Abstract
The NaChBac sodium channel from Bacillus halodurans is a homologue of eukaryotic voltage-gated sodium channels. It can be solubilized in a range of detergents and consists of four identical subunits assembled as a tetramer. Sodium channels are relatively flexible molecules, adopting different conformations in their closed, open and inactivated states. This study aimed to design and construct a mutant version of the NaChBac protein that would insert into membranes and retain its folded conformation, but which would have enhanced stability when subjected to thermal stress. Modelling studies suggested a G219S mutant would have decreased conformational flexibility due to the removal of the glycine hinge around the proposed gating region, thereby imparting increased resistance to unfolding. The mutant expressed in Escherichia coli and purified in the detergent dodecyl maltoside was compared to wildtype NaChBac prepared in a similar manner. The mutant was incorporated into the membrane fraction and had a nearly identical secondary structure to the wildtype protein. When the thermal unfolding of the G219S mutant was examined by circular dichroism spectroscopy, it was shown to not only have a Tm ∼10°C higher than the wildtype, but also in its unfolded state it retained more ordered helical structure than did the wildtype protein. Hence the G219S mutant was shown to be, as designed, more thermally stable.
Introduction
Voltage-gated sodium channels (VGSCs) are pore-forming transmembrane proteins that selectively conduct sodium ions across the plasma membrane. VGSC-mediated sodium influx constitutes the upstroke of action potentials, which propagate in the electrically-excitable cells of the nervous, muscular and cardiac systems. In bacterial systems the functions VGSCs contribute to remains to be elucidated but, as their activity modulates intra- and extra-cellular sodium concentrations in response to plasma membrane potential changes, the channels have been implicated in motility, chemotaxis and pH homeostasis Citation[1].
In members of the sodium, potassium and calcium channel super-family, four transmembrane domains assemble to form the central ion-conducting pore. In voltage-gated channels each domain is comprised of six transmembrane helices, S1-S4 and S5-S6, which form the voltage sensor and pore module, respectively. The S5-S6 linker (also called the P-loop) forms the extracellular mouth of the pore and the selectivity filter. The alpha (pore-forming) subunits of eukaryotic voltage-gated sodium and calcium channels consist of a single polypeptide chain with four pseudo-homologous domains. In contrast, potassium channels consist of four smaller polypeptide chains, each forming a single domain, which associate to form a homo-tetramer. A bacterial sodium channel, NaChBac Citation[2], isolated from Bacillus halodurans, shows strong sequence homology to potassium channels. Biophysical characterization of this channel over-expressed in Escherichia coli and isolated in the detergent dodecyl maltoside, reveals that it too is tetrameric Citation[3]. The ion selectivity of NaChBac is comparable to mammalian VGSC homologues but NaChBac activates and inactivates 10–100 times slower than its monomeric eukaryotic counterparts Citation[2].
Voltage-gated ion channels are highly flexible molecules that undergo transitions between resting-closed, activated and inactivated functional states in response to changes in the membrane potential. Major structural rearrangements are associated with these transitions, as evidenced by circular dichroism spectroscopy of NaChBac and eel VGSCs in the absence and presence of ligands which bind to specific states Citation[3], Citation[4]. Comparison of the KcsA Citation[5] and the MthK Citation[6] potassium channels crystal structures reveals the nature of the conformational transitions of their pore modules upon gating and gives insight into equivalent potential rearrangements in sodium channels. The pore-lining helices, corresponding to S6 in NaChBac, bend by 30° around a central glycine residue, splaying the helices into the open state and forming a 12Å cytoplasmic opening in MthK. Glycine can perform the role of a ‘gating-hinge’ as its achiral nature reduces the energy barrier associated with adoption of a wide range of backbone dihedral angles. Thus flexibility is conferred on the pore-lining helix. Glycine is widely conserved at the gating-hinge position in members of the ion channel super-family.
