Abstract
The mitochondrial ADP/ATP carrier is a six helix bundle membrane transport protein, which couples the exit of ATP from the mitochondrial matrix to the entry of ADP. Extended (4×20 ns) molecular dynamics simulations of the carrier, in the presence and absence of bound inhibitor (carboxyatractyloside), have been used to explore the conformational dynamics of the protein in a lipid bilayer environment, in the presence and absence of the carboxyatractyloside inhibitor. The dynamic flexibility (measured as conformational drift and fluctuations) of the protein is reduced in the presence of bound inhibitor. Proline residues in transmembrane helices H1, H3 and H5 appear to form dynamic hinges. Fluctuations in inter-helix salt bridges are also observed over the time course of the simulations. Inhibitor-protein and lipid-protein interactions have been characterised in some detail. Overall, the simulations support a transport mechanism in which flexibility about the proline hinges enables a transition between a ‘closed’ and an ‘open’ pore-like state of the carrier protein.
Introduction
Membrane proteins play a number of key roles in cells, including transport of diverse solutes across cell membranes. Their importance is reflected in the observation that ∼25% of open reading frames encode membrane proteins Citation[1]. A number of different families of membrane proteins exist, and are classified on, e.g. the TCDB website http://www.tcdb.org/. Progress in determination of structures of membrane proteins by X-ray diffraction has been slow, but is starting to accelerate Citation[2]. A number of structures for symporter and antiporter transport proteins have been determined including: (i) members of the major facilitator superfamily (LacY Citation[3], GlpT Citation[4], and EmrD Citation[5]); (ii) a neurotransmitter sodium symporter, LeuTAa Citation[6]; (iii) a sodium/protein antiporter NhaA Citation[7]; and (iv) the mitochondrial ADP/ATP carrier Citation[8].
Mitochondria of eukaryotes contain typically 30–55 different carriers homologous to the ADP/ATP carrier Citation[9]. The ADP/ATP carrier plays a key role in mitochondrial metabolism. It is located in the mitochondrial inner membrane and exchanges cytosolic ADP for ATP in the mitochondrial matrix. The ADP/ATP carrier is an antiporter, coupling the outward movement of 1 ATP molecule to the inward movement of 1 ADP molecule in an electrogenic process. Thus, the net direction of transport is driven by both the ATP and ADP concentration gradients and the voltage difference across the membrane. It is thought that the carrier functions as a homodimer, with each monomer of the dimer in one of two inter-convertible states (although it should be noted that recent studies of the yeast ADP/ATP carrier have shown monomeric behaviour and function in both mild detergents Citation[10] and mitochondrial membranes Citation[11]). These two states are termed the m- and c-states (corresponding to the matrix and cytosolic facing conformations). The carrier can be trapped in these states by two inhibitors, the m-state is trapped by Bongkrekic acid (BKA) and the c-state is trapped by carboxyatractyloside (CATR) Citation[12].
Co-crystallization with CATR enabled the X-ray structure of the c-state of the protein to be determined at 2.2 Å resolution (PDB id 1OKC) Citation[8]. Residues 2–293 of the 297 residues of the protein were resolved along with one bound CATR molecule, four lipids (including two cardiolipin molecules) and 82 water molecules.
The structure of the ADP/ATP carrier monomer reveals six TM α-helices with a threefold repeat of approximately 100 residues in the sequence, resulting in three domains packed together in a pseudo-symmetrical fashion. Each domain contains three helices: two transmembrane (TM) helices (H1 to H6) and one “linker” helix (h12, h34, and h56). The odd numbered TM helices (H1, H3, H5) line the interior of a cavity with external mouth diameter ∼20 Å and depth of ∼30 Å, providing a well-defined binding site, thought to be similar to that for ADP, in which the CATR molecule is located (see A). Despite the minimal sequence homology between the ∼100-residue repeats (only ∼15%), there is a root-mean-squared deviation (RMSD) of only 2 Å between the folds of the mainchain ().
Figure 1. Structure of 1OKC and CATR. (A) Structure of protein, viewed perpendicular to approx. 3× axis; rainbow scale N = blue, C = red. The dotted line represents the approximate position of the lipid bilayer headgroups. (B) View down the approx 3×, CATR in pink, domains: D1 (residues 2–101), D2 (residues 102–201), and D3 (residues 202–292). The transmembrane helices H1 to H6 are indicated.
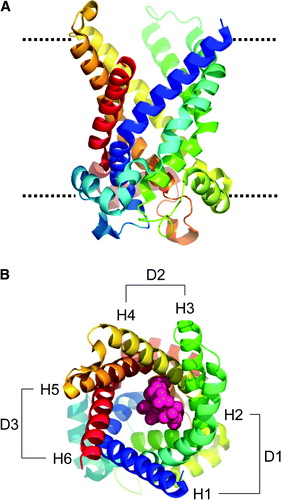
Each of the odd numbered helices (H1, H3, H5) has a pronounced kink at a proline residue. These prolines are part of a signature sequence motif PX(−)XX(+) that is conserved across all superfamily members. It is suggested that movement at the hinge point around the proline residue may be a basis for a potential transport mechanism. The charged residues in this motif form a network of charged pairs between the helices Citation[8]. The CATR inhibitor forms van der Waals contacts, π-stacking interactions, and salt bridges with the TM helices, as detailed in B and Citation[8]. The glucose moiety faces the opening of the cavity and the glycerol and sulphate groups form polar interactions with the cavity lining residues (see B) Citation[8].
