD51 to RIBE-mediated MN formation in bystander HTB94 cells, we co-cultured bystander cells with irradiated cells for 72 h in the presence of RI-1, a RAD51 inhibitor that irreversibly destabilizes the formation of RAD51 filaments on DNA by covalent binding to the surface of RAD51 protein (Budke et al. Citation2012). RI-1 treatment significantly boosted the fraction of both irradiated and bystander cells with MN (), indicating the critical role of ATR/RAD51 in regulating RIBE-mediated MN formation in bystander HTB94 cells.
Figure 6. Increase of MN induction in bystander HTB94 cells by blocking RAD51. Bystander cells were co-cultured with irradiated HTB94 cells (0–1 Gy) for 72 h in the presence of RAD51 inhibitor (RI-1). (A) Representative pictures of MN formation in cells. White arrows indicate MN. (B, C) Quantification of binucleated cells with MN in irradiated and bystander cells, respectively. Each value is the mean ± standard error of four independent experiments. **p < .01 vs 0 Gy in each group; #p < .05, ##p < .01.
Discussion
Radiation-induced bystander responses of un-irradiated cells are complex and dependent on various extrinsic and intrinsic cellular factors such as genetic constitution, DNA repair capacity, surrounding environments, or pathways activated in irradiated and bystander cells (Verma and Tiku Citation2017). In the present study, we describe the role of the DNA repair capacity of receiving cells in bystander responses by investigating DDR pathways in AG01522 and HTB94 cells. We found that MN induction by bystander signals is inhibited by ATR/RAD51 signaling in bystander HTB94 cells, whereas neither ATM nor ATR signaling influences RIBE-mediated formation of MN in bystander AG01522 cells. The proposed pathways to explain these findings are shown schematically in .
Figure 7. Proposed model of DDR-mediated regulation of MN formation caused by radiation-induced bystander signaling in bystander cells. (A) HTB94 cells. (B) AG01522 cells. (C) HTB94 cells with ATR inhibitor (ATRi) or RAD51 inhibitor (RI-1).

An immunostaining experiment using γH2AX and 53BP1 antibodies revealed that irradiated HTB94 cells resolved radiation-induced γH2AX and 53BP1 foci rapidly and most foci were cleared within 24 h, while approximately 20% of irradiated AG01522 cells still contained foci at 24 h (). Consistent with our data, Lohberger et al. showed that chondrosarcoma cells could activate efficient DNA repair signaling after photon and proton irradiation and repair most DNA damage within 24 h (Lohberger et al. Citation2021). Additionally, our previous results from the colony formation assay showed that HTB94 cells had a higher survival fraction after exposure to radiation than AG01522 cells (Wakatsuki et al. Citation2012). Supporting our immunostaining data, we found that HTB94 cells had higher baseline levels of pATR T1989 than AG01522 cells, as well as a greater responsiveness to either irradiation or HU treatment (). These data indicate a greater efficacy of DNA repair in HTB94 cells than in AG01522 cells.
Different cell types have been observed to respond differently to bystander signaling, with some cells expressing damage while others do not (Yang et al. Citation2005; Yang et al. Citation2007; Yang et al. Citation2011; Wakatsuki et al. Citation2012; Mukherjee and Chakraborty Citation2018). The lack of bystander effects could be attributed to cells’ incapacity to send or respond to signals. For example, HTB94 cells do not elicit bystander responses in the form of MN and 53BP1 foci induction, although HTB94 cells can release bystander factors after irradiation that elicit responses in another cell type (Wakatsuki et al. Citation2012). This suggests that the lack of RIBE-mediated MN and 53BP1 foci induction results from the characteristics of the receiving HTB94 cells themselves. We previously examined 53BP1 foci formation in bystander HTB94 cells at only a 5 h co-culture time based on the observation of the response of AG01522 cells at this time (Yang et al. Citation2011; Wakatsuki et al. Citation2012). Because the factors influencing cellular bystander responses may differ depending on experimental conditions, endpoints studied, or time-course analysis (Mukherjee and Chakraborty Citation2018), we tested bystander cells throughout a range of time intervals within 24 h by using both 53BP1 and γH2AX foci assays in the present work. Interestingly, we found an ATR-dependent increase in the fraction of bystander HTB94 cells with γH2AX foci that was rapidly resolved within 5 h (). We also detected the ATR-dependent increase of bystander HTB94 cells with foci of downstream ATR signaling including pRPA2 S33 and RAD51 (). Treatment with an ATR inhibitor promoted the fraction of bystander HTB94 cells with MN (). These results suggest that bystander HTB94 cells with active ATR signaling efficiently repair the RIBE-induced damage, thereby inhibiting MN formation (), whereas the fraction of bystander AG01522 cells with γH2AX and 53BP1 foci remain elevated until 24 h (). Both ATM and ATR inhibitors showed no effect on the RIBE-induced MN formation in bystander AG01522 cells (). These results suggest that the responses to bystander signals differ with different cell types and highly depend on cell characteristics such as DNA repair capacity.