In this study we aimed to produce a mutant form of NaChBac that would show enhanced thermal stability by decreasing molecular flexibility. Previous studies Citation[3] had shown that upon binding the ligand mibefradil, which is a channel blocker Citation[2], the temperature for thermal denaturation (Tm) for NaChBac was increased significantly. This suggested that a mutant that was conformationally less flexible might show similarly enhanced resistance to thermal mobility and unfolding. Hence we decided to target the gating-hinge glycine (G219) as the site of mutation. It had been shown that mutating G219 to a helix-disrupting proline produced a channel that activated more readily Citation[7]. In contrast, we chose to make a substitution might stabilise the helical conformation. In human VGSCs the equivalent residues in Domains I–III are also glycines, but in Domain IV it is a serine. This indicated that NaChBac might be able to tolerate a glycine to serine substitution at this position. Therefore, in the present study we undertook modelling studies to examine the potential effects of a G219S mutation on NaChBac, and then produced that protein and characterized its thermal stability using circular dichroism (CD) spectroscopy.
Materials and methods
Materials
The cDNA of NaChBac from Bacillus halodurans was generously provided by Prof. David Clapham of Harvard Medical School. N-Dodecyl-β-D maltopyranoside (DDM) was obtained from Anatrace and mibefradil dihydrochloride hydrate was from Sigma.
Cloning and expression of wildtype and mutant NaChBac
Full length NaChBac cDNA (274 amino acids) was amplified by PCR using Pfu DNA polymerase (Stratagene) and two oligos: F: 5′-GCCATATGAAAATGGAAGCTAGACAG-3′ with an NdeI site at the 5′ end and R: 5′-CGGATCCTCATTATTTCGATTGTTTAAGC-3′ with two stop codons followed by a BamHI site at the 5′ end. The PCR product recovered from a 1% agarose gel was digested by NdeI/BamHI and inserted into a pET15b vector (Novagen) between NdeI and BamHI sites using T4 DNA ligase. Site-directed mutagenesis of NaChBac cDNA was performed using the Quik-change protocol from Stratagene, changing the GGT codon for G219 to TCT. The methods for ligation, and transformation were as previously described Citation[3]. Mutant and wildtype colonies were confirmed by sequencing to contain a (His)6 tag followed by a thrombin digestion site at the N-terminus of the NaChBac gene. The G219S modification was also confirmed by sequencing. Competent cells, the C41(DE3)pLysS strain of E. coli, were transformed by pET15b/NaChBac as previously described Citation[3].
E. coli C41(DE3)pLysS transformants carrying the pET-15b plasmid with the NaChBac gene were grown in 6 l of Luria broth supplemented with ampicillin (100 µg/ml) and chloramphenicol (25 µg/ml) to late log phase (OD600=1.0) and then induced for 4 h in the presence of 1 mM isopropyl-β-D thiogalactopyranoside (IPTG). Cells were pelleted at 6000 g and stored at −20°C.
Monitoring the rate of growth of E. coli
Single colonies of E. coli C41(DE3)pLysS transformants carrying the pET-15b plasmid with either the wildtype or mutant NaChBac gene were grown in 10 ml of Luria broth supplemented with ampicillin (100 µg/ml) and chloramphenicol (25 µg/ml) for 16 h at 37°C in an orbital shaker. Overnight cultures (250 µl) were used to seed fresh cultures (25 ml), which were grown at 37°C. Cultures were induced with 1 mM IPTG upon reaching an absorbance at 600 nm of ca. 0.2. Following induction, absorbance readings were recorded every 30 min at 600 nm.
Purification of wildtype and mutant NaChBac
For both wildtype and mutant proteins, the same procedure was utilized: The cell pellet was thawed at room temperature and resuspended in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.6) in the presence of DNase I (2 µg/ml), 2.5 mM MgSO4 and 0.2 mM PMSF, and then lysed by three passes through a EmulsiFlex-C5 cell disrupter. The sample was spun at 100,000 g at 4°C for 40 min, and the membrane pellet was solubilized in TBS supplemented with fresh PMSF (0.2 mM) and 1% DDM at 4°C for 4 h. The sample was centrifuged at 25,000 g at 4°C for 25 min. Using a semi-batch method, the supernatant was bound to Ni-NTA beads (Qiagen) at 4°C for 4 h, and then 30 mM imidazole (final concentration) was added to reduce any non-specific binding and the beads were incubated with mixing at 4°C for an additional 15 min.