Figure 2. (A) Interaction between the side chain of R279 and the carboxylate groups of CATR and between the side chains of R279 and E29. This snapshot is taken from simulation 1OKC-CATR, at t = ∼18.5 ns of the production simulation. (B) Schematic representation of key binding site interactions of 1OKC with CATR as seen in the X-ray structure. The inhibitor forms van der Waals contacts with residues L127, V130 and I183, and the diterpene moiety stacks onto Y186. The glucose moiety faces the opening of the cavity and there are polar interactions between the sulphate groups of this moiety and the sidechains of R187, K91 and N87. A salt bridge forms between one carboxylate and R79, whilst the second carboxylate forms an interaction with R279 via a water molecule Citation[8]. There is also an interaction between the hydroxyl group directly attached to the pentyl ring of CATR and residues D231 and R234.
![Figure 2. (A) Interaction between the side chain of R279 and the carboxylate groups of CATR and between the side chains of R279 and E29. This snapshot is taken from simulation 1OKC-CATR, at t = ∼18.5 ns of the production simulation. (B) Schematic representation of key binding site interactions of 1OKC with CATR as seen in the X-ray structure. The inhibitor forms van der Waals contacts with residues L127, V130 and I183, and the diterpene moiety stacks onto Y186. The glucose moiety faces the opening of the cavity and there are polar interactions between the sulphate groups of this moiety and the sidechains of R187, K91 and N87. A salt bridge forms between one carboxylate and R79, whilst the second carboxylate forms an interaction with R279 via a water molecule Citation[8]. There is also an interaction between the hydroxyl group directly attached to the pentyl ring of CATR and residues D231 and R234.](/cms/asset/6b9f9815-bad4-4a6b-bf07-da511353daa5/imbc_a_346095_f0002_b.gif)
It should be recalled that crystal structures provide an essentially static (time and space averaged) picture of a protein. Molecular dynamics (MD) simulations Citation[13], Citation[14] enable us to extend purely structural approaches to characterise the nanosecond timescale dynamics of membrane proteins Citation[15], Citation[16]. However, in order to understand mechanisms of solute transport, it is important to determine the intrinsic flexibility of carrier subunits whilst in a lipid bilayer environment. To do this, we have simulated the dynamics of the ADP/ATP carrier protein, embedded in a 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (POPC) lipid bilayer, in the presence and absence of a ligand and crystallographic waters. The simulation results appear to support a transport model in which the proline-kinks within the odd-numbered TM helices play a key role in enabling the requisite conformational changes for transport.
Methods
Protein/lipid system
All simulations were based upon the X-ray structure of the ADP/ATP carrier (PDB id 1OKC). Those residues for which the sidechains were incompletely resolved in the crystal structure (i.e. D2, R170, K205, V207, K267 and I293) were built using SPDBV/Deepview (www.expasy.org/spdbv/). A pre-equilibrated bilayer/water system (containing 175 POPC molecules) was used in which to embed the protein, using a modification of the approach described by Citation[17]. Na+ and Cl− ions were added to the system to neutralise the charge on the protein to a salt concentration of ∼0.15 M.
The inhibitor
Parameters for the CATR inhibitor were based on those for related ligands in the GROMOS96 forcefield Citation[18]. The inhibitor molecule was subjected to in vacuo energy minimisation, starting from CATR coordinates from the 1OKC crystal structure. Comparison of the energy minimised structure and the coordinates from the crystal structure of the protein/inhibitor complex coordinates gave an RMSD of only 0.39 Å. The greatest deviation was observed within the two carboxylate groups. During 5 ns of simulation in of CATR in water the key stereochemical features of the molecule were maintained. A transient hydrogen bond was observed between the hydroxyl group on the glucose moiety and the carboxylate groups. The molecule was seen to be flexible about the glycosidic linkage. The RMSD from the initial coordinates after the 5 ns simulation in water was 3.0 Å.
Simulation protocol
Simulations were performed using GROMACS v3 Citation[19] (www.gromacs.org). The ffgmx43a1 force field was used, modified for use with lipids, along with the SPC water model Citation[20], Citation[21]. During the equilibration period, the non-H protein atoms were restrained, with a force constant of 1 000 kJ mol−1 nm−2. During equilibration periods the temperature was controlled using the Berendsen thermostat Citation[22] with a coupling constant τT=0.1 ps, and the pressure was controlled using the Berendsen barostat with a coupling constant of τP=1 ps. In the subsequent production period of the simulation, the position restraints were removed and the barostat and thermostat were changed to Parinello-Rahman and Nosé-Hoover, respectively; with τP=5 ps and τT=0.5 ps. In both the equilibration phase and the production phase of the simulation (), the CATR inhibitor was temperature coupled to the protein, in the same way that the ions were coupled to the solvent.
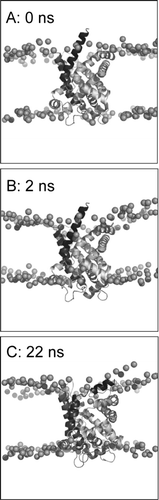
Figure 3. Set-up and progress of simulation, illustrated for simulation 1OKC. The phosphorus atoms of the lipid headgroups are shown as spheres, and the protein as a ribbon. Snapshots are shown at the start of the simulation (A, t = 0 ns), at the end of equilibration (B, t = 2 ns), and at the end of the simulation (C, t = 22 ns).
Analysis and display
Simulations were analysed using Gromacs routines and local code. Secondary structure analysis used DSSP Citation[23]. Simulations were visualized using VMD Citation[24] and images generated using Povray (http://www.povray.org/) and RasTop (http://www.geneinfinity.org/rastop/) Citation[25]. Graphs were produced using xmgrace (http://plasma-gate.weizman.ac.il/Grace/). Principal components analysis used the Dynamite server Citation[26] Proline hinge kink/swivel angle analysis used SWINK Citation[27].