The fraction of bystander HTB94 cells with pRPA2 S33 and RAD51 foci was significantly reduced by the ATR inhibitor, whereas the fraction of irradiated cells with these foci decreased negligibly in the presence of inhibitor (). This suggests that the bystander HTB94 cells rely more on ATR signaling than do the irradiated HTB94 cells to repair DNA damage.
Bystander AG01522 cells recruited both γH2AX and 53BP1 to the damage sites (); however, we could not detect any increase of the fraction of bystander HTB94 cells with 53BP1 foci at any time points evaluated (). Similar to bystander HTB94 cells, Grifalconi et al. showed that bystander human lymphoblastoid TK6 cells had a partial absence of 53BP1 colocalization in the γH2AX-induced sites (Grifalconi et al. Citation2007). The observation raises an interesting question: why do some cell lines have simultaneous bystander responses of γH2AX and 53BP1 while others do not? The genetic constitution may be one of the critical factors affecting the difference such that differing DNA repair capacity can cause distinct bystander responses. In the present work, we observed an ATR-dependent increase of the fraction of bystander HTB94 cells with γH2AX foci, but not 53BP1 foci, which occurs predominantly in EdU-positive cells (). Since bystander factors such as ROS or nitric oxide have been shown to disrupt progression of replication forks in S-phase cells and activate γH2AX foci formation in an ATR-dependent manner (Ward and Chen Citation2001; Burdak-Rothkamm et al. Citation2007), we postulate that DNA damage in bystander HTB94 cells might occur primarily in S-phase cells and may be caused by replication stress. HTB94 cells with active ATR signaling may repair these bystander-induced damage efficiently. On the other hand, bystander signaling may initially trigger stalled replication forks in bystander AG01522 cells. However, the DNA repair capacity of AG01522 cells may be insufficient to repair all stalled replication forks, resulting in secondary DSB formation that may recruit both γH2AX and 53BP1 to the damage sites. Additional studies on the nature of bystander DNA damage and its relationship with γH2AX and 53BP1 responses in each cell line are further required to elucidate this question.
In conclusion, our data suggest a critical link between DNA repair capacity and cellular responses to bystander signaling. HTB94 cells had a more proficient ability to repair bystander signaling-induced DNA damage than did AG01522 cells, thereby causing different longer-term bystander responses. Future studies on the difference in DDR signaling in bystander normal and tumor cells will hopefully reveal novel targets for optimizing the therapeutic responses to radiotherapy.
Acknowledgments
We would like to acknowledge Ms. Yoshika Machida (Gunma University) and Mr. Mark E. Essien (Emory University) for laboratory assistance. We thank Dr. Noriyuki Okonogi, Dr. Anggraeini Puspitasari, and Dr. Sandip K. Rath for many helpful discussions throughout the project.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Nho Cong Luong
Nho Cong Luong, PhD, is Postdoctoral Researcher at Department of Radiation Oncology and Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA, USA. He was a postdoc at Gunma University Initiative for Advanced Research, Gunma University, Gunma, Japan.
Hidemasa Kawamura
Hidemasa Kawamura, MD, PhD, is Professor at Gunma University Heavy Ion Medical Center, Gunma University, Gunma, Japan.
Hiroko Ikeda
Hiroko Ikeda, PhD, is Assistant Professor at Department of Life Science, Faculty of Science and Engineering, Kindai University, Osaka, Japan.
Reiko T. Roppongi
Reiko T. Roppongi, PhD, is Assistant Professor at Gunma University Initiative for Advanced Research, Gunma University, Gunma, Japan.
Atsushi Shibata
Atsushi Shibata, PhD, is Professor at Division of Molecular Oncological Pharmacy, Faculty of Pharmacy, Keio University, Tokyo, Japan.
Jiaxuan Hu
Jiaxuan Hu is a student majoring in Biology at Emory University School of Medicine, Atlanta, GA, USA.
