Abstract
The chemical solvent tetrahydrofuran (THF) increases short-circuit current (Isc) in renal epithelia endogenously expressing the cystic fibrosis transmembrane conductance regulator (CFTR). To understand how THF increases Isc, we employed the Ussing chamber and patch-clamp techniques to study cells expressing recombinant human CFTR. THF increased Isc in Fischer rat thyroid (FRT) epithelia expressing wild-type CFTR with half-maximal effective concentration (KD) of 134 mM. This THF-induced increase in Isc was enhanced by forskolin (10 µM), inhibited by the PKA inhibitor H-89 (10 µM) and the thiazolidinone CFTRinh-172 (10 µM) and attenuated greatly in FRT epithelia expressing the cystic fibrosis mutants F508del- and G551D-CFTR. By contrast, THF (100 mM) was without effect on untransfected FRT epithelia, while other solvents failed to increase Isc in FRT epithelia expressing wild-type CFTR. In excised inside-out membrane patches, THF (100 mM) potentiated CFTR Cl− channels open in the presence of ATP (1 mM) alone by increasing the frequency of channel openings without altering their duration. However, following the phosphorylation of CFTR by PKA (75 nM), THF (100 mM) did not potentiate channel activity. Similar results were obtained with the ▵R-S660A-CFTR Cl− channel that is not regulated by PKA-dependent phosphorylation and using 2′deoxy-ATP, which gates wild-type CFTR more effectively than ATP. Our data suggest that THF acts directly on CFTR to potentiate channel gating, but that its efficacy is weak and dependent on the phosphorylation status of CFTR.
| Abbreviations | ||
| ABC transporter | = | ATP-binding cassette transporter |
| CF | = | cystic fibrosis |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| 2′-dATP | = | 2′-deoxy-ATP |
| FRT cells | = | Fischer rat thyroid cells |
| H-89 | = | N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, 2HCl |
| i | = | single-channel current amplitude |
| IBI | = | interburst interval |
| ICL | = | intracellular loop |
| Isc | = | short-circuit current |
| MBD | = | mean burst duration |
| MDCK cells | = | Madin-Darby canine kidney cells |
| MSD | = | membrane-spanning domain |
| N | = | number of active channels |
| NBD | = | nucleotide-binding domain |
| NMDG | = | N-methyl-D-glucamine |
| NPPB | = | 5-nitro-2-(3-phenylpropylamino)-benzoic acid |
| Po | = | open-probability |
| RD | = | regulatory domain |
| Rt | = | transepithelial resistance |
| Tes | = | N-tris[Hydroxymethyl]methyl-2-aminoethanesulfonic acid |
| THF | = | tetrahydrofuran |
Introduction
Tetrahydrofuran (THF) is an organic oxide, consisting of a four-carbon cyclic ether Citation[1] (for the chemical structure of THF, see A). It is an aprotic, electron donating solvent that dissolves a wide range of nonpolar and polar compounds Citation[1]. As a result, THF is an important and widely used solvent in chemistry, the manufacture of dyes and adhesives.
Figure 1. THF stimulates transepithelial ion transport in FRT epithelia expressing wild-type human CFTR. Representative Isc recordings from FRT epithelia expressing wild-type human CFTR show the effects of (A) THF; (B) ethanol (EtOH); (C) methanol (MeOH); (D) DMSO (all tested at 0.8% v v−1). During the periods denoted by the solid bars the indicated chemical solvents and forskolin (Fk; 10 µM) + genistein (Gen; 50 µM) were present in the solution bathing the apical membrane. Transepithelial voltage was clamped at 0 mV and there was a large Cl− concentration gradient across the epithelium ([Cl−]basolateral=149 mM; [Cl−]apical=14.8 mM). The dotted lines indicate zero current. The chemical structure of each solvent is shown; (E) Effect of chemical solvents on Isc. Column and error bars are means + SEM (solvents, n=4; Fk + Gen, n=7).
![Figure 1. THF stimulates transepithelial ion transport in FRT epithelia expressing wild-type human CFTR. Representative Isc recordings from FRT epithelia expressing wild-type human CFTR show the effects of (A) THF; (B) ethanol (EtOH); (C) methanol (MeOH); (D) DMSO (all tested at 0.8% v v−1). During the periods denoted by the solid bars the indicated chemical solvents and forskolin (Fk; 10 µM) + genistein (Gen; 50 µM) were present in the solution bathing the apical membrane. Transepithelial voltage was clamped at 0 mV and there was a large Cl− concentration gradient across the epithelium ([Cl−]basolateral=149 mM; [Cl−]apical=14.8 mM). The dotted lines indicate zero current. The chemical structure of each solvent is shown; (E) Effect of chemical solvents on Isc. Column and error bars are means + SEM (solvents, n=4; Fk + Gen, n=7).](/cms/asset/e4b9635c-c7d0-40ad-8528-fa0113202488/imbc_a_348964_f0001_b.gif)
In previous work, we used THF as a vehicle to solubilise artificial anion transporters because it is the solvent of choice for biophysical studies of these molecules Citation[2]. To our surprise, we found that THF, itself, stimulated weakly short-circuit current (Isc; a measure of transepithelial ion transport) in type I Madin-Darby canine kidney (MDCK) epithelia Citation[2]. However, the cellular mechanisms by which THF increased Isc were not investigated in that study and we are unaware of reports in the literature of THF modulating the activity of ion channels and transporters.
We undertook the present study to identify how THF stimulates transepithelial ion transport. Type I MDCK epithelia endogenously express the ATP-binding cassette (ABC) transporter cystic fibrosis transmembrane conductance regulator (CFTR) Citation[3], a Cl− channel with complex regulation that plays an essential role in fluid and electrolyte transport across epithelia Citation[4]. CFTR is assembled from five domains: Two membrane-spanning domains (MSDs) that form an anion-selective pore, two nucleotide-binding domains (NBDs) that control channel gating and a unique regulatory domain (RD) phosphorylation of which is a prerequisite for channel activity Citation[5], Citation[6].
A large number of small molecules (termed CFTR potentiators) have been identified that interact directly with CFTR to enhance channel gating and hence, transepithelial ion transport Citation[6]. We therefore hypothesised that THF might increase Isc by enhancing CFTR-mediated transepithelial ion transport. To test this idea, we employed the Ussing chamber technique to study Fischer rat thyroid (FRT) epithelia expressing recombinant wild-type human CFTR. When we found that THF stimulates CFTR-mediated transepithelial ion transport, we employed the patch-clamp technique to investigate the effects of THF on the single-channel behaviour of CFTR. We discovered that THF acts directly on CFTR to potentiate channel gating, but that its efficacy is weak and dependent on the phosphorylation status of CFTR.
