Abstract
PagP is a bacterial outer membrane protein consisting of an 8 stranded transmembrane β-barrel and an N-terminal α-helix. It is an enzyme which catalyses transfer of a palmitoyl chain from a phospholipid to lipid A. Molecular dynamics simulations have been used to compare the dynamic behaviour in simulations starting from two different structures (X-ray vs. NMR) and in six different environments (detergent micelles formed by dodecyl phosphocholine and by octyl glucoside, vs. four species of phospholipid bilayer). Analysis of interactions between the protein and its environment reveals the role played by the N-terminal α-helix, which interacts with the lipid headgroups to lock the PagP molecule into the bilayer. The PagP β-barrel adopts a tilted orientation in lipid bilayers, facilitating access of lipid tails into the mouth of the central binding pocket. In simulations starting from the X-ray structure in lipid bilayer, the L1 and L2 loops move towards one another, leading to the formation of a putative active site by residues H33, D76 and S77 coming closer together.
Introduction
Membrane proteins play key roles in many aspects of the biology of cells, and account for ca. 25% of all genes. In Gram negative bacteria, the outer membrane contains proteins based upon an anti-parallel β-barrel transmembrane architecture. These outer membrane proteins (OMPs) have diverse roles, including active and passive transport Citation[1], and enzymatic catalysis of membrane-located reactions Citation[2]. There has been considerable progress in the structural biology of outer membrane proteins, with over 40 X-ray structures having been determined (see http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html for a summary). These studies have revealed the importance of domains additional to the core transmembrane (TM) β-barrel in the structure of OMPs. However, much remains to be learnt concerning the interplay of structure and stability with the environment, both in vivo and in vitro, of outer membrane proteins.
One important aspect of our understanding of OMPs is the role of their environment in (re)folding and stability. In this respect, OmpA has proved a valuable test case Citation[3–10]. Studies have been extended to OmpG Citation[11] and a number of other OMPs Citation[12]. For example, spectroscopic and related studies of the interactions of OMPs with lipids suggest that the number of motionally restricted lipids increases with the number of strands in the β-barrel and that selectivity in protein/lipid interactions is influenced by charged sidechains on the surface of the OMP Citation[11].
A slightly more complex example is provided by PagP, an outer membrane enzyme Citation[13], Citation[14] which plays a key role modifying lipid A under conditions of stress, e.g. in response to anti-microbial peptides Citation[15], Citation[16], transferring a palmitoyl chain from a phospholipid to lipid A. Both X-ray Citation[13] and NMR Citation[14] studies have shown PagP to have an 8-stranded TM β-barrel preceded by an amphipathic N-terminal α-helix. The barrel forms a hydrophobic pocket accessible from the extracellular mouth. In the crystal structure, this pocket is occupied by a single detergent molecule Citation[13]. A proline residue (P28) interrupts the hydrogen bonding pattern between β-strands A and B, creating a β-bulge which contributes to the highly dynamic nature of L1 loop Citation[14]. This flexibility may play a role in PagP function as H33, one of the putative catalytic residues, is in loop L1. The other two putative catalytic residues (D76 and S77) are in loop L2. All three residues are near the extracellular membrane/water interface. NMR studies have suggested that PagP alternates between two dynamic states, and that the major structural changes between these two states occur in L1 and the adjacent beta-bulge Citation[17].
There has been some discussion concerning the catalytic mechanism of PagP Citation[18]. H33 is well separated from D76 and S77, e.g. in the X-ray structure the distance between H33 and S77 is 1.3 nm. As noted above, H33 sits on loop L1 which has been shown to be highly flexible and therefore it is possible that H33 moves to allow the formation of a catalytic triad. The activity of PagP is independent of the phospholipid substrate head group, but PagP is able to select between lipid acyl chains that differ by a single methylene unit Citation[13]. This selectivity is modulated by a glycine at the bottom of hydrophobic pocket. In the crystal structure, PagP binds a single detergent (lauryldimethylamine-oxide; LDAO) molecule in its central pocket. It has been shown that the enzymatic activity of PagP is inhibited by detergents. PagP has also been investigated from the perspective of folding and stability in the bilayer. In particular, it has been shown that the N-terminal α-helix of PagP plays a key role in stabilising the folded PagP molecule within its bilayer environment Citation[19].
