
Abstract
The transcriptome of Kappaphycus alvarezii was profiled using high-throughput Solexa paired-end sequencing technology. A total of 61 million sequencing reads was generated by filtering the low-quality reads, and 28 701 unigenes with a mean length of 901 bp were obtained based on de novo assembly. In similarity alignments against the NCBI non-redundant protein sequence (NR) database, Swissprot database, Cluster of Orthologous Groups (COG) database, Gene ontology (GO) database, and Kyoto Encyclopedia of Genes and Genomes (KEGG) database, 11 996 (41.79%) unigenes were identified with significant hits (E-value < 10−5) against existing genes. Functional annotation with the KEGG pathway database identified 8975 unigenes and mapped to 125 pathways. Through functional enrichment analysis of the genes with a higher expression value than the average RPKM (reads per kilobase per million reads), we found that some important pathways were highly expressed. The substantial number of transcript sequences of K. alvarezii provides a valuable resource for potential gene identification and comparative genomic studies.
INTRODUCTION
Kappaphycus alvarezii (Doty) Doty ex P.C. Silva, a kappa-carrageenan-producing seaweed, has been cultivated for the food industry in tropical regions for 40 years. In the last decade, the world market for carrageenan has increased by about 4% annually, and the volume of sales in metric tons of these hydrocolloids was 86 100, yielding US $527 million in 2009 (Bixler & Porse, Citation2010). Depending on the market demand, K. alvarezii is now farmed in several countries including Philippines, Indonesia, Malaysia, Tanzania, Kiribati, Fiji, Kenya and Madagascar (Ask & Azanza, Citation2002). To maintain sustainable farming of K. alvarezii, considerable attention has been paid to this species, with particular focus on cultivation techniques (Hurtado et al., Citation2001; Rodrigueza & Montaño, Citation2007), variety breeding (Dawes & Koch, Citation1991; de Paula et al., Citation1999), disease control (Mendoza et al., Citation2002; Pang et al., Citation2012), photosynthesis (Schubert et al., Citation2004; Andersson et al., Citation2006), analysis of genetic diversity using molecular markers (Liu et al., Citation2009) and identification of hypo-osmotically induced genes (Liu et al., Citation2011). Limited data from cDNA libraries and ESTs have been used as a foundation for studying gene function in this species (Liu et al., Citation2011).
Next-generation sequencing based on the genome/transcriptome database can now provide an effective way to find key genes related to growth (Wang et al., Citation2010; Hao et al., Citation2012; Liu et al., Citation2013), metabolism (Sui et al., Citation2011; Chan et al., Citation2012) and stress-resistance (Hou et al., Citation2011; Lu et al., Citation2012). Transcriptome studies have been reported in several related species (Ho et al., Citation2009; Kowalczyk et al., Citation2011; Yang et al., Citation2011; Xie et al., Citation2013), but never in K. alvarezii. In this study, we sequenced the transcriptome of this species and the generated data will provide a valuable resource for further genetic and genomic research on K. alvarezii.
MATERIALS AND METHODS
cDNA library construction and mRNA sequencing
Green strain K. alvarezii was cultured using floating rafts (each raft about 10 m wide and 10 m long) in the gulf of Lian Bay, Hainan Province, China (18°27′N, 110°5′E). Seedlings were tied to polyethylene ropes (4 mm in diameter, at a depth of about 0.3 m) which were then tied to the floating rafts. The seaweed was grown at a stocking density of about 1000 g m−2, and the conditions where the samples were collected were: salinity 33‰, temperature 26°C and light 215 μmol photon m−2s−1. The seaweeds were washed with sterilized water three times to clean them of attached contaminants and dried on hygroscopic filter paper. The samples were frozen in liquid nitrogen immediately, and then stored at −80°C until use. In order to reflect the complete character of the K. alvarezii transcriptome, samples of different developmental stages (the apical, middle and basal sections of the alga) were collected. Two grams of each sample were used to extract total RNA with an E.Z.N.A. Plant RNA Kit (Omega Bio-tech, Doraville, Georgia, USA) following the manufacturer’s instructions. The quality of the total RNA samples was assessed with an Agilent 2100 bioanalyser and gel electrophoresis, while the quantity of total RNA in samples was measured with a Nanodrop 2000 spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA). Equal quantities of high-quality RNA from each sample were pooled for library construction. The mRNAs were isolated from 20 µg of total RNA with oligo (dT) magnetic beads and cleaved into short fragments in fragmentation buffer (Ambion, Austin, Texas, USA). The first strand of cDNA was synthesized from total mRNA by reverse transcription with random hexamer-primers. The cDNA was then cleaned with RNase H (Invitrogen, Carlsbad, California, USA) to remove all remaining RNA. The other strand of cDNA was synthesized using DNA polymerase I. The double-stranded cDNA was then extracted with QiaQuick PCR extraction kit (Qiagen, Chatsworth, California, USA) and eluted with EB buffer for end repair and addition of an A nucleotide base. The fragments were then ligated to the sequencing adaptors, subjected to PCR amplification and submitted to the BGI-Shenzhen facility (Shenzhen, China) for Illumina HiSeq 2000 paired-end sequencing following the manufacturer’s instructions.