Results
Overall protein conformation: drift and fluctuations
Four simulations were performed (; totalling 80 ns) using a united-atom model, in order to explore any differences in conformational dynamics of the protein in the absence vs. presence of bound inhibitor (CATR) and also to examine the possible influence of including the crystallographically determined water molecules instead of adding all waters by a standard solvation procedure. The inclusion of crystallographic waters also provides an alternative starting configuration for the system in both the presence and absence of the inhibitor, which provides additional sampling of conformations during the simulations.
Table I. Summary of simulations.
The Cα RMSD relative to the starting structure was used to measure the overall conformational drift of the protein. In order to focus on the core fold of the protein, the RMSD was evaluated separately for the core secondary structure (i.e. the α-helices) and for the surface loops of the protein. The Cα RMSD was indeed highest for regions of undefined secondary structure, i.e. the cytoplasmic and matrix loops, with a final (last 2 ns of each simulation) value of ∼4.5 Å. There were no significant differences between the loop RMSDs for the different simulations, thus indicating that the presence/absence of CATR had no effect on the overall conformational dynamics of the surface loops.
In contrast, the final Cα RMSD of the protein core (i.e. the helices) was sensitive to the presence/absence of CATR. Overall, the core Cα RMSDs ranged from 2.5–2.8 Å. A value of 2.8 Å is a little higher than might be expected, given the resolution (2.2 Å) of the X-ray structure Citation[28]. The lower value is as might be expected for a structure of this resolution. It is thus of interest that there is a clear trend in RMSDs for the three simulations (), namely 1OKC > 1OKC-W > 1OKC-CATR > 1OKC-CATR-W. It thus seems that the overall conformational drift of the protein is significantly greater in the absence of bound inhibitor. The secondary structure of the protein as a function of time was examined (see Supplementary Data, , online version only). Overall, there is no great change in secondary structure of the protein over time for any of the simulations. However, closer examination of the protein structures and secondary structure analyses suggests some local loss of α-helicity in helices H1, H3 and H5 of simulation 1OKC (i.e. without bound inhibitor), especially in the region of the key proline residues (P27, P132, P229, respectively) in these helices. We will return to this in more detail below.
Differences between the crystal structure and those obtained from the MD simulations (with inhibitor present) are observed. As noted above, a crystal structure provides a static representation of a protein (in the absence of a lipid bilayer and at cryogenic temperatures). Thus, simulation under a different set of physical conditions (i.e. at 300K in a lipid bilayer) may reasonably be expected to exhibit some deviations from the X-ray structure. However, this is not considered to be suggestive of error in either the MD simulation or the crystal structure, but rather reveals relaxation of the protein in its changed environment Citation[29], Citation[30].
We also analysed the root mean square fluctuations (RMSFs) of each Cα atom from its mean position as a function of residue number (see Supplementary Data, , online version only). This revealed the usual pattern of peaks corresponding to surface loops of the protein and troughs corresponding to TM α-helices. Comparison of the RMSF profiles of the three simulations revealed no obvious trend. In seven out of 11 residues involved in binding the inhibitor, the simulations in the absence of bound inhibitor showed the greatest RMSF value, but in most cases the differences in RMSF were very small.
Overall, these analyses suggest that the protein is generally stable over the course of three simulations, and that deviations from the conformation of the crystal structure are relatively localised. This is to be expected, as the timescale of the simulations (20 ns) is much shorter than that for the solute transport process (>1 ms Citation[31]). However, there are some interesting short timescale differences in conformational dynamics caused by the presence/absence of bound inhibitor. These were examined in more detail to see if they provided any clues as to the relationship between the intrinsic flexibility of the protein and its transport mechanism.
Domains
Given the three-fold pseudosymmetry of the structure of the ADP/ATP carrier, it was of interest to compare the conformational dynamics of the individual domains (D1, D2, D3 – see B). Comparison of the Cα RMSDs for the three domains () reveals some interesting differences in conformational drift from the X-ray structure. In the absence of inhibitor and crystallographic water (simulation 1OKC) there is relatively little difference in the RMSDs of the three domains, all of which range from ∼3 to ∼3.5 Å.
Figure 4. Conformational drift of the three domains, for simulations: 1OKC, 1OKC-W, 1OKC-CATR, and 1OKC-CATR-W RMSDs. In each case the Cα RMSD of the domain is shown as a function of time, relative to the initial structure of that domain. Black = D1 (residues 2–101), red = D2 (residues 102–201), and green = D3 (residues 202–292).