Molecular dynamics (MD) simulations provide a useful tool for exploring the dynamics of membrane proteins and their interactions with their environment Citation[20], Citation[21]. For example, OmpA they have been used to explore the behaviour of the protein in three different environments: a lipid bilayer, a detergent micelle and a (detergent-containing) crystal. Here we present a simulation study of PagP () which compares two structures (X-ray vs. NMR) and six environments (two species of detergent micelles vs. four species of lipid bilayer). A comparative analysis of interactions between the protein and its environment reveals the role played by the N-terminal α-helix and its interaction with the lipid bilayer. The dynamics of the extracellular loops may be related to the likely formation of the catalytically active site upon substrate binding.
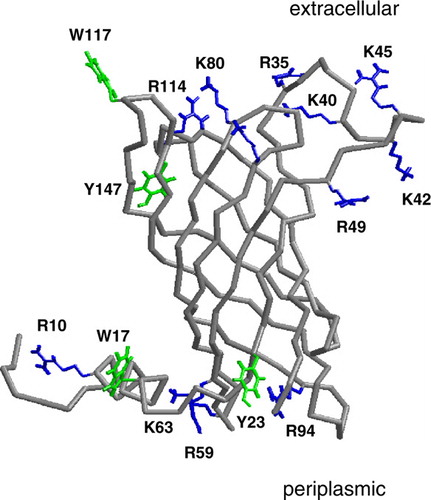
Figure 1. The Cα trace of PagP (grey) with selected residues shown in bonds format in green (Trp, Tyr) or blue (Arg, Lys) leaflet residues. The selected residues are those which form hydrogen bond to DMPC for >30% of simulation Sim1. This figure is reproduced in colour in Molecular Membrane Biology online.
Methods
Protein structures
Two PagP structures were used as the starting point for simulations: the X-ray structure (resolution 1.9 Å; PDB id 1THQ), and the NMR structure as determined in dodecyl phosphocholine (DPC) micelles (PDB id 1 MM4). Residues missing from the X-ray structure of PagP (i.e. residues 38–47 of loop L1) were built using Modeller v4 (http://salilab.org/modeller/) Citation[22], Citation[23]. The ionisation state of the sidechains in PagP were based on pKA calculations, performed using the program WhatIf Citation[24] combined with locally written code to take account of the low dielectric environment provided by a lipid bilayer. As a result of these calculations the sidechains of histidines H22, H33, and H120 were protonated (on the Nδ1 nitrogen). All other sidechains remained in their standard (i.e. default) charge states at neutral pH.
Set-up of simulation systems
For each of the bilayer simulations the protein molecule was embedded in a pre-equilibrated lipid bilayer. To orient the protein relative to the hydrophobic core of the bilayer the initial tilt and translation of the protein relative to a 3 nm thick hydrophobic slab was optimised by minimization of the membrane-protein interaction energy as calculated using empirical hydrophobicities Citation[25]. Each protein was subsequently embedded in a lipid bilayer as described in Citation[26]. After insertion of the protein, simple point charge (SPC; Citation[27]) water molecules and ions equivalent to ~0.1 M NaCl were added by superimposition. The system was energy minimized and then underwent 0.5 ns of protein restrained dynamics to allow lipid and water relaxation. This was followed by 15 ns of unrestrained MD simulations.
For the micelle simulations, PagP was placed in preformed micelles, built by creating an ‘expanded micelle like torus’ Citation[28] which consisted of semicircular planes of detergent molecules, randomly rotated about their tail axes. These detergent molecules were placed so that the terminal methyl groups of their tails were at least 0.5 nm from the outer barrel exterior and each other so as to avoid van der Waal's overlap. Each headgroup was approximately equidistant from its nearest neighbour. Once the idealised micelle was thus generated the protein was fitted into the central cavity. The protein-micelle system was then solvated through superimposition of a pre-equilibrated box of SPC water followed by removal of any water molecules too close to either the protein or detergent. Sodium and chloride ions were added to the system, via random replacement of water molecules, to an approximate concentration of ~0.1 M. As for the bilayer systems, >100 steps of energy minimisation were then performed, followed by 0.5 ns MD during which the protein was restrained and the detergent and water were allowed to relax their positions and orientations. Finally, all position restraints were removed and a production run MD simulation of duration 15 ns was performed.