Materials and methods
Cells and cell culture
For this study, we used Fischer rat thyroid (FRT) epithelial cells expressing wild-type, F508del- and G551D- human CFTR Citation[8] and mouse mammary epithelial (C127) cells stably expressing either wild-type human CFTR or the variant ▵R-S660A-CFTR Citation[9]. FRT cells were supplied by Dr. L.J.V. Galietta (Istituto Giannina Gaslini, Genova, Italy) while C127 cells were gifts of Dr C.R. O'Riordan (Genzyme Corp., Framingham, MA, USA) and Prof. M.J. Welsh (University of Iowa, Iowa, USA). C127 and FRT cells were cultured as described previously Citation[8], Citation[10], with the exception that FRT cells expressing F508del-CFTR were not incubated at reduced temperatures to augment the cell surface expression of CFTR.
To grow FRT cells as polarized epithelia, trypsinized cells were seeded directly onto permeable filter supports (Millicell-PCF culture plate inserts 0.4 µm pore size, 12 mm diameter, Millipore Corp.; Fisher Scientific UK, Loughborough, UK) at a density of 4.5×105 cells per 0.6 cm2. Apical and basolateral media were replaced every 48 h and transepithelial resistance (Rt) measured using an epithelial voltohmeter (EVOM; World Precision Instruments, Stevenage, UK). Rt increased with time and five days after seeding (when Rt=3.36±0.08 kω cm2; n=79), we used FRT epithelia for experiments. For experiments using excised inside-out membrane patches, C127 cells were seeded onto glass coverslips and used within 48 h.
Short-circuit current measurements
FRT epithelia were mounted in modified Ussing chambers (Warner Instrument Corp., Dual Channel Chamber; Harvard Apparatus Ltd., Edenbridge, UK). The basolateral membrane of FRT epithelia was bathed in a solution containing (in mM): 140 NaCl, 5 KCl, 0.36 K2HPO4, 0.44 KH2PO4, 1.3 CaCl2, 0.5 MgCl2, 10 Hepes, and 4.2 NaHCO3, pH 7.2 with Tris ([Cl−], 149 mM); osmolarity, 288±0.1 mOsm (n=4). The composition of the solution bathing the apical membrane of FRT epithelia was identical to that of the basolateral solution, with the exception that (in mM): 133.3 Na gluconate + 2.5 NaCl and 5 K gluconate replaced 140 NaCl and 5 KCl, respectively, to create a transepithelial Cl− concentration gradient ([Cl−], 14.8 mM); osmolarity, 280±0.2 mOsm, (n=4). To compensate for the Ca2 + buffering capacity of gluconate, we used 5.7 mM Ca2 + in the apical solution. All solutions were maintained at 37°C and bubbled continuously with 5% CO2.
We clamped transepithelial voltage (referenced to the basolateral solution) at 0 mV and recorded Isc continuously using an epithelial voltage-clamp amplifier (Warner Instrument Corp., model EC-825, Harvard Apparatus Ltd.), digitising data as described previously Citation[3]. The resistance of the filter and solutions, in the absence of cells, was subtracted from all measurements. Flow of current from the basolateral to the apical solution is shown as an upward deflection.
Patch-clamp experiments
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200A patch-clamp amplifier (Molecular Devices Corp., Union City, CA, USA) and pCLAMP data acquisition and analysis software (versions 6.03 and 9.0.0, Molecular Devices Corp.) as previously described Citation[9], Citation[10]. The established sign convention was used throughout; currents produced by positive charge moving from intra- to extracellular solutions (anions moving in the opposite direction) are shown as positive currents.
The pipette (extracellular) solution contained (mM): 140 N-methyl-D-glucamine (NMDG), 140 aspartic acid, 5 CaCl2, 2 MgSO4 and 10 N-tris[Hydroxymethyl]methyl-2-aminoethanesulfonic acid (Tes), adjusted to pH 7.3 with Tris ([Cl−], 10 mM; osmolarity, 279±0.5 mOsm; n=7). The bath (intracellular) solution contained (mM): 140 NMDG, 3 MgCl2, 1 CsEGTA and 10 Tes, adjusted to pH 7.3 with HCl, ([Cl−], 147 mM; free [Ca2 + ],<10−8 M; osmolarity, 281±0.5 mOsm; n=4) and was maintained at 37°C; voltage was −50 mV.
After excision of inside-out membrane patches, CFTR Cl− channels were activated by the addition of ATP (1 mM) and the catalytic subunit of PKA (75 nM) to the intracellular solution within 5 min of patch excision. To test the effects of THF, the solvent was added to the intracellular solution either before or after PKA addition. We recorded, filtered and digitised data as described previously Citation[9]. For the purposes of illustration, data were filtered at 500 Hz and digitised at 1 kHz.
To investigate the effects of THF on fully phosphorylated CFTR Cl− channels, we used membrane patches containing multiple active channels as previously described Citation[11]. To measure single-channel current amplitude (i), Gaussian distributions were fit to current amplitude histograms. To study the effects of THF on CFTR channel gating prior to phosphorylation by PKA, we calculated burst duration and frequency of channel gating using membrane patches that contained only bursts of single level openings with no superimposed openings that were separated from one another by a minimum of 20 ms in the presence of ATP (1 mM) as described previously Citation[10]. In membrane patches containing between 1 and 5 channels NPo was calculated using the equation:1
where N is the number of channels, Ttot is the total time analysed and T1 is the time that one or more channels are open, T2 is the time two or more channels are open and so on. However, when membrane patches contained large numbers of active channels, NPo was calculated by dividing I by i.
Reagents
PKA was purchased from Promega Ltd. (Southampton, UK). ATP (disodium salt), 2′-deoxyadenosine 5′-triphosphate disodium salt (2′-dATP), forskolin, Hepes, Tes, THF and vitamin C were obtained from the Sigma-Aldrich Company, Ltd. (Gillingham, UK). Genistein was purchased from LC Laboratories (Woburn, MA, USA), N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, 2HCl (H-89; Calbiochem) from Merck Biosciences, Ltd. (Nottingham, UK), 5-nitro-2-(3-phenylpropylamino)-benzoic acid (NPPB) from Semat Technical (UK) Ltd. (St Albans, UK) and CFTRinh-172 from Asinex Ltd. (Moscow, Russia) and Calbiochem. All other chemicals were of reagent grade.
ATP, 2′-dATP and vitamin C were dissolved in distilled water, forskolin in methanol and all other reagents in DMSO. Stock solutions were stored at −20°C with the exception of those of (i) vitamin C, which was stored at ∼23°C and (ii) ATP and 2′-dATP, which were prepared directly before each experiment. Immediately before use, stock solutions were diluted to achieve final concentrations. Precautions against light-sensitive reactions were observed when using genistein and vitamin C.
Statistics
Results are expressed as means±SEM of n observations. To test for differences between two groups of data, we used paired and unpaired t tests, and for non-parametric data the Wilcoxon signed rank test (paired) and Mann-Whitney rank sum test (unpaired). To test for differences between multiple sets of data, we used a one-way analysis of variance (ANOVA) and the Dunn's and Holm Sidak method post tests where appropriate. Differences were considered statistically significant when p<0.05. Tests were performed using SigmaStatTM (version 2.03, Jandel Scientific GmbH, Erkrath, Germany).