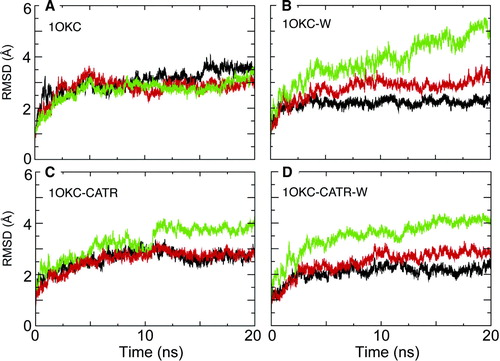
In contrast, in the other three simulations, an asymmetry in the behaviour of the domains emerges. In all simulations, the conformational drift of domain D2 remains relatively unchanged. In the presence of crystallographic waters (1OKC-W and 1OKC-CATR-W), the conformational drift of D1 is reduced to approximately 2 Å, thus, crystallographic waters lower the drift of D1 relative to D2, and relative to itself in the absence of crystallographic water (1OKC). For both simulations 1OKC-CATR and 1OKC-CATR-W, the conformational drift of D1 is lower in presence of inhibitor (∼2.8 Å) relative to in its absence (simulation 1OKC, ∼3.5 Å). There also appears to be a significant increase in the conformational drift of D3 in presence of inhibitor, and in the presence of crystallographic water, such that 1OKC-W > 1OKC-CATR-W≈1OKC-CATR ∼4 Å > >1OKC ∼3.5 Å. This asymmetry is of interest given that the interactions of CATR in the X-ray structure (and in the simulations) are predominantly with D1 and D2 (see B). Analysis of the RMSDs for just the transmembrane helices (see Supplementary Data, , online version only) reveals a slightly different pattern. However, in the case of simulation 1OKC-CATR-W, the transmembrane helix RMSDs for D1 and D2 are much lower than for D3, again suggesting that interactions of the inhibitor with D1 and D2 seem to stabilise these helices relative to those of D3.
Principal components analysis
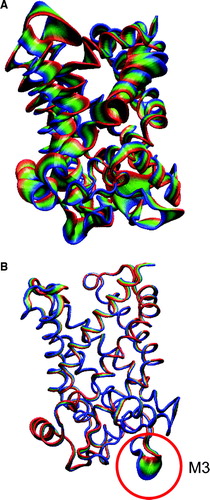
The results discussed above suggest a general “tightening” of the structure of the ADP/ATP carrier in the presence of CATR. This was confirmed by using PCA to identify the main motions of the protein during the simulations. The motions of each simulation were projected along the corresponding first eigenvector determined by PCA of the latter 10 ns of the simulation. It can be seen (A) that for simulation 1OKC (i.e. no inhibitor) the motions of the first eigenvector are distributed across the whole protein. In contrast, for e.g. simulation 1OKC-CATR-W (i.e. with both inhibitor and crystallographic water) the first eigenvector motions are restricted to the loop regions, and in particular loop M3. Interestingly, the interactions between the M3 loop and lipid headgroups (see below) are lower in the simulations in the presence of CATR than in the 1OKC simulation. The other two simulations, 1OKC-W and 1OKC-CATR, indicated motions intermediate between the two shown here.
Figure 5. Principal components analysis of simulations: A 1OKC, and B 1OKC-CATR-W. In each case the simulated motions are projected along eigenvector 1, with each trajectory fitted to the t=0 ns structure. The protein is drawn in tube representation and coloured to shown the range of the motions. For 1OKC-CATR-W the major motions are restricted to the loop regions, in particular loop M3 (highlighted by the red circle).
Role of prolines
One of the most striking features of the structure of the ADP/ATP carrier Citation[8] is that helices of the inner bundle (i.e. H1, H3, H5) are kinked so as to close the ‘pore’ that would otherwise be formed across the bilayer at the inner (matrix) mouth. The kink in each helix is associated with the a conserved proline in the PX(-)XX(+) motif of the odd-numbered TM helices (A). Given the importance of proline kinks in a number of membrane proteins Citation[32], and indeed the presence of such a structural motif in a large number of membrane proteins Citation[27], Citation[33] we have examined the conformational dynamics of this motif in the ADP/ATP carrier in some detail.
Figure 6. (A) The inner helix bundle formed by H1, H3, and H5, illustrated by the X-ray structure. Left to right: Side view; top view from cytosol. The white spheres indicate the positions of the key prolines (P27 in H1 [white], P132 in H3 [black], and P229 in H5 [grey]). (B) and (C) Snapshots of H1 (B) and H5 (C) from all four simulations. In each case structures saved every 2 ns are shown, superimposed in on the residues C-terminal to the key proline (indicated by the arrows).
![Figure 6. (A) The inner helix bundle formed by H1, H3, and H5, illustrated by the X-ray structure. Left to right: Side view; top view from cytosol. The white spheres indicate the positions of the key prolines (P27 in H1 [white], P132 in H3 [black], and P229 in H5 [grey]). (B) and (C) Snapshots of H1 (B) and H5 (C) from all four simulations. In each case structures saved every 2 ns are shown, superimposed in on the residues C-terminal to the key proline (indicated by the arrows).](/cms/asset/672d7aaa-b5d3-4955-adab-41618403c909/imbc_a_346095_f0006_b.gif)
To visualize the conformational dynamics of H1, H3, and H5 about the proline-induced kink, snapshots of the helices were saved every 2 ns from each simulation, and each helix fitted onto the Cα atoms of the region C-terminal to the proline. In each case the proline was seen to induce a dynamic hinge in the helix. Examining the superimposed snapshots in more detail for, e.g. H1 and H5 (B, C), it is evident that there is a trend in dynamic flexibility such that 1OKC≈1OKC-W > 1OKC-CATR≈1OKC-CATR-W. This clearly reflects the trend in overall conformational drift noted above. Overall, it appears that H1 is most flexible about the proline residue in the absence of the inhibitor.
This can be quantified via analysis of the helix-hinge behaviour in terms of the kink and swivel angles Citation[27], Citation[34] (). From this is can be seen that for, e.g. H1 the range of swivel angles is ∼120–150° without CATR (simulation 1OKC, 1OKC-W), compared to ∼100° in the presence of CATR inhibitor (simulations 1OKC-CATR and 1OKC-CATR-W). There does not seem to be a corresponding difference in kink angles. For H3 there is considerable flexibility in all simulations, and the presence/absence of the inhibitor appears to have little effect on the flexibility of the helix. The flexibility of H5 is somewhat less than for H1 or H3.