Simulation protocols
Simulations were performed using GROMACS v3.14 Citation[29] (www.gromacs.org). The GROMOS96 force field Citation[30] was used for all simulations. During protein restrained runs, harmonic position restraints with force constants of 1000 kJ mol−1 nm−2 per atom were applied to all non hydrogen atoms of the protein. Electrostatic energies were calculated using particle mesh Ewald Citation[31] with a 1 nm cut-off for the real space calculations. A cut-off of 1 nm was also used for van der Waals interactions. All the simulations were performed at constant temperature, pressure and number of particles. The temperatures of the lipid, solvent (i.e. water and ions), and proteins were coupled separately. The Berendsen temperature coupling algorithm Citation[32] was applied at 310 K, i.e. above the main phase transition temperature of dimyristoyl phosphatidylcholine (DMPC; TM=297 K), with a coupling constant of τT=0.1 ps. The system pressure in the micelle simulations was isotropically coupled using a Berendsen barostat Citation[27] at 1 bar with coupling constant τP=1 ps. In the bilayer simulations the pressure was semi-isotropically coupled, i.e. the pressure along the z-axis (the bilayer normal) was allowed to vary independently of that in the xy (i.e. bilayer) plane. The timestep for integration was 2 fs, and coordinates and velocities were saved every 5 ps. The LINCS algorithm was used to restrain bond lengths Citation[33]. Secondary structure analysis used DSSP Citation[34]. VMD Citation[35] and Rasmol Citation[36] were used for visualization.
Results
Simulations and their progress
We have performed a number of simulations (each of 15 ns duration, see ) starting from either the X-ray structure (1THQ) or from the NMR structure in DPC micelles (1 MM4). These simulations have been performed with the protein embedded in either a lipid bilayer or in a detergent micelle. A simple measure of the degree of conformational drift is provided by the root mean square deviation (RMSD) from the starting structure of the Cα atoms. As is generally the case for OMPs Citation[37], for the TM domain this RMSD is low, reflecting the conformational stability of the β-barrel domain (despite absence of a bound detergent molecule in the central pocket – see below). Thus, for the X-ray based simulations (Sim1 to Sim6) the TM domains Cα RMSD is ca. 0.1 nm whereas for the NMR based simulations it is a little higher (0.15 nm), echoing previous studies Citation[38]. The Cα RMSD is significantly higher for the non-TM regions, ranging from ca. 0.4 nm for simulations based on the X-ray structure to up to 1.1 nm for the NMR structure in an octyl-β-glucoside (OG) micelle. The higher RMSD for the non-TM regions reflects two factors: the relatively high conformational mobility of the extracellular loops (as seen in many OMP simulations Citation[39]), and also the behaviour of the N-terminal α-helix (discussed in more detail below).
Table I. Simulations.
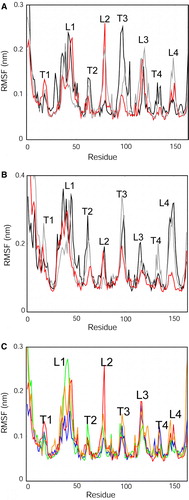
This distinct behaviour of the extracellular loops and of the N-terminal helix can be verified if one examines the residue-by-residue root mean square fluctuations (RMSFs; ). In all of these a common pattern is observed, with high RMSF values for the extracellular (EC) loops, especially loop L1 (which contains residue H33 of the putative active site). The RMSFs for the N-terminal α-helix can also be seen to be high, especially in the case of the NMR structure based simulations (B). Some differences between the individual micelle and bilayer based profiles can be observed but it is not clear that there is a consistent simple difference between the bilayer and micelle environments (unlike, e.g., the case with OmpA Citation[28]), although the RMSFs of the periplasmic turn T3 (and to a lesser extent of T2 and T4) are generally higher for the micelle than for the bilayer simulations. We also compared the behaviour of the X-ray structure of the protein in different bilayer environments (C). The overall profile seems to be conserved between the different bilayers. Interestingly, the fluctuations of the EC loops seem generally to be lower in the palmitoyl-oleoyl phosphatidylglycerol/palmitoyl-oleoyl phosphatidylethanolamine (POPG/POPE) bilayer. However, in all cases elevated fluctuations of the N-terminal α-helix and of L1 are observed.