Results
THF causes a Cl−-dependent increase in Isc
To investigate the effects of the chemical solvent THF on transepithelial ion transport, we grew FRT cells expressing wild-type human CFTR as polarised epithelia, mounted them in Ussing chambers and recorded Isc, a measure of transepithelial ion transport. To study Cl−-dependent transepithelial ion transport, we imposed a large Cl− concentration gradient ([Cl−]basolateral=148 mM; [Cl−]apical=14.8 mM) across epithelia without permeabilizing the basolateral membrane and clamped transepithelial voltage at 0 mV. As controls, we tested the effects of other chemical solvents including ethanol, methanol and DMSO. A and 1E demonstrate that addition of THF (0.8% v v−1; 100 mM) to the solution bathing the apical surface of FRT epithelia expressing wild-type CFTR increased Isc. By contrast, ethanol, methanol and DMSO (all tested at 0.8% v v−1) failed to increase Isc (B–1E). Following treatment of FRT epithelia with chemical solvents, addition to the apical solution of the cAMP agonist forskolin (10 µM) and the CFTR potentiator genistein (50 µM) generated a substantial Isc, 5-fold greater than that achieved by THF (100 mM) ().
Next we explored the Cl−-dependence of the THF-induced Isc. When FRT epithelia expressing wild-type CFTR were bathed in symmetrical 148 mM Cl− solutions, the Isc elicited by THF (100 mM) decreased to 7% (4±1 µA cm−2; n=3) of that with the Cl− gradient (G; p=0.006). Similarly, the response to forskolin (10 µM) in the presence of symmetrical Cl− concentrations was only 5% (12±3 µA cm−2; n=5) of that under gradient conditions (G; p<0.001). In the presence of a Cl−-concentration gradient, addition of THF (100 mM) to the solution bathing the basolateral membrane elicited an Isc of similar magnitude to that evoked when THF was added to the apical solution (n=3; data not shown). Finally, both THF (100 mM) and forskolin (10 µM) were without effect on Isc in mock transfected FRT epithelia (E, 2F). These experiments demonstrate that THF increases Isc in FRT epithelia expressing recombinant CFTR and suggest that this Isc is Cl−-dependent. They also argue that the increase in Isc is not a consequence of the solvent acting non-specifically.
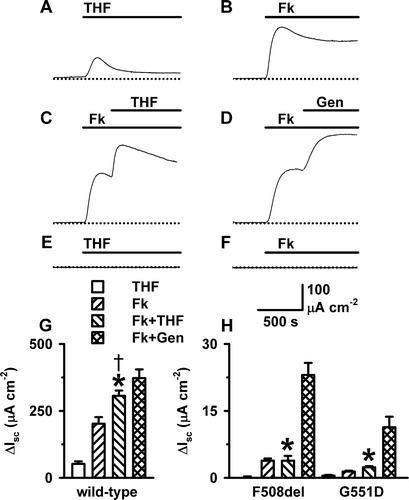
Figure 2. THF stimulation of transepithelial ion transport is cAMP-dependent. Representative Isc recordings from FRT epithelia expressing wild-type CFTR show the effects of (A) THF (100 mM); (B) forskolin (Fk; 10 µM); (C) forskolin (10 µM) and THF (100 mM); and (D) forskolin (10 µM) and genistein (Gen; 50 µM); (E, F) The effects of THF (100 mM) and forskolin (10 µM), respectively, on Isc in mock transfected FRT epithelia (n=5). During the periods denoted by the solid bars the indicated agents were present in the solution bathing the apical membrane. (G, H) The change in Isc evoked in wild-type, F508del- and G551D-CFTR FRT epithelia by different agents. Columns and error bars are means + SEM (n=6–23). The asterisks and cross denote values that are significantly different from those of THF alone and forskolin alone, respectively, (p<0.05). Note the change in ordinate scale between G and H. Other details as in .
THF augments cAMP-stimulated Isc
To characterise the Isc elicited by THF, we compared the effects of THF (100 mM) and forskolin (10 µM) on transepithelial ion transport. A demonstrates that the THF-induced Isc was biphasic, with an initial transient peak followed by a sustained plateau 6-fold lower than the peak. By contrast, the Isc response of FRT epithelia expressing wild-type CFTR to forskolin was characterised by a large initial peak followed by a high, sustained plateau (B). Thus, the magnitude of the THF-elicited Isc was only one quarter that of the forskolin-induced Isc (G).
Many agents have been identified that potentiate cAMP-stimulated Isc mediated by the CFTR Cl− channel Citation[6]. To investigate whether THF potentiates cAMP-stimulated Isc, we applied THF (100 mM) to FRT epithelia expressing wild-type CFTR after first stimulating Isc with forskolin (10 µM). A, 2C and 2G demonstrates that following the stimulation of Isc by forskolin (10 µM), the Isc evoked by THF (100 mM) was enhanced compared with that elicited by THF (100 mM) alone. As a result, the peak change in Isc produced by forskolin (10 µM) and THF (100 mM) approached that generated by forskolin (10 µM) and genistein (50 µM; D, 2G). We interpret these data to suggest that CFTR is involved in the THF-induced increase in Isc.
To provide further evidence that THF increases Isc by acting on CFTR, we tested its effects on the cystic fibrosis (CF) mutants F508del and G551D, both of which are located in NBD1 of CFTR and associated with a severe disease phenotype. F508del causes CF principally by disrupting the processing of CFTR protein and its delivery to the cell surface Citation[12]. However, like G551D Citation[13], F508del also perturbs severely CFTR channel gating Citation[13]. Both mutations attenuate markedly cAMP-stimulated Isc Citation[14]. H demonstrates that THF (100 mM), itself, had little or no effect on Isc in FRT epithelia expressing ?F508del- and G551D-CFTR, whereas forskolin (10 µM) elicited a modest increase in Isc in F508del- and G551D-expressing FRT epithelia. Moreover, THF (100 mM) failed to potentiate Isc following treatment of F508del- and G551D-expressing FRT epithelia with forskolin (10 µM; H). By contrast, genistein (50 µM) enhanced robustly cAMP-stimulated Isc in FRT epithelia expressing F508del- and G551D-CFTR (H). These data suggest that THF increases Isc by potentiating the activity of CFTR. However, unlike genistein, THF is ineffective at restoring CFTR function to CF mutants.
Blockers of cAMP-stimulated transepithelial ion transport attenuate the THF-induced Isc
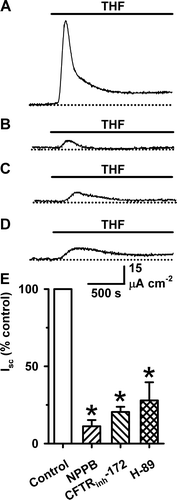
As a final test to determine whether the CFTR Cl− channel mediates the THF-induced Isc, we examined the effects on the THF-induced Isc of agents that inhibit CFTR by different mechanisms. We used the arylaminobenzoate NPPB (50 µM), the thiazolidinone CFTRinh-172 (10 µM) and H-89 (10 µM), a membrane-permeant selective inhibitor of PKA. NPPB is an open-channel blocker of the CFTR Cl− channel Citation[15], CFTRinh-172 likely acts by an allosteric mechanism Citation[16], while H-89 impedes the phosphorylation of CFTR by PKA Citation[17]. Because the THF-induced Isc is transient, we added inhibitors to the apical solution bathing FRT epithelia expressing wild-type CFTR prior to the application of THF (100 mM). demonstrates that each of the agents tested markedly attenuated the Isc elicited by THF (100 mM), diminishing the magnitude of both peak and steady-state Isc. Taken together, our data argue that the THF-induced Isc is mediated by the CFTR Cl− channel.