The beads were packed into a polypropylene column (Qiagen) and washed with three lots of TBS containing 30 mM imidazole supplemented with 0.1% DDM. The protein was then eluted in TBS containing 0.1% DDM and 300 mM imidazole. The eluate was buffer-exchanged into 20 mM Tris and 20 mM NaCl containing 0.1% DDM and concentrated in an Amicon concentrator (100 kDa cut-off). Following concentration the sample was centrifuged at 100,000 g at 4°C for 20 min.
CD spectroscopy
CD spectra were obtained using an Aviv 215 spectropolarimeter equipped with a detector acceptance angle of > 90° (for membrane/scattering samples) that had been calibrated using camphour sulfonic acid for optical rotation and benzene vapour for wavelength. Spectra of 1.2 mg/ml solutions of the proteins in 0.1% DDM were measured in a 0.01 cm pathlength Suprasil quartz cuvette, and collected at 1 nm intervals over the wavelength range from 280–185 nm. Three scans were collected for each protein sample and its corresponding baseline (which contained buffer plus 0.1% DDM) in the same cuvette. Using the CDtools processing package Citation[8], the averaged baseline spectra were subtracted from the corresponding averaged protein spectra, and the spectra smoothed with a Savitsky-Golay filter. For measurements at the high temperature, the samples were equilibrated for 3 min before the first of three spectra were obtained. As all three repeat scans (taken over a period of more than an hour) were identical, it was assumed that the sample had reached thermal equilibrium prior to the first measurement at the high temperature.
For thermal scans, measurements were made on samples in 0.1% DDM (1.2 mg/ml protein in a 0.01 cm pathlength cell) by monitoring the CD signal at 222 nm using temperature steps of 10°C over the set temperature range from 20–110°C; an equilibration time of 3 min was used at each temperature point. The actual temperatures (as opposed to the set temperature) were determined by a pre-calibration using a thermal probe that was embedded in the sample cell. Thermal scans also were made in the presence of 200 µM mibefradil by adding 5 µl of a 2 mM stock solution to 50 µl of protein.
Data were normalized to a common value at 222 nm at the initial temperature point to allow comparison between samples. Sigmoidal fits and calculations of Tm were undertaken in Origin 6.0.
Molecular modelling
Sequence alignments were carried out using the ClustalW algorithm Citation[9] (a). Homology models of the closed- and open-state NaChBac pore were generated using KcsA [PDB code 1BL8] Citation[5] and MthK [PDB code 1 LNQ] Citation[6] potassium channel crystal structures, respectively. Residues were mutated to the NaChBac wildtype or G219S mutant sequence using the Biopolymer module of SYBYL (Version 7.3, Tripos Inc., St Louis, MO, USA). Previously our modeling of NaChBac Citation[3] had included the voltage-sensor region, but as the gating-hinge residue is located in the S6 helix, modeling in this study has focused on different conformational states of the pore module (S5-S6, including the selectivity filter P-loop). A model of the NaV1.4 mammalian VGSC selectivity filter Citation[10] provided the template for modeling the NaChBac selectivity filter. Domain I coordinates were used as this domain displays the greatest sequence homology with the corresponding section of NaChBac (b). The pore-pointing helices of the modelled selectivity filter were superimposed onto the pore-pointing helices of the KcsA or MthK structures using the SUPERIMPOSE program Citation[11].