Figure 2. Cα atom root mean square fluctuations (RMSFs) vs. residue number compared between simulations. RMSFs were calculated over the final 10 ns of each simulation: (A) X-ray structure: micelle vs. bilayer, i.e. simulations Sim1, Sim5 and Sim6. (B) NMR structure: micelle vs. bilayer, i.e. simulations Sim7, Sim8 and Sim9. The black lines correspond to the DPC micelle simulations; the grey lines to the OG micelle simulations; and the red lines to the DMPC bilayer simulations. (C) RMSF vs. residue profiles for the different bilayer simulations of the X-ray structure (Sim1 to Sim4). The red line corresponds to the DMPC simulation (Sim1); green to the POPC simulation (Sim2); orange to DPPC (Sim3); blue to the POPG/POPE simulation (Sim4). Note that in this graph and the associated discussion in the text, L1 to L4 correspond to the four extracellular loops, T1 corresponds to the turn linking the N-terminal α-helix to the first β-strand, and T2 to T4 correspond to the periplasmic turns linking β2 to β3 etc. This figure is reproduced in colour in Molecular Membrane Biology online.
Interactions with lipids and letergents
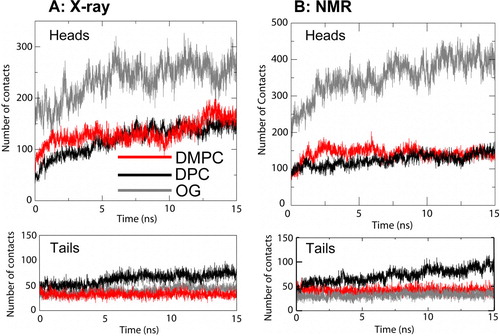
In order to characterise the interactions with the lipid bilayer/detergent micelle environment, the detergent micelles and DMPC bilayer simulations were analysed in terms of the number of contacts made to the protein by the hydrophobic tails and the polar headgroups of the lipid/detergent molecules (). The main difference is that the glucose headgroup of OG forms substantially more contacts than does the headgroup of either DPC or DMPC. For DMPC and DPC there seem to be more equivalent numbers of contacts made whether the simulation starts from the X-ray or NMR structure. The greater number of tail contacts for DPC seems to reflect entry of the detergent molecule into the ‘binding pocket’ of the barrel (see below). Results for the other lipid bilayers (data not shown) are similar to those for DMPC, showing no strong dependence on lipid species in the bilayers.
Figure 3. Numbers of contacts (defined using a 0.3 nm cut-off) by PagP to lipid/detergent molecules. The analysis is divided into lipid/detergent headgroups and tails. (A) X-ray structure: micelle vs. bilayer simulations, Sim1 (DMPC; red), Sim5 (DPC; black) and Sim6 (OG, grey). (B) NMR structure: micelle vs. bilayer simulations, Sim7 (DMPC, red), Sim8 (DPC, black) and Sim9 (OG, grey). This figure is reproduced in colour in Molecular Membrane Biology online.
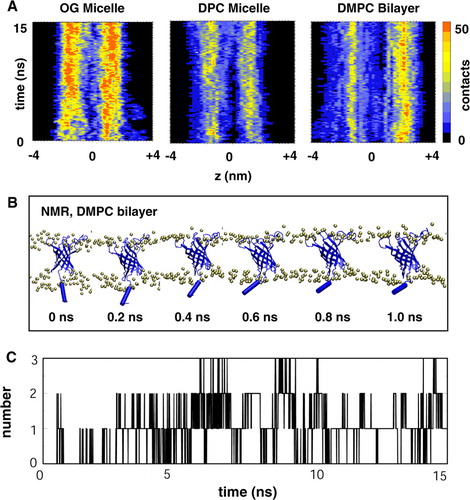
The distribution of the headgroup interactions along the length of the PagP molecule was analysed in more detail by displaying contours of the number of interactions as a function of time and z axis position (). From this it is evident that the numbers and patterns of interactions have equilibrated within the duration (15 ns) of the simulations. For both of the micelles it can be seen that there are approximately equal numbers of interactions at both the periplasmic (z negative) and extracellular (z positive) ends of PagP. In contrast, for the DMPC bilayer, more headgroups interact with the EC loops (as is also seen, for e.g., OmpA Citation[40]). However, there is a broader and more diffuse (on z) pattern of interactions at the periplasmic end of PagP for the DMPC simulations. This reflects interactions with the N-terminal helix (see below for more detail).