Jinmeng G. Jiang
Jinmeng G. Jiang is a student majoring in Biology at Emory University School of Medicine, Atlanta, GA, USA.
David S. Yu
David S. Yu, MD, PhD, is the Jerome Landry MD Chair in Cancer Research and Professor at Department of Radiation Oncology and Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA, USA, where he also serves as director of the Division of Cancer Biology.
Kathryn D. Held
Kathryn D. Held, PhD, is President of the National Council on Radiation Protection and Measurements (NCRP), Bethesda, MD, USA; Associate Professor and Radiation Biologist at Department of Radiation Oncology, Massachusetts General Hospital/Harvard Medical School, Boston, MA, USA; and Distinguished Visiting Professor at Gunma University Initiative for Advanced Research, Gunma University, Gunma, Japan.
References
- Awasthi P, Foiani M, Kumar A. 2015. ATM and ATR signaling at a glance. J Cell Sci. 128(23):4255–4262.
- Azzam EI, De Toledo SM, Little JB. 2001. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from alpha-particle irradiated to nonirradiated cells. Proc Natl Acad Sci U S A. 98(2):473–478. doi:10.1073/pnas.011417098
- Azzam EI, De Toledo SM, Spitz DR, Little JB. 2002. Oxidative metabolism modulates signal transduction and micronucleus formation in bystander cells from alpha-particle-irradiated normal human fibroblast cultures. Cancer Res. 62(19):5436–5442.
- Budke B, Logan HL, Kalin JH, Zelivianskaia AS, McGuire WC, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. 2012. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 40(15):7347–7357. doi:10.1093/nar/gks353
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJL, Dellaire G, Haas W, Xia B, et al. 2017. Coupling of homologous recombination and the checkpoint by ATR. Mol Cell. 65(2):336–346. doi:10.1016/j.molcel.2016.12.007
- Burdak-Rothkamm S, Prise KM. 2009. New molecular targets in radiotherapy: DNA damage signalling and repair in targeted and non-targeted cells. Eur J Pharmacol. 625(1-3):151–155. doi:10.1016/j.ejphar.2009.09.068
- Burdak-Rothkamm S, Rothkamm K, McClelland K, Al Rashid ST, Prise KM. 2015. BRCA1, FANCD2 and Chk1 are potential molecular targets for the modulation of a radiation-induced DNA damage response in bystander cells. Cancer Lett. 356(2 Pt B):454–461. doi:10.1016/j.canlet.2014.09.043
- Burdak-Rothkamm S, Rothkamm K, Prise KM. 2008. ATM acts downstream of ATR in the DNA damage response signaling of bystander cells. Cancer Res. 68(17):7059–7065. doi:10.1158/0008-5472.CAN-08-0545
- Burdak-Rothkamm S, Short SC, Folkard M, Rothkamm K, Prise KM. 2007. ATR-dependent radiation-induced γH2AX foci in bystander primary human astrocytes and glioma cells. Oncogene. 26(7):993–1002. doi:10.1038/sj.onc.1209863
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. 1998. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17(1):159–169. doi:10.1093/emboj/17.1.159
- Dhuppar S, Roy S, Mazumder A. 2020. γH2AX in the S phase after UV irradiation corresponds to DNA replication and does not report on the extent of DNA damage. Mol Cell Biol. 40(20):e00328–00320.