Figure 3. Inhibition of THF-stimulated Isc by blockers of cAMP-stimulated transepithelial Cl− transport. (A–D) Representative Isc recordings show the effects of blockers of cAMP-stimulated transepithelial Cl− transport on THF-stimulated Isc in FRT epithelia expressing wild-type CFTR. During the periods indicated by the bars THF (100 mM) was present in the solution bathing the apical membrane. (A) Control; (B) NPPB (50 µM) added immediately prior to THF; (C) CFTRinh-172 (10 µM) added 1 h prior to THF (D) H-89 (10 µM) added 2 h prior to THF; (E) The effects of inhibitors of transepithelial ion transport on THF-stimulated Isc. Peak Isc values measured in the presence of inhibitors are expressed as a percentage of the peak Isc recorded in the presence of THF (100 mM) alone. Data are means + SEM (NPPB, n=11; CFTRinh-172, n=5; H-89, n=8). The asterisks indicate values that are significantly different from the control value (p<0.05). Other details as in .
THF causes a concentration-dependent increase in Isc
To determine the relationship between THF concentration and Isc, we added increasing concentrations of THF to the apical solution bathing FRT epithelia expressing wild-type CFTR in the absence of forskolin and measured peak Isc. THF (50–400 mM) caused a concentration-dependent increase in Isc, whereas THF (800 mM), the highest concentration tested, decreased Isc and CFTRinh-172 (10 µM) subsequently abolished Isc (A). B demonstrates that the relationship between THF concentration and Isc is fit by a Sigmoidal Hill equation (3 parameter) (R2=0.97) with Isc max of 141 µA cm−2, KD of 134 mM and Hill coefficient of 2.4. Two conclusions can be drawn from these data: (i) THF weakly potentiates CFTR-mediated Isc and (ii) THF might interact at multiple sites to stimulate Isc.
Figure 4. THF stimulation of transepithelial ion transport is concentration-dependent. (A) Representative Isc recording from an FRT epithelium expressing wild-type CFTR shows the effects of THF (50–800 mM) on peak Isc. During the periods indicated by the solid bars the indicated concentrations of THF and CFTRinh-172 (10 µM) were present in the solution bathing the apical membrane. (B) Effect of THF concentration on Isc in FRT epithelia expressing wild-type human CFTR. Data are means±SEM (n=8, except THF (800 mM), where n=6). The continuous line is the fit of the Hill equation to the data. Other details as in .

THF alters the gating behaviour of the CFTR Cl− channel
To understand how THF enhances CFTR-mediated transepithelial ion transport, we investigated the effects of THF on the single-channel activity of CFTR using the excised inside-out configuration of the patch-clamp technique. Excised membrane patches were clamped at −50 mV and a large Cl− concentration gradient imposed across the membrane to magnify the size of CFTR Cl− currents. To determine how the chemical solvent acts on CFTR, we added THF (100 mM) to the intracellular solution of freshly excised membrane patches with unknown numbers of channels present. In the absence of both ATP (1 mM) and PKA (75 nM), THF did not activate CFTR Cl− channels in those patches that subsequently displayed channel activity in the presence of ATP and PKA (n=6; data not shown).
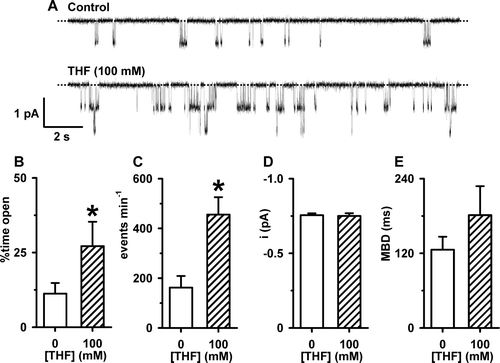
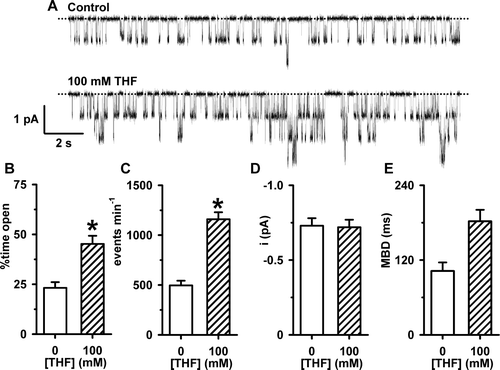
Because THF, by itself, increased Isc in FRT epithelia expressing wild-type CFTR, we investigated whether the solvent might enhance the activity of CFTR prior to its phosphorylation with PKA. A shows representative channel recordings of wild-type CFTR Cl− channels in the absence and presence of THF (100 mM) in the solution bathing the intracellular face of the excised membrane patch; ATP (1 mM) was continuously present in the solution. Upon addition of the chemical solvent, there was a noticeable change in CFTR channel gating. THF (100 mM) increased the frequency of channel openings without appearing to alter their duration (A).
Figure 5. The effects of THF on the single-channel activity of wild-type human CFTR prior to PKA-dependent phosphorylation. (A) Representative recordings of CFTR Cl− current in an excised inside-out membrane patch from a C127 cell expressing wild-type human CFTR. During the periods denoted by the bars ATP (1 mM), THF (100 mM) and PKA (75 nM) were present in the intracellular solution. Voltage was −50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal=147 mM; [Cl−]external=10 mM). For the purpose of illustration, the time-course has been inverted so that upward deflections represent inward currents. The inserts show the sections of the record labelled 1 and 2 on an expanded time scale. The dotted lines indicate where channels are closed and downward deflections of the traces correspond to channel openings. (B, C, D and E) The effects of THF (100 mM) on percent time open, events per minute, single-channel current amplitude (i) and mean burst duration (MBD). Columns and error bars are means + SEM (percent time open, events per minute and MBD n=5; i, n=12). The asterisks indicate values that are significantly different from control values (p<0.05).