Figure 4. (A) Number of contacts from PagP to the detergent/lipid headgroups as a function of time and z coordinate for Sim7 (NMR, DMPC), Sim8 (NMR, DPC) and Sim9 (NMR, OG). Note that a negative z coordinate corresponds to the periplasmic end and a positive z coordinate to the extracellular end of PagP. (B) Snapshots taken from Sim7 (NMR, DMPC) showing the movement of the amphipathic helix towards the bilayer interface. (C) Number of hydrogen bonds between the amphipathic helix and the lipid molecules as a function of time for Sim7. This figure is reproduced in colour in Molecular Membrane Biology online.
The N-terminal α-helix
An unusual feature of the PagP structure is the presence of an amphipathic α-helix N-terminal to the β-barrel. It has been suggested that this may play a key role in the folding/stability of the protein Citation[19]. The amphipathic nature of the helix suggests that it might play a role in protein/bilayer interactions, as is the case for comparable helices in, e.g., K channels and in certain monotopic membrane proteins Citation[41]. In the NMR structure(s) of PagP the location of the α-helix is not heavily restrained relative to the β-barrel. In the X-ray structure it packs close to the β-barrel, in part mediated via a detergent (LDAO) molecule bound in the crystal structure.
We analysed the motion and interactions of the α-helix with lipid molecules, especially in the NMR structure based simulations (B). As might be anticipated for an amphipathic helix, it was seen to move towards the bilayer/water interface within the first nanosecond of the simulation. Analysis of the number of hydrogen bonds between the helix and the lipid as a function of time (C) indicates that the movement of the helix to the bilayer/water interface is accompanied by formation of H-bonds to the lipid headgroups. In the bilayer simulations based on the X-ray structure (which started with the helix closer to the interface initially) the number of such H-bonds was a little higher (ranging from ~4 for DMPC to ~10 for DPPC (dipalmitoyl phosphatidylcholine); data not shown). Thus, the N-terminal helix seems to play a significant role in interactions between PagP and its membrane environment.
The specific residues of PagP which form H-bonds to DMPC molecules for at least 30% of the simulation time (averaged across all DMPC simulations) are indicated in . It can be seen that a number of basic and amphipathic aromatic (i.e. Trp and Tyr) sidechains are involved, as might be expected based on the known general rules concerning membrane protein interactions with bilayers Citation[42]. In particular, R10 and W17 from the N-terminal helix form H-bonds to lipid molecules. H-bonds to lipids are also formed by R59 and Y23 of the periplasmic end of the β-barrel. Interestingly, W17 and R59 have been implicated in the folding/stability of PagP Citation[19].
Barrel tilt and access to the binding pocket
Not all OMP β-barrels sit perpendicular to the bilayer plane. For example, infrared spectroscopic studies of OmpA, OmpG and FhuA indicate that the β-barrel axis may be tilted at an angle of from 38° (OmpA) to 21° (FhuA) relative to the bilayer normal Citation[11]. Visualization and analysis of the distribution of exposed surface residues of the X-ray structure of PagP has been used to suggest a ~25° tilt of the barrel axis with respect to the membrane normal Citation[13]. We have therefore explored the tilt angle of the β-barrel of PagP in the bilayer simulations, which ranges between a mean (over the last 5 ns of simulation) of 33±2° in Sim4 (X-ray, POPE/POPG) to 46±2° in Sim2 (X-ray, POPC [palmitoyl-oleoyl phosphatidylcholine]). We thus may conclude that, as can be seen in, e.g., B, the PagP molecule is tilted to a significant extent when located in a lipid bilayer.