Figure 7. Swivel and kink angles shown as polar plots (see e.g. Citation[27], Citation[34]), for motions about the proline-induced molecular hinges in columns: (A) helix H1, (B) helix H3, and (C) helix H5. In each case the plots are shown for simulations 1OKC, 1OKC-W, 1OKC-CATR and 1OKC-CATR-W.
![Figure 7. Swivel and kink angles shown as polar plots (see e.g. Citation[27], Citation[34]), for motions about the proline-induced molecular hinges in columns: (A) helix H1, (B) helix H3, and (C) helix H5. In each case the plots are shown for simulations 1OKC, 1OKC-W, 1OKC-CATR and 1OKC-CATR-W.](/cms/asset/dbd3719d-ee59-4b4a-9bff-0f410c7580bc/imbc_a_346095_f0007_b.gif)
There is some correlation of these differences with the dimensions of the cavity/pore in the interior of the carrier (the latter evaluated using HOLE Citation[35]). Thus, ‘pore’ radius analysis (data not shown) reveals that there is a degree of flexibility in the pore in the absence of CATR over the duration of the simulation. Indeed, the cavity seems to narrow slightly along its complete length. In contrast, in the presence of CATR, any narrowing is largely confined to the mouth of the cavity, whilst the inhibitor-binding region remained relatively unchanged.
Salt bridges
The conserved (+) and (-) residues in the PX(-)XX(+) motif of H1, H3 and H5 (i.e. the R/K and D/E residues) form a salt bridge network between these helices in the region C-terminal to the proline residues. The integrity of this network was examined by determining the distances between the sidechains of the residues involved, namely: E29-R137 (H1-H3), D134-R234 (H3-H5), and D231-K32 (H5-H1) (). In the X-ray structure Citation[8], these sidechains are all within ∼0.3 nm of one another. As expected, given the dynamic hinge behaviour about the proline, there were substantial fluctuations in the distances between the residues. We have seen above that the hinge dynamics are most pronounced for the H1 and H3 helices in the 1OKC simulation (i.e. in the absence of bound inhibitor). This correlates with large fluctuations in the interhelix salt-bridges for H1-H3 in this simulation. These fluctuations are only reduced to a small extent in the presence of the inhibitor.
Figure 8. Interdomain salt-bridges. Interatomic distances as a function of time are shown for the charged pair interactions (E29(H1)-R137(H3); D134(H3)-R234(H5); and D231(H5)-K29(H1)) between the PXX(-)XX(+) motifs of adjacent helices: (A) H1-H3, (B) H3-H5, and (C) H5-H1. Black: 1OKC; red: 1OKC-W; green: 1OKC-CATR; blue: 1OKC-CATR-W. (Running averages were applied every 10 data points to reduce the effect of noise and thus simplify the plots. The distances computed are those between the centre of mass of the relevant groups of atoms, e.g. the C, O, O atoms of a carboxylate.)
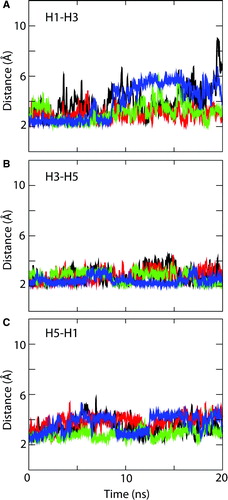
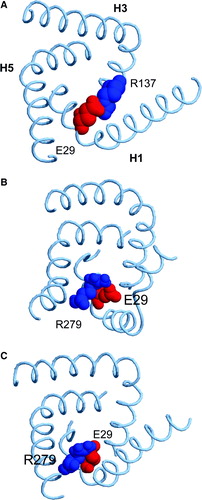
In each of the three different systems, it appears that the salt bridge between E29 and R137 is not maintained over the course of the simulation. In each case, the sidechain of R137 switches to interact with that of D134 (also on H3). As a consequence of this switch, the bridge between E29 (in H1) and R137 (in H3) is lost. Instead, E29 switches to interact with R279 (which is in H6).
The snapshots shown in indicate the effect of the E29/R137 switch on the overall structure of the protein. It can be seen how, especially in simulation 1OKC, the loss of the E29/R137 interaction is associated with local distortion of the secondary structure of helices H1 and H5 in the absence of the inhibitor.
Figure 9. Snapshots of the H1-H3 salt bridge interactions in (A) the crystal structure, (B) at the end of simulation 1OKC, and (C) at the end of simulation 1OKC-CATR-W. The key acidic (E29, red) and basic (R137 or R279, blue) residues are shown as van der Waals radius spheres.
Inhibitor binding interactions
An interaction between R279 and E29 was observed during each of the three simulations (as shown in and discussed above). In simulation 1OKC-CATR there are interactions present between the sidechains of R279, R79 and K22 and the carboxylate groups of CATR for the duration of the simulation. Similarly, in simulation 1OKC-CATR-W, there are interactions between the carboxylate groups of CATR and residues R279, K22 and R79 over the first 8.5 ns of the simulation. After approximately the first 7 ns of the simulation, there are interactions between the CATR carboxylate groups and residues R279, K22 and R137. This correlates with the loss of the salt bridge between E29 and R137, indicating a change of conformation after ∼7 ns for the sidechain of R137.