Sequence data analysis and de novo assembly
All the sequencing reads were filtered, discarding low-quality reads, reads with adaptors, and reads with ambiguous bases greater than 5% of the sequence. The Trinity program (Grabherr et al., Citation2011) was used to assemble the sequences derived from the Illumina sequencing into longer contigs by using overlaps of these sequences. We then aligned the Illumina sequences against the contigs. As paired-ends sequencing was used in Illumina sequencing, different versions of contigs of the same transcript could be observed and therefore the distance determined between these partial contigs. Again, with Trinity, the contigs were combined into un-extendable sequences, which we defined here as unigenes. As only one pooled cDNA sample was sequenced, the unigene sequences can also be considered as non-redundant unigenes. Non-redundant unigenes were then used to cluster to large EST datasets with TIGR Gene Indices clustering tools (TGICL) (Pertea et al., Citation2003). Finally, the sequence direction of non-redundant unigenes was determined by BlastX alignment (Altschul et al., Citation1997; Cameron et al., Citation2004) using a maximum E-value of 10−5 as cut-off. After searching the unigenes against the NCBI non-redundant (Nr), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and Cluster of Orthologous Groups (COG) database, the sequence direction of the unigenes was determined based on the best search results (Xiao et al., Citation2013). When there was no significant hit for a unigene within these databases, ESTScan software (Iseli et al., Citation1999) was used to decide the sequence direction. All unigenes with a sequence length of no less than 200 bp were submitted into the Transcriptome Shotgun Assembly (TSA) database at the NCBI with TSA master record: SRP040669.
Similarity search and functional annotation
The functional annotation of unigenes was determined by a similarity search using the BlastX algorithm against the NR, Swiss-Prot, KEGG and COG databases with the E-value <10−5. The functions of the best homologous sequences were assigned to the corresponding unigenes. For well-annotated sequences against the NR database, gene ontology (GO) annotation was carried out to obtain the molecular function, cellular component and biological process terms with the Blast2GO program (Conesa et al., Citation2005) with the E-value < 10−5. Species distribution analysis was further carried out to confirm the identity of the organism through BlastX analysis followed by unigenes annotation. For all the unigenes, WEGO software (Ye et al., Citation2006) was used for GO functional classification to find the distribution of gene functions of K. alvarezii. The functions of the unigenes were also predicted by aligning them against the COG database. Comparison with the KEGG database was carried out to assign pathway annotations to the unigenes. This pathway annotation was also based on the BlastX algorithm with an E-value < 10−5. Each annotated sequence was assigned with an enzyme commission (EC) number, which was then mapped to the KEGG pathway to get the KEGG Pathway Maps.
RPKM calculation
To evaluate gene expression, a reads per kilobase per million reads (RPKM) method (Mortazavi et al., Citation2008) i.e. sequencing reads per kb transcriptome per million mapped sequencing reads was used, which is based on the following equation:
Assuming a gene ‘A’, C is the count of sequencing reads that can uniquely align to the gene ‘A’, N is the total count of sequencing reads that can uniquely align to any gene out of the whole gene set, and L is the length (bp) of gene ‘A’. The RPKM method takes the influence of gene length into consideration, when calculating gene expression levels.
Enrichment analysis
The analysis first maps all target genes (TGs) to KEGG within the database, calculating the number of TGs associated with each term. Compared with the whole gene set, the ultra-geometric test was used to identify pathway nodes significantly enriched in TGs. The formula is:
(http://smd.stanford.edu/help/GO-TermFinder/GO_TermFinder_help.shtml),
N is the count of genes with any pathway, n is the count of TGs in N, M is the count of genes with a specific KEGG pathway, and m is the count of TGs in M. The Bonferroni correction is then applied to the calculated P-value, and the final significance threshold is a corrected P-value ≤ 0.05.