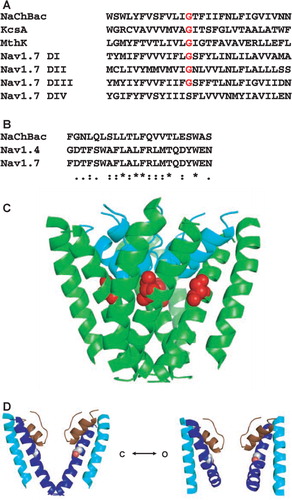
Figure 1. Modelling of wildtype and mutant NaChBac. (a) Sequence alignment of the S6 helices of NaChBac, KcsA, and domains I–IV of human NaV1.7, showing the position of the equivalent glycines (or in the case of domain IV, the equivalent serine). (b) Sequence alignment of NaChBac and the NaV1.4 and NaV1.7 selectivity filters (P-loops) showing the conservation of key residues, including an aspartate that aligns with the critical glutamate of the specificity-defining ‘LESWAS’ sequence motif in NaChBac. Substitutions are (*) identical, (:) conserved and (.) semi-conserved. (c) Homology model for the closed-state mutant NaChBac showing the location of G219 (the selectivity filter backbone is in cyan, the glycine residue is in red space-fill). (d) Comparison of closed (left) and open (right) homology models of wildtype NaChBac, indicating the potential flexibility and role in gating of the ‘hinge’ glycine at position G219S (shown in space-fill). Only two of the four subunits are shown in each case, for clarity.
Models were subjected to 250 rounds of steepest-descent minimization followed by 750 rounds of conjugate-gradient minimization in SYBYL using the Tripos force-field Citation[12]. The AREAIMOL program Citation[11] was used to calculate differences in buried surface area within a single domain in the open and closed channel models. Figures were produced using the PyMOL molecular graphics system (DeLano Scientific, San Carlos, CA, USA).
Results
Modelling
Modelling studies were employed to inform the decision of which residue to substitute experimentally at the gating-hinge position. Homology models of closed- and open-state NaChBac wildtype and mutant pore modules were generated based on the KcsA and MthK potassium channel structures. The selectivity filter region was also included in the models because the side chain of the gating-hinge residue is situated in close proximity to the pore-pointing helix, which could influence the nature of the mutated structure (c).
Surface area calculations showed that the buried surface area (as a measure of the extent of inter-domain interactions) within the pore module is reduced from 1,743 Å3 to 1,288 Å3 upon transition from the closed (d left) to the open state (d right). Thus a mutation that favoured the closed state might be expected to be more thermally stable. Therefore it was decided to create a mutant that was likely to lock the protein in the closed state, and G219, which is proposed to be the gating-hinge, was the obvious target. Limiting the conformational flexibility of this residue by replacing it with a larger, chiral residue could inhibit gating of the channel. In homologous members of the bacterial VGSC family, the gating-hinge glycine position is occupied by a threonine Citation[13], but as their kinetics and voltage dependence differ significantly from those of NaChBac, so it was decided that the threonine mutant would not be ideal. In addition, modelling studies suggested that in NaChBac there is potential for steric hindrance with the pore-pointing helix with a threonine substitution, so this was another reason it was discarded as a potential mutant. In contrast, a serine residue can be suitably accommodated in the pore. Although glycines are found in the homologous gating-hinge position in Domains I–III of mammalian sodium channels, serine is found in Domain IV (a), the movement of which correlates with the slow component of activation Citation[14]. Hence, it was decided that a G219S mutant may produce the inflexibility required, whilst not disrupting the integrity of the protein fold.
Production and characterization of wildtype and mutant proteins
The aim of this study was to produce a mutant protein having a subtle structural change that would lead to increased thermal stability but not significantly alter the protein structure, and that would express in the membrane to a comparable extent as wildtype protein.