![Figure 5. The effects of THF on the single-channel activity of wild-type human CFTR prior to PKA-dependent phosphorylation. (A) Representative recordings of CFTR Cl− current in an excised inside-out membrane patch from a C127 cell expressing wild-type human CFTR. During the periods denoted by the bars ATP (1 mM), THF (100 mM) and PKA (75 nM) were present in the intracellular solution. Voltage was −50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal=147 mM; [Cl−]external=10 mM). For the purpose of illustration, the time-course has been inverted so that upward deflections represent inward currents. The inserts show the sections of the record labelled 1 and 2 on an expanded time scale. The dotted lines indicate where channels are closed and downward deflections of the traces correspond to channel openings. (B, C, D and E) The effects of THF (100 mM) on percent time open, events per minute, single-channel current amplitude (i) and mean burst duration (MBD). Columns and error bars are means + SEM (percent time open, events per minute and MBD n=5; i, n=12). The asterisks indicate values that are significantly different from control values (p<0.05).](/cms/asset/9ba94eb0-94a0-47f9-bc78-0b7c18825109/imbc_a_348964_f0005_b.gif)
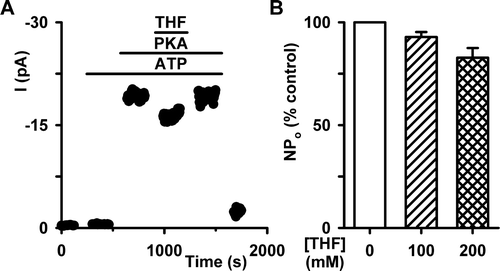
To begin to analyse the effects of THF (100 mM) on channel gating, we measured NPo because the number of active channels in the membrane patch was unknown. Under control conditions (1 mM ATP) NPo was 0.06±0.04 (n=9), whereas following the addition of THF (100 mM) NPo increased to 0.13±0.08 (n=9), an amplification of 2.2-fold (p<0.05). However, following phosphorylation with PKA (75 nM), NPo increased to 12.11±3.83 (n=9), an amplification of 94-fold (p<0.01). Next, we measured the frequency of channel gating in membrane patches where only a single CFTR Cl− channel was active. (We did not calculate interburst interval (IBI) because the number of active channels in the membrane patch was unknown in the absence of PKA). THF (100 mM) increased markedly the percent time open and the frequency of channel gating (B, 5C). However, THF was without effect on mean burst duration (MBD) (E) and depressed slightly single-channel current amplitude (i; D). Taken together, these results suggest that THF increases the activity of CFTR Cl− channels that are not fully phosphorylated by PKA by increasing the frequency of channel opening.
To determine whether THF potentiates the activity of CFTR after its phosphorylation by PKA, we added THF to the intracellular solution following channel activation by PKA (75 nM) and ATP (1 mM). In both membrane patches containing large (≥6) or small numbers of active channels (<6), THF (100 mM) was without effect on CFTR Cl− current, whereas THF (200 mM) depressed albeit not significantly current magnitude (). We interpret these data to suggest that the effects of THF on CFTR channel gating are dependent on the phosphorylation status of CFTR.
Figure 6. THF is without effect on CFTR following PKA-dependent phosphorylation in an excised membrane patch. (A) Time-course of CFTR Cl− current in an excised inside-out membrane patch from a C127 cell expressing wild-type CFTR. During the periods indicated by the bars ATP (1 mM), PKA (75 nM) and THF (100 mM) were present in the intracellular solution. (B) NPo of wild-type CFTR following phosphorylation with PKA in the absence and presence of THF (100–200 mM). NPo values measured in the presence of THF are expressed as a percentage of control values recorded in the absence of the drug. Columns and error bars are means + SEM (n=6). Other details as in .
Addition of THF (100 mM) to the intracellular solution increases osmolarity from 280±0.5 mOsm (n=5) to 396±7.4 mOsm (n=5). To exclude the possibility that a change in solution osmolarity accounts for the observed effects of THF, we mimicked the THF-induced change in osmolarity of the intracellular solution using D-mannitol (180 mM). Elevating the osmolarity of the intracellular solution did not enhance CFTR activity. Instead, high solution osmolarity caused small, but not statistically significant decreases in i and NPo (i: control, −0.79±0.04 pA (n=3); D-mannitol (180 mM), −0.73±0.03 (n=3; p>0.05); NPo: control, 0.47±0.3 (n=3); D-mannitol (180 mM), 0.27±0.15 (n=3; p>0.05). These results argue that the effects of THF on the CFTR Cl− channel are not caused by a change in osmolarity of the intracellular solution.
THF potentiates the ▵R-S660A-CFTR Cl− channel and wild-type CFTR gated by 2′-deoxy-ATP
The lack of effect of THF on the CFTR Cl− channel after phosphorylation by PKA, suggests that the RD might be a target for the actions of THF. To test this hypothesis, we used the CFTR construct ▵R-S660A Citation[8], performing the same experiments on this CFTR variant as those with wild-type CFTR. The CFTR mutant ▵R-S660A lacks much of the RD (residues 708-835) and disables the dibasic phosphorylation site at serine 660. This CFTR construct no longer requires PKA-dependent phosphorylation to open in the presence of intracellular ATP Citation[9]. Supplementary Information (SI) (online version only) demonstrates that THF (100 mM) augments the activity of ▵R-S660A-CFTR Cl− channels by increasing the frequency of channel openings. These data suggest that THF might potentiate CFTR channel gating by interacting with a site distinct from the RD.
SI Figure 1. THF increases the opening rate of the ▵R-S660A-CFTR Cl− channel. (A) Representative recordings show the single-channel activity of ▵R-S660A-CFTR in an excised inside-out membrane patch from a C127 cell. The upper and lower traces were recorded in the absence and presence of THF (100 mM), respectively, in the intracellular solution; ATP (1 mM) was continuously present in the intracellular solution. Voltage was −50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal=147 mM; [Cl−]external=10 mM). The dotted lines indicate where channels are closed and downward deflections of the traces correspond to channel openings. (B, C, D and E) Percent time open, events per min, i and MBD of ▵R-S660A-CFTR in the absence and presence of THF (100 mM). For percent time open, events per minute and MBD columns and error bars are means + SD (n=2), for i columns and error bars are means + SEM (n=5). The asterisks indicate values that are significantly different from control values (p<0.05).
![SI Figure 1. THF increases the opening rate of the ▵R-S660A-CFTR Cl− channel. (A) Representative recordings show the single-channel activity of ▵R-S660A-CFTR in an excised inside-out membrane patch from a C127 cell. The upper and lower traces were recorded in the absence and presence of THF (100 mM), respectively, in the intracellular solution; ATP (1 mM) was continuously present in the intracellular solution. Voltage was −50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal=147 mM; [Cl−]external=10 mM). The dotted lines indicate where channels are closed and downward deflections of the traces correspond to channel openings. (B, C, D and E) Percent time open, events per min, i and MBD of ▵R-S660A-CFTR in the absence and presence of THF (100 mM). For percent time open, events per minute and MBD columns and error bars are means + SD (n=2), for i columns and error bars are means + SEM (n=5). The asterisks indicate values that are significantly different from control values (p<0.05).](/cms/asset/75975683-3b8d-4b72-be47-d87272470cba/imbc_a_348964_f0007_b.gif)
Previous work has demonstrated that many agents interact directly with the NBDs to enhance CFTR channel gating (for review, see Citation[6]). We therefore speculated that THF might exert its effects on CFTR channel gating by modulating ATP-dependent channel gating Citation[18]. To test this idea, we employed the hydrolysable ATP analogue 2′-deoxy ATP (2′-dATP), which interacts with the two ATP-binding sites at the NBD dimer interface to gate CFTR more effectively than ATP Citation[10]. We reasoned that THF might be ineffective at potentiating wild-type CFTR Cl− channels gated by 2′-dATP. However, SI (online version only) demonstrates that prior to channel activation by PKA, THF (100 mM) augmented strongly the single-channel activity of wild-type CFTR gated by 2′-dATP (1 mM). We interpret these results to suggest that THF potentiates CFTR channel gating by interacting with a site distinct from CFTR's two ATP-binding sites.