RESULTS AND DISCUSSION
Sequence analysis and de novo assembly
A total of 77 463 312 raw reads were generated from whole transcriptome sequencing of K. alvarezii. After stringent quality assessment and data filtering, 61 168 402 clean reads with 93.49 % Q20 bases (an error probability for base calling of less than 1%) were selected as high-quality reads for further analysis (). The high-quality reads were assembled into 84 827 contigs using the Trinity program, with stringent quality checks. The length of contigs ranged from 100 to 10 318 bp, with a mean length of 333 bp (N50 = 723 bp). Based on TGICL clustering, 28 701 unigenes, ranging from 200 to 10 581 bp, were generated, with a total length of 25.87 Mb and GC content of 52.66%. The mean length of unigenes was 901 bp, and the N50 value was 1697 bp. Among the 28 701 unigenes we obtained, 13 365 unigenes (46.57%) had a length of more than 500 bp, while 8119 unigenes (28.29%) had a length of more than 1000 bp. Illumina HiSeq 2000 produces shorter reads (with mean read length of 100 bp) when compared with the Roche 454 Genome Sequencer (with mean read length of 800 bp). However, with the vast number of short reads and the assembling program, the length of unigenes obtained in this study was similar to those produced by a Roche 454 Genome Sequencer (e.g. Ulva linza (Zhang et al., Citation2012b), Patinopecten yessoensis (Hou et al., Citation2011) and Apostichopus japonicus (Du et al., Citation2012)). Thus the sequences we obtained from the Illumina HiSeq 2000 can be used for further analysis.
Table 1. Summary of transcriptome sequencing and assembly for K. alvarezii unigenes.
Table 2. Functional annotation of non-redundant unigenes.
Functional annotation analysis
In order to understand the transcriptome profiling of K. alvarezii, an assessment of the putative identities of the assembled unigenes was performed, based on sequence similarity search. All unigenes were aligned against the Nr, Swiss-prot, KEGG and COG databases. The results showed that out of 28 701 unigenes, 11 456 (NR), 8970 (Swiss-prot), 8975 (KEGG) and 7296 (COG) unigenes had significant similarity to each database. Altogether, 11 996 unigenes (41.8%) matched well with the sequences in these protein databases (). The remaining 16 705 (58.2%) unigenes showed no significant match. Species distribution analysis showed that 35.2% of annotated unigenes could be assigned with a best score to the sequences from the top seven species, i.e. Volvox carteri f. nagariensis (9.6%), Ectocarpus siliculosus (5.9%), Physcomitrella patens subsp. patens (4.5%), Selaginella moellendorffii (4.3%), Glycine max (4.0%), Phytophthora sojae (3.5%) and Chlamydomonas reinhardtii (3.4%). To our knowledge, genomic information for only a few algae (e.g. Cyanidioschyzon merolae (Matsuzaki et al., Citation2004), Chondrus crispus (Collen et al., Citation2013), Porphyridium purpureum (Bhattacharya et al., Citation2013), Galdieria sulphuraria (Schonknecht et al., Citation2013) and Pyropia yezoensis (Nakamura et al., Citation2013)) and several transcriptomic studies (e.g. Porphyra yezoensis (Yang et al., Citation2011), Chondrus crispus (Kowalczyk et al., Citation2011), Gracilaria changii (Ho et al., Citation2009), and Pyropia haitanensis (Xie et al., Citation2013)) are available. Thus, there are insufficient genome or transcriptome sequences available in public databases for the genus Kappaphycus and phylogenetically closely related species. This may account for the relatively low annotation rate. On the other hand, short sequences frequently fail to match database entries due to the significance level required for the BlastX similarity search, which depends partly on the length of the query sequence (Altschul et al., Citation1997; Cameron et al., Citation2004). In the present study, 8435 unigenes had a length of less than 300 bp, of which only 1273 (15.1%) had a Blast match. In contrast, 7025 of 8747 unigenes (80.3%) with a length over 900 bp found a match (). Some of the unannotated unigenes may be species-specific genes of K. alvarezii.
Fig. 1. The annotated unigenes (hit) of all unigenes correlates to the length of the unigene. The white bar shows the number of unigenes and the grey bar shows the number of the unigene that can be annotated. By comparing the proportion of the annotated gene within all the unigenes with different length, the result shows that longer unigenes were more likely to have annotation against protein databases.