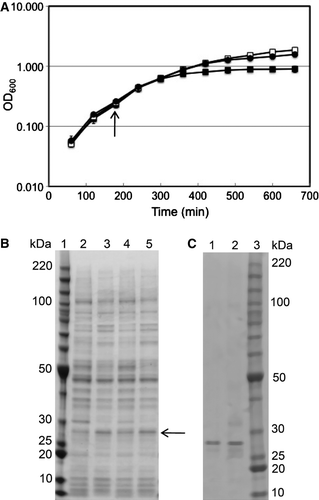
Although constructs that placed the (His)6 tag at the C-terminal end of the protein resulted in significantly different amounts of protein produced and different growth characteristics for wildtype and mutant-containing cells (G. Nurani, unpublished results), for the N-terminal (His)6 tag constructs discussed in this paper, both the growth rates (a) and amount of wildtype and mutant protein produced (b) were roughly comparable. The induced mutant growth and uninduced growths achieved somewhat higher OD600 levels than did the induced wildtype growth, suggesting that expression of the wildtype protein might be inhibitory. This could be because E. coli do not have an y native sodium channels, hence the presence of a readily-gating (wildtype form) of the NaChBac protein may be inhibitory to the growth of these cells. Both proteins were localized in the membrane fraction, extracted efficiently with 1% DDM, and isolated to comparable degrees of purity after chromatography on Ni-NTA beads (c).
Figure 2. Growth and purification of wildtype and G219S mutant NaChBac. (a) Growth curves for uninduced and induced wildtype (open squares and closed squares, respectively) and mutant (closed circles) proteins; the arrow indicates the point of induction with IPTG. Data for G219S without IPTG were almost identical to those for wildtype without IPTG and are omitted for clarity. (b) SDS gel comparing the expression of wildtype and the G219S mutant. Lane 1 contains BenchMark molecular mass standards (Invitrogen). Lanes 2 and 3 contain the whole cell protein for wildtype NaChBac without and with IPTG induction, respectively. Lanes 4 and 5 contain the whole cell protein for G219S NaChBac without and with IPTG induction, respectively. The arrow indicates the expected position of NaChBac. (c) SDS gel comparing the wildtype and mutant proteins purified in 0.1% DDM. Lanes 1 and 2 contain wildtype and G219S mutant proteins, respectively. The samples were solubilized in SDS buffer and loaded on a gradient SDS gel (NuPAGE 4–12% Bis-Tris gel w/MOPS). Lane 3 contains BenchMark molecular mass standards (Invitrogen).
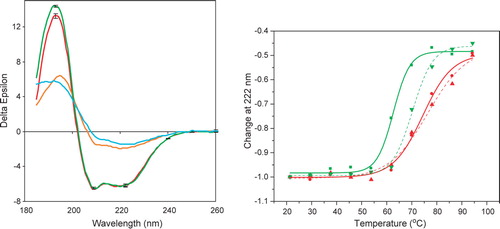
CD spectroscopy was then utilized to examine whether the proteins were folded and, if so, whether there were detectable secondary structural changes between the mutant and wildtype protein. The CD spectra of both proteins (a) were very similar; the spectra were consistent with proteins that were both folded and mostly helical and similar to those previously reported for wildtype NaChBac Citation[3]. There was a small but significant difference in the spectra at ∼ 193 nm, with the wildtype protein spectrum having a magnitude at this wavelength approximately 5% larger than that of the mutant protein although in all other regions the spectra were identical. This suggests the wildtype NaChBac may be very slightly (∼1–2%) more helical than the G219S mutant.
Figure 3. Circular dichroism spectroscopy of wildtype and mutant NaChBac. (a) Spectra of wildtype (green) and mutant (red) proteins at 25°C, and wildtype (cyan) and mutant (orange) protein at 94°C. Error bars shown for the 25° spectra represent one standard deviation in the measurements. (b) Thermal (melt) scans of wildtype (green, solid line▾), wildtype plus mibefradil (green, dashed line▴), mutant (red, solid line ▪) and mutant plus mibefradil (red, dashed line •) proteins, monitored at 222 nm. The sigmoidal fits were produced using Origin 6.0.
Thermal stability
To test if the mutant protein was more thermally stable than the wildtype protein, the CD signals of the proteins were monitored at 222 nm (a wavelength at which the signal is almost entirely due to helical structure) as a function of temperature (b). The Tm of the wildtype protein was approximately 63°C, whilst the Tm of the mutant was roughly 10°C higher (∼74°C), suggesting a substantial stabilization. The threshold for the thermal unfolding of the mutant also was about 10°C higher than that of the wildtype.