A bridging water molecule is observed in the crystal structure, between R279 and the CATR carboxylate group. This interaction is lost early on in the simulations (after ∼30 ps, i.e. during the equilibration period). Despite losing the bridging water molecule, the sidechain of R279 is able to maintain an interaction with the characteristic carboxylate of the CATR molecule for the duration of the simulation. This interaction is surrounded by water molecules, although none of these water molecules bridge the interaction in the same way as is observed in the crystal structure. The sidechain of K22 also maintains an interaction with CATR for almost the entirety of the simulation. The snapshot in A shows the interaction between the cationic sidechain of R279 and anionic CATR carboxylate groups and sidechain of E29.
The contacts observed between CATR and the protein in the crystal structure are summarised in B. As previously discussed, there are a number of polar interactions, between the sulphate groups and the side-chains of N87, K91 and R187. The carboxylate moieties interact with R79 and with R279 via a water molecule (although it is noted that the presence of the water molecule in this interaction is short lived in the simulations) and the hydroxyl group interacts with D231. The diterpene moiety of CATR stacks onto the aromatic ring of Y186 and there are van der Waals contacts with residues L127, V130 and I183.
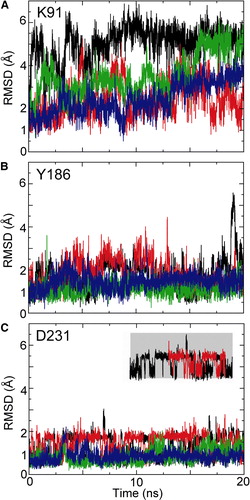
Of these contacts to the bound inhibitor, a number of sidechains show a change in mobility in simulations of the ligand-free protein (simulation 1OKC) compared with those simulations in which bound CATR is present. Thus, for example K91 (electrostatic interaction with the sulphate), Y186 (stacking interaction with the diterpene ring), and R231 (H-bond to the hydroxyl) all display enhanced conformational fluctuations in the absence of CATR (see Supplementary Data , online version only). In particular, D231 indicates the presence of two stable conformations in the absence of the inhibitor, switching between two RMSD values; whilst, when CATR is present, the sidechain is restricted to one conformation. This is shown in the inset of Figure 10, D231.
Lipid-protein interactions
In the X-ray study of the ADP/ATP carrier it is suggested that the “linker” helices (h12, h34 and h56, corresponding to residues 52-63, 155–166, and 253–264, respectively) may interact with the membrane Citation[8]. Such interactions are evident in the simulations. Visualisation of the protein/lipid system at the end of the 2 ns equilibration period () shows that the POPC lipid bilayer undergoes a degree of local distortion to accommodate the protein, allowing interaction of the non-TM helices (i.e. h12, h34 and h56) with the headgroups of the lipids.
The crystal structure also reveals that there are four lipids and two detergent molecules bound to the protein. The interactions mainly occur at the matrix face of the protein. This asymmetry is also seen in the simulations, with a greater number of interactions (data not shown) between the lipid headgroups and each of the M loops, and between the headgroups and the linker helices, than with the C loops.
Cysteine scanning experiments Citation[36] have suggested a role in gating for loops LM1 and LC1 in the yeast homologue of the ADP/ATP carrier. These loops are approximately equivalent to M1 and C1 in the bovine carrier, although recently, structural differences have been pinpointed using a Copper-o-phenanthroline inter-monomer cross-linker Citation[37]. In the crystal structure, loop C1 interacts with the only lipid molecule attached to the cytosolic facing region of the protein. An interaction between C1 and the lipid headgroups of the POPC bilayer is present for the duration of all three simulations. This interaction may be important to maintaining the opening of the channel in the cytosolic facing conformation of the protein.
The C2 loop has generally fewer interactions with the bilayer headgroups than the C1 loop and behaves differently in the presence and absence of CATR. In particular, the C2 loop folds in towards the centre of the protein in the presence of the inhibitor, such that N206 and K205 reach down into the cavity. The movement of this loop in the presence of inhibitor may allude to the initial stages of the conformational change which takes place upon nucleotide binding. Cross linking behaviour in the M loops was shown by Kihira and co-workers Citation[37], to be affected by the binding of ADP vs. BKA, thus the presence of ligand in the binding site can be assumed to affect the conformation and behaviour of the loops. It is reasonable to conjecture that a similar effect might be observed for the C-loops when binding CATR vs. ATP is considered. However, that is beyond the scope of the study presented here.
Discussion and conclusion
From a biological perspective, perhaps the key result is the ‘tightening’ of structure by the inhibitor (CATR). This decrease in (local) flexibility may help to explain the stabilization of the c-state of the carrier, which was important in enabling crystallization of the protein. In particular, the tight closure of the cavity at the inner end of the helix bundle is a key feature of the c-state structure. It is likely that, on a substantially longer timescale than addressed in the current simulations (see below), the increased flexibility in the absence of the inhibitor may facilitate the conformation transition to the m-state.
Another mechanistically relevant aspect of the simulation results is the role of the proline residues in the dynamic flexibility H1, H3, and H5 helices. It has been suggested that flexibility about these proline hinges may enable a conformational change whereby the protein forms a “channel” through the membrane Citation[8], Citation[12]. The results of our simulations would support such a mechanism. It is perhaps significant that the presence of CATR reduces the flexibility of H1 in particular. Interestingly, Proline-kinked helices are also present in a number of other transport proteins, e.g. lactose permease Citation[3], and indeed in other major families of membrane proteins Citation[32]. A recent simulation study of the antiporter NhaA has suggested a functional role for a GxP-mediated helix kink Citation[38]. Furthermore, Parelvarez-Marin Citation[39] and colleagues demonstrate that proline to alanine mutations (at P50, P91 and P186) in bacteriorhodopsin significantly perturb a number of functional properties of the protein in both the resting and light activated states, even though the X-ray structure of bacteriorhodopsin remains largely unchanged by these mutations Citation[40]. Thus the functional differences may be attributed to changes in flexibility of the transmembrane helices upon mutation of prolines. This study suggests a dynamic role for proline-mediated kinks in TM helices, consistent with our findings for the ADP/ATP carrier.