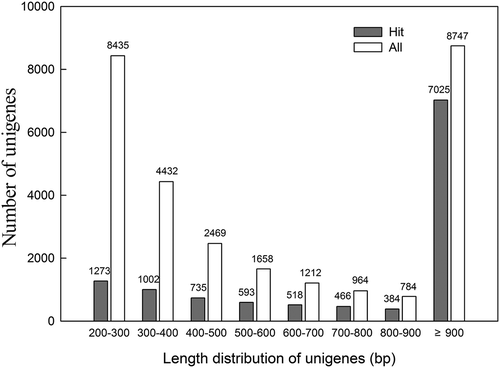
Gene Ontology (GO) analysis was conducted for functional classification of the K. alvarezii unigenes. Of 11 456 unigenes with Blast matches to the NR database, 5394 were assigned to 34 882 GO terms with an average of one unigene assigned to 6.5 GO terms. The assigned 34 882 GO terms were then summarized into three main GO categories, with 15 283 (43.8%) for biological processes, 13 990 (40.1%) for cellular components, and 5609 (16.1%) for molecular functions (Supplementary File 1). The distribution of 52 sub-categories in each main category is shown in and Supplementary File 1. Under the category of biological process, proteins for cellular process (3331; 21.8%) and metabolic process (3015; 19.7%) were dominant GO terms. For the category of cellular components, the majority GO terms were proteins involved in the cell (3378; 24.1%) and cell part (3373; 24.1%) categories, accounting for almost half of this category. Regarding the category of molecular functions, proteins involved in catalytic activity (2890; 51.5%) were the most represented GO terms, suggesting that some significant catalytic activities occurred in K. alvarezii. GO assignments for biological process showed that 480 and 473 unigenes (Supplementary File 1) were assigned to the GO terms reproduction and reproductive process, respectively.
Fig. 2. Gene Ontology classification of the K. alvarezii transcriptome. A total of 11,456 unigenes with BlastX matches against the Nr database were classified into three main GO categories and 52 sub-categories. The scale on the y-axis indicates the number of unigenes in each category.
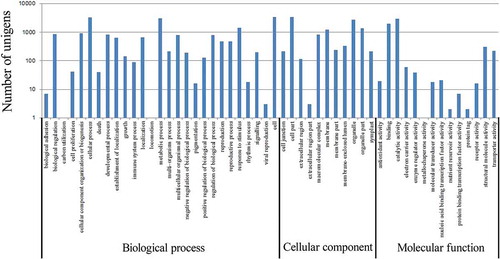
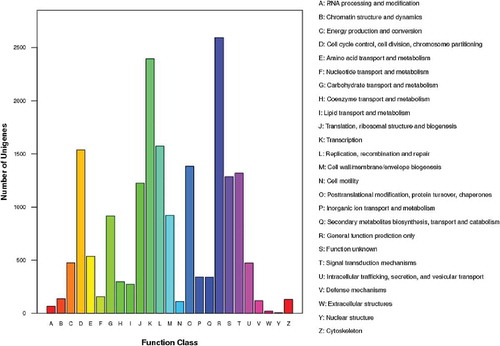
To further evaluate the completeness of the analysis and to classify possible functions of the transcriptome of K. alvarezii, all the unigenes were compared with the COG database. In total, 18 616 unigenes were assigned to the 25 COG classifications ( and Supplementary File 2) and 85 473 COG functional annotations were obtained because some unigenes were assigned to multiple COG functions. The cluster of general function prediction only (2592, 13.9%) represented the largest group, followed by transcription (2393, 12.9%), replication, recombination and repair (1574, 8.5%), cell cycle control, cell division, chromosome partitioning (1538, 8.3%), posttranslational modification, protein turnover, chaperones (1384, 7.4%) and signal transduction mechanisms (1318, 7.1%). The cluster of nuclear structure (four unigenes) and extracellular structures (20 unigenes) represented the smallest groups.
Fig. 3. COG functional classification of the K. alvarezii unigenes. A total of 18 616 unigenes were assigned to the 25 COG classifications.
KEGG pathway analysis was also performed for all the annotated unigenes to understand the interaction of genes and to further identify active biological pathways in K. alvarezii. The Enzyme Commission (EC) numbers of the annotated unigenes were obtained based on comparison with the KEGG databases and then assigned to KEGG pathways. In this study, 8795 unigenes were assigned to 125 pathways (Supplementary File 3). A total of 3874 unigenes were grouped into the metabolic pathways and most of these unigenes were involved in energy metabolism, carbohydrate metabolism, and/or amino acid metabolism. Unigenes involved in metabolic pathways were enriched for proteins that maintain the essential functions of K. alvarezii, which was consistent with the transcriptome of Arachis hypogaea (Zhang et al., Citation2012a), Gentiana macrophylla (Hua et al., Citation2014) and Hevea brasiliensi (Li et al., Citation2012). The other top five enriched pathways included endocytosis (2062 unigenes), glycerophospholipid metabolism (2011 unigenes), ether lipid metabolism (1969 unigenes) and starch and sucrose metabolism (813 unigenes).