SI Figure 2. THF enhances the single-channel activity of wild-type CFTR gated by 2′-dATP prior to PKA-dependent phosphorylation. (A) Representative recordings show the single-channel activity of CFTR in an excised inside-out membrane patch from a C127 cell. The upper and lower traces were recorded in the absence and presence of THF (100 mM), respectively, in the intracellular solution; 2′d-ATP (1 mM) was continuously present in the intracellular solution. One CFTR Cl− channel was active under control conditions and two in the presence of THF (100 mM). (B, C, D and E) Percent time open, events per minute, i and MBD of wild-type CFTR in the absence and presence of THF (100 mM). Columns and error bars are means + SEM (n=7). The asterisks indicate values that are significantly different from control values (p<0.05). Other details as in SI .
THF potentiates wild-type CFTR in the presence of the reducing agent vitamin C
We considered the possibility that the chemical properties of THF might provide insight into its mechanism of action. Because THF is an aprotic, electron donating solvent Citation[1], it might act as a reducing agent. This characteristic is significant in two respects. First, the intracellular redox status influences CFTR channel gating Citation[19]. Second, the reducing agent vitamin C, which shares some similarities in chemical structure to THF, has analogous effects on CFTR channel gating as those of the solvent Citation[20]. These comparisons suggest that THF might potentiate CFTR channel gating by altering the redox status of CFTR. To explore this possibility, we investigate whether prior to channel activation by PKA, pre-treatment of CFTR Cl− channels with vitamin C (100 µM) might prevent the potentiation of CFTR channel gating by THF. However, SI (0nline version only) demonstrates that THF (100 mM) enhanced robustly the single-channel activity of wild-type CFTR potentiated by vitamin C (100 µM). These results suggest that potentiation of CFTR channel gating by THF likely does not involve modulation of the redox status of CFTR.
SI Figure 3. THF enhances CFTR channel gating in the presence of the reducing agent vitamin C. (A) Representative recordings show the single-channel activity of CFTR in an excised inside-out membrane patch from a C127 cell. The upper and lower traces were recorded in the absence and presence of THF (100 mM), respectively, in the intracellular solution; ATP (1 mM) and vitamin C (100 ?M) were continuously present in the intracellular solution. Two CFTR Cl− channels were active under control conditions and three in the presence of THF (100 mM). (B, C, D and E) Percent time open, events per minute, i and MBD of wild-type CFTR in the absence and presence of THF (100 mM). Columns and error bars are means + SEM (n=4). The asterisks indicate values that are significantly different from control values (p<0.05). Other details as in SI . In two experiments, we tested the effects of vitamin C (100 µM) on CFTR channel gating in the presence of ATP (1 mM), but absence of PKA (75 nM). Data in the absence and presence of vitamin C (100 µM) are as follows: (i) percent time open: control, 6.3±1.4%; vitamin C, 26.4±2.3%; (ii) events per min: control, 213.6±46.6 min−1; vitamin C, 777.1±70.0 min−1; (iii) i: control, −0.77±0.05 pA; vitamin C, −0.76±0.05 pA and (iv) MBD: control, 65.1±43 ms; vitamin C, 85.2±24.0 ms (all data are means±SD; n=2).
Discussion
In this study, we investigated the effects of the chemical solvent THF on CFTR-mediated transepithelial ion transport. Using Isc measurements and single-channel recording, we demonstrated that THF interacts directly with CFTR to potentiate channel gating, but that its efficacy is weak and dependent on the phosphorylation status of CFTR.
Our data demonstrate that the potentiation of CFTR Cl− currents by THF is enhanced by forskolin in FRT epithelia expressing wild-type human CFTR, but not by PKA in membrane patches excised from C127 cells expressing the same CFTR construct. One possible explanation for these conflicting results is the different cell types, which we used to study CFTR-mediated transepithelial ion transport and single-channel behaviour. However, a more likely explanation is the phosphorylation status of CFTR and hence, the level of channel activity. In excised membrane patches from C127 cells expressing wild-type CFTR bathed in a Cl−-rich intracellular solution containing ATP (1 mM), PKA (75 nM) robustly stimulates the CFTR Cl− channel (Po=0.52±0.04; n=18; Citation[10]). However, in cell-attached recordings from the same cells, forskolin (20 µM) is less effective at stimulating CFTR (Po=0.24±0.02; n=16; Z Xu & DN Sheppard, unpublished observation). Based on these data, we suggest that THF robustly enhances the activity of weakly phosphorylated CFTR Cl− channels, but has little or no effect on the activity of strongly phosphorylated CFTR Cl− channels. Support for this idea is provided by previous work on the best-studied CFTR potentiator genistein. The effect of genistein on channel activity depends on the phosphorylation status of CFTR: Genistein strongly enhances channel gating when CFTR is weakly phosphorylated by PKA, but has little or no effect on channel gating when CFTR is highly phosphorylated by PKA Citation[12], Citation[22].
A further difference between the effects of THF in FRT epithelia and excised membrane patches is the transient nature of the THF response in FRT epithelia compared with the solvent's sustained effects in excised membrane patches (present results and data not shown). In the presence of a Cl− concentration gradient, there is unlikely to be a change in the driving force for Cl− exit across the apical membrane as a result of changes in the activity of basolateral membrane K+ channels. Instead, the transient nature of the THF response in FRT epithelia likely reflects the deactivation of CFTR Cl− channels in intact cells by protein kinases (e.g., AMP kinase; Citation[22]) and phosphatases (e.g., PP2A and PP2C; Citation[23]).
Many CFTR potentiators (e.g., genistein; Citation[21]) enhance the gating behaviour of CFTR by (i) accelerating channel opening and (ii) slowing dramatically channel closure. Other CFTR potentiators (e.g., phloxine B; Citation[24]) augment CFTR channel gating solely by retarding channel closure. By contrast, THF enhanced CFTR channel gating by increasing the rate of channel opening without altering the duration of bursts. This effect of THF is reminiscent of agents that modulate CFTR activity by altering the phosphorylation status of the RD. For example, phosphorylation of the RD by PKA and PKC enhances CFTR Cl− currents by increasing the frequency of channel openings without altering their duration Citation[25], Citation[26]. These considerations suggest that the simplest interpretation of our data is that PKA-dependent phosphorylation and THF binding might elicit similar changes in the conformation of the CFTR Cl− channel, albeit with markedly different efficacy. Thus, the effects of THF (the weaker agonist) are masked in fully phosphorylated channels by the overriding effects of phosphorylation on CFTR conformation.