The changes in patterns of salt-bridges during the simulations are of also of interest. These salt bridges maintain the ‘closed’ conformation of the c-state in the X-ray structure. It is likely that dynamic changes in these patterns of salt-bridges will occur during the transport mechanism. This has been discussed in the context of the X-ray structure of the carrier Citation[8], Citation[9], Citation[12]. Such changes may be promoted by binding of the highly charged nucleotide solutes. However, in the absence of a structure with bound ATP or ADP it is unwise to speculate as to the exact nature of the induced change. A recent simulation study Citation[41] provides a model of ADP binding to the ADP/ATP carrier.
It is important also to consider the limitations of the current methodology. The simulations are short (∼10 ns) compared with the transport kinetics (∼1 ms) Citation[31] of the protein. Thus we must remember that we are trying to extrapolate from observations on local flexibility to large scale conformational transitions. It is conceivable that more approximate approaches (e.g. network models Citation[42]) may help in this process of extrapolation.
Another limitation of the simulation methodology is the use of fixed ionisation states for acidic and basic sidechains. If studies are to be extended to explore possible changes in salt-bridges on binding nucleotides, it may be necessary to treat sidechain ionisation states dynamically during the course of a simulation Citation[43].
The current simulations employ a relatively simple (phosphatidyl choline) lipid bilayer. This does not reflect the complexities of the lipids of the mitochondrial inner membrane. Given the role of cardiolipin in stabilising the dimeric form of the ADP/ATP carrier Citation[44], and given the ongoing debate about the relative functional importance of the monomeric and dimeric forms of the protein (see e.g. Citation[9] and Citation[12], Citation[44]), it would be of some interest to model and simulate the protein dimer in more complex lipid environments.
Figure S1. Secondary structure as a function of time for simulations: (A) 1OKC; (B) 1OKC-CATR; and (C) 1OKC-CATR-W. The colour code for secondary structure is as follows: blue = α-helix; yellow = turn; green = bend; purple = Π-helix; grey = 310-helix; and white = coil.
Figure S2. Root mean square fluctuation (RMSF) of Cα atoms, averaged over the final 5 ns of each simulation. Black line = 1OKC; red line = 1OKC-W; green line = 1OKC-CATR; and blue line = 1OKC-CATR-W. The labels are of contacts between the protein and the CATR molecule.
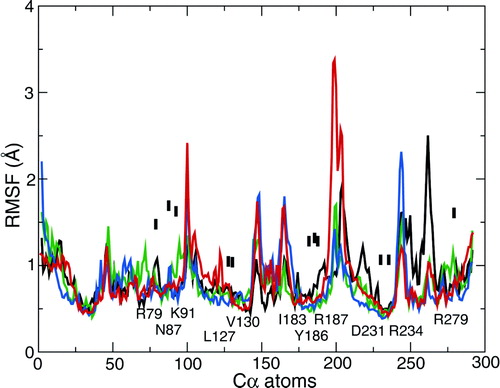
Figure S3. Conformational drift of the transmembrane helices of three domains, for simulations: (A) 1OKC, (B) 1OKC-W, (C) 1OKC-CATR, and (D) 1OKC-CATR-W RMSDs. In each case the Cα RMSD of the domain is shown as a function of time, relative to the initial structure of that domain. Black = D1, red = D2, and green = D3.
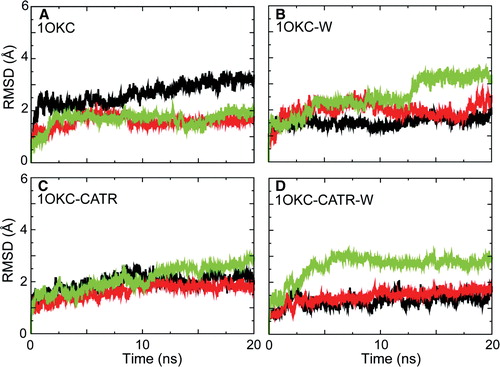
Figure S4. RMSDs of key CATR-binding sidechains, in each case for simulations 1OKC (black), 1OKC-W (red), 1OKC-CATR (green), and 1OKC-CATR-W (blue). The inset indicates two states observed for the sidechain of D231.