Comparison with other red algae and functional analysis
Firstly, the GC content of the unigenes of K. alvarezii was compared with the coding sequences of three other red algae: Chondrus crispus, Cyanidioschyzon merolae and Porphyridium purpureum. The result showed that the GC content of K. alvarezii followed the Poisson distribution, and the mean GC content was 51.70%, similar to that of Chondrus crispus (53.50%), but less than that of Cyanidioschyzon merolae (56.60%) and Porphyridium purpureum (56.50%) (Supplementary Fig. 4). Then, the unigenes of K. alvarezii were clustered with these three species, and 14 697 out of 28 701 (51.21%) could be clustered into gene families with the three species. In addition, the expression values of these unigenes were evaluated using RPKM method and 26 622 (92.76%) unigenes were expressed (RPKM > 1) with the average RPKM value of 25.39. Among all the expressed unigenes, 3628 unigenes had a RPKM value greater than the mean, and 24 952 unigenes had a RPKM value less than the mean. Interestingly, 83.88% of the clustered unigenes and 98.47% of the unclustered unigenes were expressed, suggesting that these unclustered unigenes were probable specific to K. alvarezii.
Furthermore, all the unigenes were classified into two classes including 24 952 unigenes with relative higher read mapping (Class A) and 3628 unigenes with relative lower read mapping (Class B). There were 1407 unigenes out of Class B that were involved in the first six pathway maps, including mismatch repair, DNA replication, folate biosynthesis, pentose and glucuronate interconversions, starch and sucrose metabolism and ubiquinone and other terpenoid-quinone biosynthesis. The KEGG pathways mentioned above were housekeeping gene families, which indicated that the housekeeping genes tend to express below average.
There were 355 unigenes out of Class A involved in the first 10 pathways, including selenocompound metabolism (ko00450), sulphur metabolism (ko00920) and two photosynthesis-related pathways (ko00710 and ko00196). For selenocompound metabolism, previous studies (Novoselov et al., Citation2002; Kim et al., Citation2006) suggested that algal species might require selenium (Supplementary Fig. 5). In general, converting selenate to selenide/hydrogen selenide needs a series of reduction steps for its assimilation into the seleno-amino acids L-selenocysteine and seleno-L-methionine (Sors et al., Citation2009). Although it has importance as a micronutrient, excessive selenium is toxic to plants, largely owing to the fact that transporters and enzymes involved in sulphur and sulpho-compound metabolism can mis-incorporate seleno-amino acids into proteins (Brown & Shrift, Citation1981; Ellis & Salt, Citation2003; Zwolak & Zaporowska, Citation2012). However, selenium accumulation can protect plants against attacks by insects, and fungi (McClung, Citation2006). In K. alvarezii, unigenes of class A were significantly enriched in the selenocompound metabolism pathway and sulphur metabolism, which were probably related to the alga’s resistance to grazers and expelling of toxins (Lobanov et al., Citation2007). Furthermore, it is interesting that the genes for sulphur metabolism were expressed above average, considering the high yields of carrageenan in the cell walls that includes sulphated sugars.
DISCLOSURE STATEMENT
No potential conflict of interest was reported by the author(s).
SUPPLEMENTARY INFORMATION
The following supplementary material is accessible via the Supplementary Content tab on the article’s online page at http://dx.doi.org/10.1080/09670262.2015.1069403
Supplementary File 1. Unigenes involved in the three main GO categories.
Supplementary File 2. Unigenes assigned to the 25 COG classifications.
Supplementary File 3. Categorization of Kappaphycus alvarezii unigenes to KEGG pathways.
Supplementary Fig. 4. Comparison of the GC content of K. alvarezii and Chondrus crispus, Cyanidioschyzon merolae and Porphyridium purpureum.
Supplementary Fig. 5. The pathway of the Selenocompound metabolism containing the unigenes with larger RPKM than mean value.
ACKNOWLEDGEMENTS
Special thanks to Dr John van der Meer (Pan-American Marine Biotechnology Association) for his assistance with proofreading.
Additional information
Funding
Notes on contributors
Zhen Zhang
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
Tong Pang
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
Qianqian Li
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
Litao Zhang
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
Ling Li
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
Jianguo Liu
Z. Zhang: original concept, analysis of original data, drafting and editing manuscript; T. Pang: culture experiments and analysis of original data; Q.Q. Li and L.T. Zhang: analysis of original data and drafting; L. Li: analysis of original data; J.G. Liu: original concept, analysis of original data and editing manuscript.
REFERENCES
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, 25: 3389–3402.
- Andersson, M., Schubert, H., Pedersen, M. & Snoeijs, P. (2006). Different patterns of carotenoid composition and photosynthesis acclimation in two tropical red algae. Marine Biology, 149: 653–665.
- Ask, E.I. & Azanza, R.V. (2002). Advances in cultivation technology of commercial eucheumatoid species: a review with suggestions for future research. Aquaculture, 206: 257–277.