Where might THF interact with CFTR to exert its effects? Our observation that THF potentiates the ▵R-S660A-CFTR Cl− channel argues that the solvent interacts with a site distinct from the RD. Because the NBDs are the location of binding sites for a number of CFTR potentiators Citation[6], THF might exert its effects by binding to the NBDs. Using the ATP-driven NBD dimerisation model of CFTR channel gating Citation[18], we suggest that the interaction of THF with the NBDs accelerates channel opening by providing binding energy to drive NBD dimerisation. Because THF did not substitute for ATP in supporting channel gating, we consider it unlikely that THF interacts with site 1, where ATP binds tightly. Similarly, because THF was without effect on the duration of channel openings, it is unlikely to compete with ATP for binding to site 2, where ATP is hydrolysed rapidly. Consistent with this idea, THF potentiated CFTR Cl− channels gated by 2′-dATP, which exerts its effects by acting at sites 1 and 2 Citation[10]. Instead, the THF-binding site might be located at the NBD1:NBD2 interface based on the interaction of CFTR potentiators with a molecular model of the NBD dimer Citation[27].
However, two further THF binding sites are worthy of consideration. First, structural models of CFTR based on the ABC transporter Sav1866 Citation[28–30] suggest that the intracellular loops (ICLs) play a critical role in communication between the NBDs and MSDs. This raises the intriguing possibility that CFTR potentiators, such as THF, might exert their effects by altering the conformation of the ICLs. Second, THF might potentiate CFTR channel gating by influencing the conformation of the MSDs, which form the CFTR pore. THF might interact directly with the MSDs to alter the packing of the transmembrane segments. Alternatively, the solvent might influence the conformation of the MSDs indirectly by altering the mechanical properties of the lipid bilayer. In support of this latter mechanism, Hwang et al. Citation[31] demonstrated that the best-studied CFTR potentiator genistein modulates the activity of gramicidin A channels in planar lipid bilayers by altering bilayer mechanics, while the data of Artigas et al. Citation[32] suggest that 2,3-butanedione monoxime might affect CFTR function by a similar mechanism.
In conclusion, we demonstrated that THF potentiates CFTR-mediated transepithelial ion transport by accelerating channel opening. THF has the simplest chemical structure of any CFTR potentiator identified to date. Moreover, the chemical structure of a number of CFTR potentiators identified by high-throughput screening incorporate the structure of THF (e.g., CFTRact-07; Citation[14]). The effects of THF on CFTR channel gating suggest that it might interact directly with the NBDs to promote their dimerization. The data also highlight the importance of the phosphorylation status of CFTR for the action of CFTR potentiators.
Supplementary information
We undertook a series of experiments to understand better how the chemical solvent THF potentiates CFTR channel gating. Below, we present these data and discuss the results.
THF potentiates the ▵R-S660A-CFTR Cl− channel and wild-type CFTR gated by 2′-deoxy-ATP
The lack of effect of THF on the CFTR Cl− channel after phosphorylation by PKA, suggests that the R domain (RD) might be a target for the actions of THF. To test this hypothesis, we used the CFTR construct ▵R-S660A Citation[33], performing the same experiments on this CFTR variant as those with wild-type CFTR. The CFTR mutant ▵R-S660A lacks much of the R domain (residues 708–835) and disables the dibasic phosphorylation site at serine 660. This CFTR construct no longer requires PKA-dependent phosphorylation to open in the presence of intracellular ATP Citation[33].
Supplementary Information (SI) A shows representative recordings of a single ▵R-S660A-CFTR Cl− channel in the absence and presence of THF (100 mM). Visual inspection of these single-channel records suggests that THF (100 mM) increases markedly the frequency of channel openings. In membrane patches with only a single active ▵R-S660A-CFTR Cl− channel, THF enhanced strongly the percent time open and the frequency of channel gating, was without effect on MBD and depressed slightly i (SI ). We interpret these results to suggest that THF potentiates CFTR channel gating by interacting with a site distinct from the RD.
Previous work has demonstrated that many agents interact directly with the NBDs to enhance CFTR channel gating (for review, see Citation[34]). We therefore speculated that THF might exert its effects on CFTR channel gating by modulating ATP-driven NBD dimerisation and hence conformation changes in the MSDs, which open and close the channel pore Citation[35]. To test this idea, we employed the hydrolysable ATP analogue 2′-deoxy ATP (2′-dATP), which interacts with the two ATP-binding sites at the NBD dimer interface to gate CFTR more effectively than ATP Citation[36]. We reasoned that THF might be ineffective at potentiating wild-type CFTR Cl− channels gated by 2’-dATP. However, SI A demonstrates that THF (100 mM) augmented strongly the gating behaviour of wild-type CFTR prior to channel activation by PKA when ATP (1 mM) was substituted by 2′-dATP (1 mM) in the intracellular solution. Like its effects on wild-type CFTR gated by ATP (), THF (100 mM) increased the percent time open and frequency of channel gating, but was without effect on MBD (SI B, 8C and 8E). The only difference between ATP and 2′-dATP was that in the presence of 2′-dATP, THF (100 mM) did not decrease i (SI D). We interpret these results to suggest that THF potentiates CFTR channel gating by interacting with a site distinct from CFTR's two ATP-binding sites.
THF potentiates wild-type CFTR in the presence of the reducing agent vitamin C
We considered the possibility that the chemical properties of THF might provide insight into its mechanism of action. Because THF is an aprotic, electron donating solvent Citation[37], it might act as a reducing agent. Previous work has demonstrated that the intracellular redox status influences CFTR channel gating. For example, Harrington et al. Citation[38] demonstrated that reducing agents accelerate and oxidising agents slow channel gating. Because the non-hydrolysable ATP analogue ATPγS attenuated the effects of the reducing agent β-mercaptoethanol, Harrington et al. Citation[38] speculated that the NBDs are the site of action of redox reagents. Recently, Fischer et al. Citation[39] demonstrated that the reducing agent vitamin C potentiates the CFTR Cl− channel. The authors’ data are significant in two respects: First, the effects of vitamin C on CFTR channel gating are similar to those of THF. Second, the chemical structure of vitamin C contains a furan ring, the parent molecule of THF. These comparisons suggest that THF might potentiate CFTR channel gating by altering the redox status of CFTR.
To explore whether THF potentiates CFTR channel gating by altering the redox status of CFTR, we investigated whether pre-treatment of CFTR Cl− channels with vitamin C might prevent the potentiation of CFTR channel gating by THF. For these studies, we selected a concentration of vitamin C (100 µM), three-fold greater than the vitamin C concentration required for half-maximal potentiation of CFTR-mediated transepithelial ion transport in Calu-3 epithelia Citation[39]. Like THF (100 mM; ), addition of vitamin C (100 µM) to the intracellular solution in the presence of ATP (1 mM), but absence of PKA (75 nM) enhanced CFTR channel gating. Vitamin C (100 µM) increased percent time open and frequency of channel gating, but was without effect on MBD (see legend of SI ). However, unlike THF (100 mM; D) vitamin C did not decrease i (SI legend).