The presence of the channel-blocking drug mibefradil also increases the stability of the wildtype protein, shifting the Tm by ∼7°C (b), as previously reported Citation[3]. It was assumed that this was due to decreasing the conformational flexibility by shifting the equilibrium towards a single state. Not surprisingly, then, the effect of binding the drug on the mutant protein is substantially less (resulting a shift of only ∼2°C) (b). This is likely because the equilibrium mixture of conformations has already been shifted toward one state, so further ligand effects on conformational flexibility are expected to be only local and much less in the mutant.
The spectra of the ‘unfolded’ proteins were then compared (a). Both indicated that the proteins retained substantial helical content, even when heated to 94°C, in accordance with previous observations on NaChBac Citation[3] and its eukaryotic homologue VGSC from electric eels (Charalambous et al., submitted). However, an interesting observation is that the spectrum of the mutant at this very high temperature was significantly different than that of wildtype, with all mutant peaks larger, but especially the peak at around 193 nm, indicating that the denatured mutant protein retained a higher helical content in the ‘unfolded’ state. Also the ratio of the 193/222 peaks, another indicator of helical content, was considerably higher for the mutant.
These results together demonstrate that although the mutant and wildtype have very similar folds at room temperature, as the temperature is increased, the wildtype protein unfolds more readily than does the mutant and the ‘final’ unfolded form of the mutant retains more ordered structure than does the wildtype. Both of these characteristics indicate that, as designed, the mutant is more thermally-stable than the wildtype.
Discussion
It has been possible to design a mutant of the VGSC from B. halodurans that retains it ability to adopt a folded conformation and insert into membranes, but which is significantly more thermally stable.
Voltage gated ion channels appear to be highly flexible molecules. The crystal structures of open and closed potassium channels Citation[5], Citation[6], which are homologues of these sodium channels, have been shown to have significantly different structures from each other. Spectroscopic studies of sodium channels from both prokaryotic and eukaryotic sources Citation[3], Citation[4] show that they also undergo significant conformational changes upon binding ligands, shifting the equilibrium balances towards inactive, open and blocked states with different ligands. This demonstrates a particularly useful application of CD spectroscopy in studies of membrane proteins: not only can CD demonstrate that the ligands bind (something many other techniques can do), but it can also identify the changes induced with specific conformational states of the protein (something not easily attainable by other methods). In this case, the flexibility demonstrated is key to the functioning of the sodium channels in vivo.
The present studies were done on a sample containing the equilibrium mixture of conformations present in the absence of ligands and showed that when subjected to substantial thermal stress, the proteins resisted unfolding and maintained a folded structure even at high temperatures. Although these observations of thermal stability but functional flexibility may seem contradictory, they can be rationalized. Increasing the temperature increases the thermal motion of proteins to the point of unfolding. However, proteins with conformational flexibility intrinsic for their activity require greater tolerance to dynamic changes, and so may display increased resistance to unfolding by thermal stress.
The proposed effect of the serine substitution at the gating-hinge position was reduction of S6 helix flexibility through elimination of the central glycine around which the helix bends, and whilst the resulting protein had the biophysical characteristics expected, whether the closed- or open-state predominates in this mutant channel remains to be determined. If, as expected, the closed-state predominates, then it is possible that greater inter-domain interactions (25% greater inter-domain van der Waals contacts in the pore module in the closed relative to the open state) stabilise the tetrameric conformation and thereby contribute to the observed increase in thermal stability.
To demonstrate functionally that the mutant protein is proper folded, especially in the region of the selectivity filter, is difficult because the very nature of the design was to impair the protein flexibility and therefore the protein function by making subtle structural changes in this region. There are, however, a number of pieces of evidence that indicate this has been achieved:
(i) The mutant protein appears to be still capable of binding mibefradil, as demonstrated by the slight shift in the thermal scan; however, the magnitude of the shift is greatly decreased from that of the wildtype protein, as would be expected if its conformation was already biased towards a single state before adding the drug.