- Bhattacharya, D., Price, D.C., Chan, C.X., Qiu, H., Rose, N., Ball, S., Weber, A.P.M., Arias, M.C., Henrissat, B., Coutinho, P.M., Krishnan, A., Zauner, S., Morath, S., Hilliou, F., Egizi, A., Perrineau, M.M. & Yoon, H.S. (2013). Genome of the red alga Porphyridium purpureum. Nature Communications, 4: 1941.
- Bixler, H.J. & Porse, H. (2010). A decade of change in the seaweed hydrocolloids industry. Journal of Applied Phycology, 23: 321–335.
- Brown, T.A. & Shrift, A. (1981). Exclusion of selenium from proteins of selenium-tolerant astragalus species. Plant Physiology, 67: 1051–1053.
- Cameron, M., Williams, H.E. & Cannane, A. (2004). Improved gapped alignment in BLAST. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 1: 116–129.
- Chan, C.X., Blouin, N.A., Zhuang, Y.Y., Zauner, S., Prochnik, S.E., Lindquist, E., Lin, S.J., Benning, C., Lohr, M., Yarish, C., Gantt, E., Grossman, A.R., Lu, S., Muller, K., Stiller, J.W., Brawley, S.H. & Bhattacharya, D. (2012). Porphyra (Bangiophyceae) transcriptomes provide insights into red algal development and metabolism. Journal of Phycology, 48: 1328–1342.
- Collen, J., Porcel, B., Carre, W., Ball, S.G., Chaparro, C., Tonon, T., Barbeyron, T., Michel, G., Noel, B., Valentin, K., Elias, M., Artiguenave, F., Arun, A., Aury, J.M., Barbosa-Neto, J.F., Bothwell, J.H., Bouget, F.Y., Brillet, L., Cabello-Hurtado, F., Capella-Gutierrez, S., Charrier, B., Cladiere, L., Cock, J.M., Coelho, S.M., Colleoni, C., Czjzek, M., Da Silva, C., Delage, L., Denoeud, F., Deschamps, P., Dittami, S.M., Gabaldon, T., Gachon, C.M.M., Groisillier, A., Herve, C., Jabbari, K., Katinka, M., Kloareg, B., Kowalczyk, N., Labadie, K., Leblanc, C., Lopez, P.J., McLachlan, D.H., Meslet-Cladiere, L., Moustafa, A., Nehr, Z., Collen, P.N., Panaud, O., Partensky, F., Poulain, J., Rensing, S.A., Rousvoal, S., Samson, G., Symeonidi, A., Weissenbach, J., Zambounis, A., Wincker, P., & Boyen, C. (2013). Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the archaeplastida. Proceedings of the National Academy of Sciences USA, 110: 5247–5252.
- Conesa, A., Gotz, S., Garcia-Gomez, J.M., Terol, J., Talon, M. & Robles, M. (2005). Blast2Go: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21: 3674–3676.
- Dawes, C.J. & Koch, E.W. (1991). Branch, micropropagule and tissue-culture of the red algae Eucheuma denticulatum and Kappaphycus alvarezii farmed in the Philippines. Journal of Applied Phycology, 3: 247–257.
- de Paula, E.J., Pereira, R.T.L. & Ohno, M. (1999). Strain selection in Kappaphycus alvarezii var. alvarezii (Solieriaceae, Rhodophyta) using tetraspore progeny. Journal of Applied Phycology, 11: 111–121.
- Du, H., Bao, Z., Hou, R., Wang, S., Su, H., Yan, J., Tian, M., Li, Y., Wei, W., Lu, W., Hu, X., Wang, S. & Hu, J. (2012). Transcriptome sequencing and characterization for the sea cucumber Apostichopus japonicus (Selenka, 1867). PloS ONE, 7: e33311.
- Ellis, D.R. & Salt, D.E. (2003). Plants, selenium and human health. Current Opinion in Plant Biology, 6: 273–279.
- Grabherr, M.G., Haas, B.J., Yassour, M., Levin, J.Z., Thompson, D.A., Amit, I., Adiconis, X., Fan, L., Raychowdhury, R., Zeng, Q.D., Chen, Z.H., Mauceli, E., Hacohen, N., Gnirke, A., Rhind, N., di Palma, F., Birren, B.W., Nusbaum, C., Lindblad-Toh, K., Friedman, N. & Regev, A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29: 644–652.
- Hao, D.C., Ma, P., Mu, J., Chen, S.L., Xiao, P.G., Peng, Y., Huo, L., Xu, L.J. & Sun, C. (2012). De novo characterization of the root transcriptome of a traditional Chinese medicinal plant Polygonum cuspidatum. Science China – Life Sciences, 55: 452–466.
- Ho, C.-L., Teoh, S., Teo, S.-S., Rahim, R. & Phang, S.-M. (2009). Profiling the transcriptome of Gracilaria changii (Rhodophyta) in response to light deprivation. Marine Biotechnology, 11: 513–519.