SI A demonstrates that THF (100 mM) potentiated robustly the gating behaviour of wild-type CFTR in the presence of ATP (1 mM) and vitamin C (100 µM), but absence of PKA (75 nM) in the intracellular solution. THF (100 mM) increased the percent time open and frequency of channel gating, but was without effect on i and MBD (SI B–9E). We interpret these results to suggest that THF interacts with CFTR at a site distinct from that of vitamin C to potentiate CFTR channel gating. The data also suggest that potentiation of CFTR channel gating by THF likely does not involve modulation of the redox status of CFTR.
Acknowledgements
We thank Professor A.P. Davis, Dr. G. Magro and our departmental colleagues for valuable discussions and assistance, especially Dr. H. Li. We also thank Drs. L.J.V. Galietta (Istituto Giannina Gaslini), C.R. O'Riordan (Genzyme Corp.) and Professor M.J. Welsh (University of Iowa) for generous gifts of cells expressing wild-type and mutant CFTRs. This work was supported by the Biotechnology and Biological Sciences Research Council [grant no. BBS/B/11044] and the Cystic Fibrosis Trust. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Loudon GM. Organic chemistry4th ed. Oxford University Press, New York 2002; 318
- Koulov AV, Lambert TN, Shukla R, Jain M, Boon JM, Smith BD, Li H, Sheppard DN, Joos J-B, Clare JP, Davis AP. Chloride transport across vesicle and cell membranes by steriod-based receptors. Angew Chem Int Ed Engl 2003; 42: 4931–4933
- Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl− secretion, and renal cyst growth: A study using CFTR inhibitors. Kidney Int 2004; 66: 1926–1938
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. The metabolic and molecular basis of inherited disease, CR Scriver, AL Beaudet, WS Sly, D Valle. McGraw-Hill Inc, New York 2001; 5121–5188
- Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006; 440: 477–483
- Cai Z, Chen J-H, Hughes LK, Li H, Sheppard DN. The physiology and pharmacology of the CFTR Cl( channel. Chloride movements across cellular membranes, M Pusch. Elsevier Ltd, San Diego 2007; 109–143
- Fischer H, Schwarzer C, Illek B. Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci USA. 2004; 101: 3691–3696
- Zegarra-Moran O, Romio L, Folli C, Caci E, Becq F, Vierfond J-M, Mettey Y, Cabrini G, Fanen P, Galietta LJV. Correction of G551D-CFTR transport defect in epithelial monolayers by genistein but not by CPX or MPB-07. Br J Pharmacol 2002; 137: 504–512
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by negative charge in the R domain. J Biol Chem 1993; 268: 20259–20267
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J Physiol 1997; 503: 333–346
- Cai Z, Taddei A, Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem 2006; 281: 1970–1977
- Lansdell KA, Cai Z, Kidd JF, Sheppard DN. Two mechanisms of genistein inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in murine cell line. J Physiol 2000; 524: 317–330
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63: 827–834
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq J-P, Lazdunski M. Altered chloride ion channel kinetics associated with the ▵F508 cystic fibrosis mutation. Nature 1991; 354: 526–528
- Ma T, Vetrivel L, Yang H, Pedemonte N, Zegarra-Moran O, Galietta LJV, Verkman AS. High-affinity activators of cystic fibrosis transmembrane conductance regulator (CFTR) chloride conductance identified by high-throughput screening. J Biol Chem 2002; 277: 37235–37241
- Zhang Z-R, Zeltwanger S, McCarty NA. Direct comparison of NPPB and DPC as probes of CFTR expressed in Xenopus oocytes. J Membr Biol 2000; 175: 35–52
- Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJV. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett 2004; 558: 52–56
- Yurko-Mauro KA, Reenstra WW. Prostaglandin F2α stimulates CFTR activity by PKA- and PKC-dependent phosphorylation. Am J Physiol 1998; 275: C653–660
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005; 433: 876–880
- Harrington MA, Gunderson KL, Kopito RR. Redox reagents and divalent cations alter the kinetics of cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem 1999; 274: 27536–27544
- Fischer H, Schwarzer C, Illek B. Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci USA 2004; 101: 3691–3696
- Wang F, Zeltwanger S, Yang ICH, Nairn AC, Hwang T-C. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating: Evidence for two binding sites with opposite effects. J Gen Physiol 1998; 111: 477–490
- Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest 2000; 105: 1711–1721
- Gadsby DC, Nairn AC. Control of cystic fibrosis transmembrane conductance regulator channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 1999; 79: S77–107
- Cai Z, Sheppard DN. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J Biol Chem 2002; 277: 19546–19553
- Winter MC, Welsh MJ. Stimulation of CFTR activity by its phosphorylated R domain. Nature 1997; 389: 294–296
- Jia Y, Mathews CJ, Hanrahan JW. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J Biol Chem 1997; 272: 4978–4984
- Moran O, Galietta LJV, Zegarra-Moran O. Binding site of activators of the cystic fibrosis transmembrane conductance regulator in the nucleotide binding domains. Cell Mol Life Sci 2005; 62: 446–460
- Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature 2006; 443: 180–185
- Serohijos AWR, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA 2008; 105: 3256–3261
- Mornon J-P, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: Membrane-spanning domains and coupling interfaces. Cell Mol Life Sci 2008; 65: 2594–2612
- Hwang T-C, Koeppe RE II, Andersen OS. Genistein can modulate channel function by a phosphorylation-independent mechanism: Importance of hydrophobic mismatch and bilayer mechanics. Biochemistry 2003; 42: 13646–13658
- Artigas P, Al'aref SJ, Hobart EA, Díaz LF, Sakaguchi M, Straw S, Andersen OS. 2,3-butanedione monoxime affects cystic fibrosis transmembrane conductance regulator channel function through phosphorylation-dependent and phosphorylation-independent mechanisms: The role of bilayer material properties. Mol Pharmacol 2006; 70: 2015–2026
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl( channel by negative charge in the R domain. J Biol Chem 1993; 268: 20259–20267
- Cai Z, Chen J-H, Hughes LK, Li H, Sheppard DN. The physiology and pharmacology of the CFTR Cl( channel. Chloride movements across cellular membranes, M Pusch. Elsevier Ltd, San Diego 2007; 109–143
- Vergani P, Lockless SW, Nairn AC, Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 2005; 433: 876–880
- Cai Z, Taddei A, Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl( channel. J Biol Chem 2006; 281: 1970–1977
- Loudon GM. Organic chemistry4th ed. Oxford University Press, New York 2002; 318
- Harrington MA, Gunderson KL, Kopito RR. Redox reagents and divalent cations alter the kinetics of cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem 1999; 274: 27536–27544
- Fischer H, Schwarzer C, Illek B. Vitamin C controls the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci USA 2004; 101: 3691–696