Acknowledgements
Research in MSPS's group is supported by grants from the BBSRC, the EPSRC, the EU, and from the Wellcome Trust. JMJ was an MRC research student. Our thanks to all of our colleagues for their interest in this work. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archean, and eukaryotic organisms. Prot Sci 1998; 7: 1029–1038
- White SH. The progress of membrane protein structure determination. Prot Sci 2004; 13: 1948–1949
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003; 301: 610–615
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003; 301: 616–620
- Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 2006; 312: 741–744
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 2005; 437: 215–223
- Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H. Structure of a Na + /H+ antiporter and insights into mechanism of action and regulation by pH. Nature 2005; 435: 1197–1202
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003; 426: 39–44
- Kunji ERS. The role and structure of mitochondrial carriers. FEBS Lett 2004; 564: 239–244
- Bamber L, Slotboom DJ, Kunji ERS. Yeast mitochondrial ADP/ATP carriers are monomeric in detergents as demonstrated by differential affinity purification. J Mol Biol 2007; 371: 388–395
- Bamber L, Harding M, Monne M, Slotboom DJ, Kunji ERS. The yeast mitochondrial ADP/ATP carrier functions as a monomer in mitochondrial membranes. Proc Natl Acad Sci USA 2007; 104: 10830–10834
- Nury H, Dahout-Gonzalez C, Trezeguet V, Lauquin CJM, Brandolin G, Pebay-Peyroula E. Relations between structure and function of the mitochondrial ADP/ATP carrier. Ann Rev Biochem 2006; 75: 713–741
- Karplus MJ, McCammon JA. Molecular dynamics simulations of biomolecules. Nature Struct Biol 2002; 9: 646–652
- Adcock SA, McCammon JA. Molecular dynamics: survey of methods for simulating the activity of proteins Chem Rev 2006; 106: 1589–1615
- Ash WL, Zlomislic MR, Oloo EO, Tieleman DP. Computer simulations of membrane proteins. Biochim Biophys Acta 2004; 1666: 158–189
- Gumbart J, Wang Y, Aksimentiev A, Tajkhorshid E, Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr Opin Struct Biol 2005; 15: 423–431
- Faraldo-Gómez JD, Smith GR, Sansom MSP. Setup and optimisation of membrane protein simulations. Eur Biophys J 2002; 31: 217–227
- van Gunsteren WF, Kruger P, Billeter SR, Mark AE, Eising AA, Scott WRP, Huneberger PH, Tironi IG 1996. Biomolecular simulation: The GROMOS96 Manual and User Guide. Groning and Zurich: Biomos & Hochschulverlag AG an der ETH Zurich.
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Molec Model 2001; 7: 306–317
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. Intermolecular forces, B Pullman. Reidel, Dordrecht 1981; 331–342
- van der Spoel D, van Maaren PJ, Berendsen HJC. A systematic study of water models for molecular simulations. J Chem Phys 1998; 108: 10220–10230
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81: 3684–3690
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22: 2577–2637
- Humphrey W, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Molec Graph 1996; 14: 33–38
- Sayle RA, Milner-White EJ. RasMol: Biomolecular graphics for all. Trends Biochem Sci 1995; 20: 374–376
- Barrett CP, Hall BA, Noble MEM. Dynamite: a simple way to gain insight into protein motions. Acta Cryst D 2004; 60: 2280–2287
- Cordes FS, Bright JN, Sansom MSP. Proline-induced distortions of transmembrane helices. J Mol Biol 2002; 323: 951–960
- Law RJ, Capener C, Baaden M, Bond PJ, Campbell J, Patargias G, Arinaminpathy Y, Sansom MSP. Membrane protein structure quality in molecular dynamics simulation. J Mol Graph Mod 2005; 24: 157–165
- Bond PJ, Sansom MSP. Membrane protein dynamics vs. environment: simulations of OmpA in a micelle and in a bilayer. J Mol Biol 2003; 329: 1035–1053
- Bond PJ, Faraldo-Gómez JD, Deol SS, Sansom MSP. Membrane protein dynamics and detergent interactions within a crystal: a simulation study of OmpA. Proc Natl Acad Sci USA 2006; 103: 9518–9523
- Gropp T, Brustovetsky N, Klingenberg M, Müller V, Fendler K, Bamberg E. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys J 1999; 77: 714–726
- Sansom MSP, Weinstein H. Hinges, swivels & switches: the role of prolines in signalling via transmembrane a-helices. Trends Pharm Sci 2000; 21: 445–451
- Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr Opin Struct Biol 2004; 14: 465–479
- Bright JN, Sansom MSP. The flexing/twirling helix: exploring the flexibility about molecular hinges formed by proline and glycine motifs in transmembrane helices. J Phys Chem B 2003; 107: 627–636
- Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP. Hole: a program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph 1996; 14: 354–360
- Hashimoto M, Majima E, Goto S, Shinohara Y, Terada H. Fluctuation of the first loop facing the matrix of the mitochondrial ADP/ATP carrier deduced from intermolecular cross-linking of Cys56 residues by bifunctional dimaleimides. Biochem 1999; 38: 1050–1056
- Kihira Y, Ueno M, Terada H. Difference between yeast and bovine mitochondrial ADP/ATP carriers in terms of conformational properties of the first matrix loop as deduced by use of copper-o-phenanthroline. Biol Pharma Bull 2007; 30: 885–890
- Olkhova E, Padan E, Michel H. The influence of protonation states on the dynamics of the NhaA antiporter from Escherichia coli. Biophys J 2007; 92: 3784–3791
- Perálvarez-Marín A, Bourdelande J-L, Querol E, Padrós E. The role of proline residues in the dynamics of transmembrane helices: the case of bacteriorhodopsin. Molec Memb Biol 2006; 23: 127–135
- Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. The evolution of transmembrane helix kinks and the structural diversity of G protein-coupled receptors. Proc Natl Acad Sci USA 2004; 101: 959–963
- Dehez F, Pebay-Peyroula E, Chipot C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J Am Chem Soc 2008; 130: 12725–12733
- Keskin O, Jernigan RL, Bahar I. Proteins with similar architecture exhibit similar large-scale dynamic behavior. Biophys J 2000; 78: 2093–2106
- Lee MS, Salsbury FR, Brooks CL. Constant-pH molecular dynamics using continuous titration coordinates. Proteins: Struct Func Bioinf 2004; 56: 738–752
- Nury H, Dahout-Gonzalez C, Trezeguet V, Lauquin CJM, Brandolin G, Pebay-Peyroula E. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett 2005; 579: 6031–6036