- Hou, R., Bao, Z.M., Wang, S., Su, H.L., Li, Y., Du, H.X., Hu, J.J. & Hu, X.L. (2011). Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PloS ONE, 6: e21560.
- Hua, W., Zheng, P., He, Y., Cui, L., Kong, W. & Wang, Z. (2014). An insight into the genes involved in secoiridoid biosynthesis in Gentiana macrophylla by RNA-Seq. Molecular Biology Reports, 41: 4817–4825.
- Hurtado, A.Q., Agbayani, R.F., Sanares, R. & de Castro-Mallare, M.T.R. (2001). The seasonality and economic feasibility of cultivating Kappaphycus alvarezii in Panagatan Cays, Caluya, Antique, Philippines. Aquaculture, 199: 295–310.
- Iseli, C., Jongeneel, C.V. & Bucher, P. (1999). ESTscan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proceedings of the International Conference on Intelligent Systems – Molecular Biology, 199: 138–148.
- Kim, H.Y., Fomenko, D.E., Yoon, Y.E. & Gladyshev, V.N. (2006). Catalytic advantages provided by selenocysteine in methionine-s-sulfoxide reductases. Biochemistry, 45: 13697–13704.
- Kowalczyk, N., Carre, W., Brillet, L., Corre, E., Porcel, B., Da Silva, C., Boyen, C. & Collen, J. (2011). Transcriptomic analysis of life stages of the red alga Chondrus crispus. European Journal of Phycology, 46: 96–97.
- Li, D.J., Deng, Z., Qin, B., Liu, X.H. & Men, Z.H. (2012). De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics, 13: 192.
- Liu, C.L., Huang, X.H. & Liu, J.G. (2009). The primary application of ISSR analysis on strains identification of Kappaphycus alvarezii. Marine Sciences, 33: 50–53.
- Liu, C.L., Wang, X.L., Huang, X.H. & Liu, J.G. (2011). Identification of hypo-osmotically induced genes in Kappaphycus alvarezii (Solieriaceae, Rhodophyta) through expressed sequence tag analysis. Botanica Marina, 54: 557–562.
- Liu, T.M., Zhu, S.Y., Tang, Q.M., Chen, P., Yu, Y.T. & Tang, S.W. (2013). De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud). BMC Genomics, 14: 125.
- Lobanov, A., Fomenko, D., Zhang, Y., Sengupta, A., Hatfield, D. & Gladyshev, V. (2007). Evolutionary dynamics of eukaryotic selenoproteomes: large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biology, 8: 1–16.
- Lu, M.W., Ngou, F.H., Chao, Y.M., Lai, Y.S., Chen, N.Y., Lee, F.Y. & Chiou, P.P. (2012). Transcriptome characterization and gene expression of Epinephelus spp. in endoplasmic reticulum stress-related pathway during betanodavirus infection in vitro. BMC Genomics, 13: 651.
- Matsuzaki, M., Misumi, O., Shin-I, T., Maruyama, S., Takahara, M., Miyagishima, S.Y., Mori, T., Nishida, K., Yagisawa, F., Yoshida, Y., Nishimura, Y., Nakao, S., Kobayashi, T., Momoyama, Y., Higashiyama, T., Minoda, A., Sano, M., Nomoto, H., Oishi, K., Hayashi, H., Ohta, F., Nishizaka, S., Haga, S., Miura, S., Morishita, T., Kabeya, Y., Terasawa, K., Suzuki, Y., Ishii, Y., Asakawa, S., Takano, H., Ohta, N., Kuroiwa, H., Tanaka, K., Shimizu, N., Sugano, S., Sato, N., Nozaki, H., Ogasawara, N., Kohara, Y. & Kuroiwa, T. (2004). Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature, 428: 653–657.
- McClung, C.R. (2006). Plant circadian rhythms. Plant Cell, 18: 792–803.
- Mendoza, W.G., Montano, N.E., Ganzon-Fortes, E.T. & Villanueva, R.D. (2002). Chemical and gelling profile of ice-ice infected carrageenan from Kappaphycus striatum (Schmitz) Doty “sacol” strain (Solieriaceae, Gigartinales, Rhodophyta). Journal of Applied Phycology, 14: 409–418.
- Mortazavi, A., Williams, B.A., McCue, K., Schaeffer, L. & Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5: 621–628.
- Nakamura, Y., Sasaki, N., Kobayashi, M., Ojima, N., Yasuike, M., Shigenobu, Y., Satomi, M., Fukuma, Y., Shiwaku, K., Tsujimoto, A., Kobayashi, T., Nakayama, I., Ito, F., Nakajima, K., Sano, M., Wada, T., Kuhara, S., Inouye, K., Gojobori, T. & Ikeo, K. (2013). The first symbiont-free genome sequence of marine red alga, susabi-nori (Pyropia yezoensis). PloS ONE, 8: e57122.