- Grifalconi M, Celotti L, Mognato M. 2007. Bystander response in human lymphoblastoid TK6 cells. Mutat Res. 625(1-2):102–111. doi:10.1016/j.mrfmmm.2007.06.004
- Kashino G, Prise KM, Schettino G, Folkard M, Vojnovic B, Michael BD, Suzuki K, Kodama S, Watanabe M. 2004. Evidence for induction of DNA double strand breaks in the bystander response to targeted soft X-rays in CHO cells. Mutat Res. 556(1-2):209–215. doi:10.1016/j.mrfmmm.2004.08.009
- Klammer H, Mladenov E, Li FH, Iliakis G. 2015. Bystander effects as manifestation of intercellular communication of DNA damage and of the cellular oxidative status. Cancer Lett. 356(1):58–71. doi:10.1016/j.canlet.2013.12.017
- Liu S, Shiotani B, Lahiri M, Maréchal A, Tse A, Leung CCY, Glover JNM, Yang XH, Zou L. 2011. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell. 43(2):192–202. doi:10.1016/j.molcel.2011.06.019
- Liu SQ, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, Oakley GG. 2012. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 40(21):10780–10794. doi:10.1093/nar/gks849
- Lohberger B, Glänzer D, Eck N, Kerschbaum-Gruber S, Mara E, Deycmar S, Madl T, Kashofer K, Georg P, Leithner A, et al. 2021. Activation of efficient DNA repair mechanisms after photon and proton irradiation of human chondrosarcoma cells. Sci Rep. 11(1):24116. doi:10.1038/s41598-021-03529-9
- Maréchal A, Zou L. 2015. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 25(1):9–23. doi:10.1038/cr.2014.147
- Mothersill C, Rusin A, Fernandez-Palomo C, Seymour C. 2018. History of bystander effects research 1905-present; what is in a name? Int J Radiat Biol. 94(8):696–707. doi:10.1080/09553002.2017.1398436
- Mothersill C, Seymour C. 1997. Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int J Radiat Biol. 71(4):421–427. doi:10.1080/095530097144030
- Mothersill C, Seymour RJ, Seymour CB. 2004. Bystander effects in repair-deficient cell lines. Radiat Res. 161(3):256–263. doi:10.1667/rr3136
- Mukherjee S, Chakraborty A. 2018. Radiation-induced bystander phenomenon: insight and implications in radiotherapy. Int J Radiat Biol. 95(3):243–263. doi:10.1080/09553002.2019.1547440
- Nagasawa H, Huo L, Little JB. 2003. Increased bystander mutagenic effect in DNA double-strand break repair-deficient mammalian cells. Int J Radiat Biol. 79(1):35–41. doi:10.1080/0955300021000019230
- Nam EA, Zhao RX, Glick GG, Bansbach CE, Friedman DB, Cortez D. 2011. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem. 286(33):28707–28714. doi:10.1074/jbc.M111.248914
- Prise KM, Burdak-Rothkamm S, Folkard M, Kashino G, Shao C, Tartier L. 2007. New insights on radiation-induced bystander signalling and its relationship to DNA repair. Int Congr Ser. 1299:121–127. doi:10.1016/j.ics.2006.10.018
- Prise KM, O’Sullivan JM. 2009. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. 9(5):351–360. doi:10.1038/nrc2603
- Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 105(7):2415–2420. doi:10.1073/pnas.0712168105
- Shiotani B, Nguyen HD, Håkansson P, Maréchal A, Tse A, Tahara H, Zou L. 2013. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 3(5):1651–1662. doi:10.1016/j.celrep.2013.04.018
- Shiotani B, Zou L. 2009. ATR signaling at a glance. J Cell Sci. 122(Pt 3):301–304. doi:10.1242/jcs.035105
- Tu W, Dong C, Fu J, Pan Y, Kobayashi A, Furusawa Y, Konishi T, Shao C. 2019. Both irradiated and bystander effects link with DNA repair capacity and the linear energy transfer. Life Sci. 222:228–234. doi:10.1016/j.lfs.2019.03.013
- Tutt A, Ashworth A. 2002. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med. 8(12):571–576. doi:10.1016/s1471-4914(02)02434-6
- Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. 2009. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 122(Pt 22):4070–4080. doi:10.1242/jcs.053702
- Verma N, Tiku AB. 2017. Significance and nature of bystander responses induced by various agents. Mutat Res Rev Mutat Res. 773:104–121. doi:10.1016/j.mrrev.2017.05.003
- Wakatsuki M, Magpayo N, Kawamura H, Held KD. 2012. Differential bystander signaling between radioresistant chondrosarcoma cells and fibroblasts after X-ray, proton, iron ion and carbon ion exposures. Int J Radiat Oncol Biol Phys. 84(1):E103–E108. doi:10.1016/j.ijrobp.2012.02.052
- Ward IM, Chen JJ. 2001. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 276(51):47759–47762. doi:10.1074/jbc.C100569200
- Yang H, Anzenberg V, Held KD. 2007. The time dependence of bystander responses induced by iron-ion radiation in normal human skin fibroblasts. Radiat Res. 168(3):292–298. doi:10.1667/RR0864.1
- Yang H, Asaad N, Held KD. 2005. Medium-mediated intercellular communication is involved in bystander responses of X-ray-irradiated normal human fibroblasts. Oncogene. 24(12):2096–2103. doi:10.1038/sj.onc.1208439
- Yang H, Magpayo N, Rusek A, Chiang IH, Sivertz M, Held KD. 2011. Effects of very low fluences of high-energy protons or iron ions on irradiated and bystander cells. Radiat Res. 176(6):695–705. doi:10.1667/rr2674.1