(ii) Preliminary studies (K. Charalambous, unpublished results) done on vesicles reconstituted with mutant protein using the dye sodium green to monitor sodium flux showed that some flux was detected, but because this method is qualitative rather than quantitative, it can neither assure that all proteins are functional, nor that whilst macroscopic activity was detected, there are not subtle differences in function between the mutant and wildtype proteins.
(iii) The mutant cells exhibit a small but significant gain of function (a) with respect to the growth curve of the wildtype. This would be expected if the native protein inhibits growth, and the mutant is less capable of doing so because it has an impaired ability to gate sodium entry into the host cell. Hence, all of these studies together tend to suggest that G219S is a folded but slightly functionally-impaired protein, thus achieving the design aim.
The increased thermal stability of a protein can have a number of advantages for functional and biophysical studies of its structure, including the ability to crystallize the protein Citation[15]. This is particularly valuable in the case of membrane proteins embedded in detergents or lipid bilayers, hence our interest in creating a mutant protein with enhanced resistance to thermal unfolding. In this study, we have been able to engineer the NaChBac protein structure to make a subtle change that still allows the protein to fold and insert into membranes, but that increases its thermal stability significantly.
Acknowledgements
We thank Dr Bob Sarra of Birkbeck College for the thermal calibration plot. This work was supported by a BBSRC grant to the MPSI consortium (N. Isaacs, PI), a BBSRC grant and a Heptagon fund award (to BAW), and a joint project grant from the Wellcome Trust to BAW and P. Bullough (Sheffield University). Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ito M, Xu H, Guffanti AA, Wei Y, Zvi L, Clapham DE, Krulwich TA. The voltage-gated Na+ channel NaVBP has a role in motility, chemotaxis, and pH homeostasis of an alkaliphilic Bacillus. Proc Nat Acad Sci USA 2004; 101: 10566–10571
- Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE. A prokaryotic voltage-gated sodium channel. Science 2001; 294: 2372–2375
- Nurani G, Radford M, Charalambous K, O'Reilly AO, Cronin NB, Haque S, Wallace BA. Tetrameric bacterial sodium channels: Characterization of structure, stability, and drug binding. Biochemistry 2008; 47: 8114–8121
- Cronin NB, O'Reilly A, Duclohier H, Wallace BA. Binding of the anticonvulsant drug lamotrigine and the neurotoxin batrachotoxin to voltage-gated sodium channels induces conformational changes associated with block and steady-state activation. J Biol Chem 2003; 278: 10675–10682
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998; 280: 69–77
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002; 417: 515–522
- Zhao Y, Yarov-Yarovoy V, Scheuer T, Catterall WA. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron 2004; 41: 859–865
- Lees JG, Smith BR, Wien F, Miles AJ, Wallace BA. CDtool – an integrated software package for circular dichroism spectroscopic data processing, analysis and archiving. Analyt Biochem 2004; 332: 285–289
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680
- Tikhonov DB, Zhorov BS. Modeling P-loops domain of sodium channel: Homology with potassium channels and interaction with ligands. Biophys J 2005; 88: 184–197
- CCP4. 1994. The CCP4 suite: Programs for protein crystallography. Collaborative Computational Project: Number 4. Acta Crystallographica, Section D50:760–763.
- Clark M, Cramer RD, Van Opdenbosch N. Validation of the general purpose Tripos 5.2 force field. J Comp Chem 1989; 10: 982–1012
- Koishi R, Xu H, Ren D, Navarro B, Spiller BW, Shi Q, Clapham DE. superfamily of voltage-gated sodium channels in bacteria. J Biol Chem 2004; 279: 9532–9538
- Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol 2002; 120: 629–645
- Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty P J, Jr, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Nat Acad Sci USA 2006; 103: 15835–15840