- Novoselov, S.V., Rao, M., Onoshko, N.V., Zhi, H., Kryukov, G.V., Xiang, Y., Weeks, D.P., Hatfield, D.L. & Gladyshev, V.N. (2002). Selenoproteins and selenocysteine insertion system in the model plant cell system, Chlamydomonas reinhardtii. EMBO Journal, 21: 3681–3693.
- Pang, T., Liu, J.G., Liu, Q., Zhang, L.T. & Lin, W. (2012). Impacts of glyphosate on photosynthetic behaviors in Kappaphycus alvarezii and Neosiphonia savatieri detected by JIP-test. Journal of Applied Phycology, 24: 467–473.
- Pertea, G., Huang, X.Q., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., Lee, Y., White, J., Cheung, F., Parvizi, B., Tsai, J. & Quackenbush, J. (2003). TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics, 19: 651–652.
- Rodrigueza, M.R.C. & Montaño, M.N.E. (2007). Bioremediation potential of three carrageenophytes cultivated in tanks with seawater from fish farms. Journal of Applied Phycology, 19: 755–762.
- Schonknecht, G., Chen, W.H., Ternes, C.M., Barbier, G.G., Shrestha, R.P., Stanke, M., Brautigam, A., Baker, B.J., Banfield, J.F., Garavito, R.M., Carr, K., Wilkerson, C., Rensing, S.A., Gagneul, D., Dickenson, N.E., Oesterhelt, C., Lercher, M.J. & Weber, A.P.M. (2013). Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science, 339: 1207–1210.
- Schubert, H., Gerbersdorf, S., Titlyanov, E., Titlyanova, T., Granbom, M., Pape, C., & Luning, K. (2004). Circadian rhythm of photosynthesis in Kappaphycus alvarezii (Rhodophyta): independence of the cell cycle and possible photosynthetic clock targets. European Journal of Phycology, 39: 423–430.
- Sors, T.G., Martin, C.P. & Salt, D.E. (2009). Characterization of selenocysteine methyltransferases from astragalus species with contrasting selenium accumulation capacity. Plant Journal, 59: 110–122.
- Sui, C., Zhang, J., Wei, J.H., Chen, S.L., Li, Y., Xu, J.S., Jin, Y., Xie, C.X., Gao, Z.H., Chen, H.J., Yang, C.M., Zhang, Z. & Xu, Y.H. (2011). Transcriptome analysis of Bupleurum chinense focusing on genes involved in the biosynthesis of saikosaponins. BMC Genomics, 12: 539.
- Wang, Z.Y., Fang, B.P., Chen, J.Y., Zhang, X.J., Luo, Z.X., Huang, L.F., Chen, X.L. & Li, Y.J. (2010). De novo assembly and characterization of root transcriptome using illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics, 11: 726.
- Xiao, J., Jin, X., Jia, X., Wang, H., Cao, A., Zhao, W., Pei, H., Xue, Z., He, L., Chen, Q. & Wang, X. (2013). Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics, 14: 197.
- Xie, C.T., Li, B., Xu, Y., Ji, D.H. & Chen, C.S. (2013). Characterization of the global transcriptome for Pyropia haitanensis (Bangiales, Rhodophyta) and development of cSSR markers. BMC Genomics, 14: 107.
- Yang, H., Mao, Y.X., Kong, F.N., Yang, G.P., Ma, F. & Wang, L. (2011). Profiling of the transcriptome of Porphyra yezoensis with solexa sequencing technology. Chinese Science Bulletin, 56: 2119–2130.
- Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., Wang, J., Li, S., Li, R., Bolund, L. & Wang, J. (2006). WEGO: a web tool for plotting go annotations. Nucleic Acids Research, 34: W293–W297.
- Zhang, J.A., Liang, S., Duan, J.L., Wang, J., Chen, S.L., Cheng, Z.S., Zhang, Q., Liang, X.Q., & Li, Y.R. (2012a). De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.).BMC Genomics, 13: 90.
- Zhang, X.W., Ye, N.H., Liang, C.W., Mou, S.L., Fan, X., Xu, J.F., Xu, D. & Zhuang, Z.M. (2012b). De novo sequencing and analysis of the Ulva linza transcriptome to discover putative mechanisms associated with its successful colonization of coastal ecosystems. BMC Genomics, 13: 565.
- Zwolak, I. & Zaporowska, H. (2012). Selenium interactions and toxicity: a review. Selenium interactions and toxicity. Cell Biology and Toxicology, 28: 31–46.