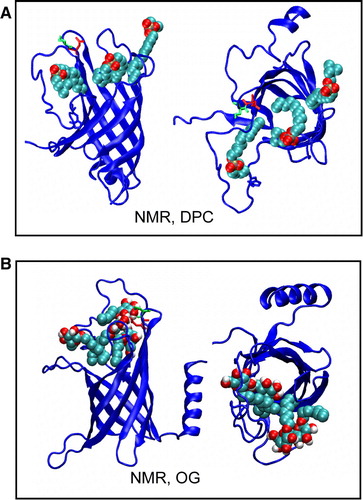
The tilt of PagP relative to the bilayer may have a functional role, aiding access of bilayer-located substrate molecules to the binding pocket in the centre of the β-barrel. It has been observed that detergents (e.g. LDAO in the 1THQ X-ray structure Citation[13]) may bind within the pocket formed by the barrel. This has been used to propose a substrate selectivity mechanism based on the depth of the binding pocket Citation[13], Citation[18]. Examination of structures from the detergent micelle simulations of PagP, in both the DPC and OG micelles (), reveals that these detergents access the mouth of the hydrophobic pocket. However, they were not observed to contact the glycine at the bottom of the binding pocket. This correlates with the observation that when PagP is in DPC or OG it is enzymatically inactive Citation[13], whereas when in CYFOS-7, which possess a bulky tail group preventing entry to the pocket, activity is restored Citation[17]. This has been interpreted as indicating that detergents, such as DPC and OG block the binding pocket to access by the natural substrate of the enzyme, in agreement with our simulation results. Interestingly, lipid tails were observed to enter the mouth of the binding pocket in a number of the bilayer simulations (data not shown) in a manner that seems to correspond to the presence of β-bulges in the PagP β-barrel structure.
Figure 5. Snapshots from (A) Sim8 (NMR, DPC) showing movement of DPC detergent molecules into mouth of the binding pocket and from (B) Sim9 (NMR, OG), showing movement of OG detergent molecules into mouth of the binding pocket. This figure is reproduced in colour in Molecular Membrane Biology online.
Integrity of the proposed active site
In both the X-ray and NMR structures the three putative active site residues (H33, D76, and S77) are relatively distant from one another. Thus in the X-ray structure (1THQ) the distances are S77:Oγ to H33:Cγ = 1.3 nm and D76:Cγ to H33:Cγ = 1.8 nm. In the DPC micelle structure (1 MM4; averaged over the first five structures in the ensemble) the distances are S77:Oγ to H33:Cγ = 2.6±0.6 nm and D76:Cγ to H33:Cγ = 2.8±0.6 nm. (For reference, in the OG micelle structure (1 MM5) the corresponding average distances are 2.1±0.3 nm and 2.1±0.2 nm respectively.) To explore this further we therefore examined whether these residues approached one another more closely during the simulations. It was seen in the simulations based on the X-ray structure that the three residues (H33, D76, and S77) were able to form a sustained H-bonding interaction provided that H33 was assumed to be in a protonated (i.e. charged) state (as was suggested to be the case by pKA calculations, see above) and that the lipid bilayer tails were completely saturated (i.e. DMPC and DPPC lipids were used). In order to study how changing environment affects the active site, scatter plots of the D76:Cγ to H33:Cγ vs. S77:Oγ to H33:Cγ distances () were generated from the seven different simulations; allowing comparison of the behaviour of the active site in both the micelle and bilayer environments.
Figure 6. Scatter plot of the potential active site geometry as defined by the pair of distances: (i) from D76 Cγ to H33 Cγ; and (ii) from S77 Oγ to H33 Cγ. Distances were measured every 0.025 ns. (A) X-ray structure: micelle vs. bilayer simulations, i.e. Sim1 (DMPC, red), Sim5 (DPC, black) and Sim6 (OG, grey); (B) NMR structure: micelle vs. bilayer simulations, i.e. Sim7 (DMPC, red), Sim8 (DPC, black) and Sim9 (OG, grey). The broken horizontal and vertical lines represent the corresponding pair of distances in the X-ray structure (i.e. 1THQ). This figure is reproduced in colour in Molecular Membrane Biology online.
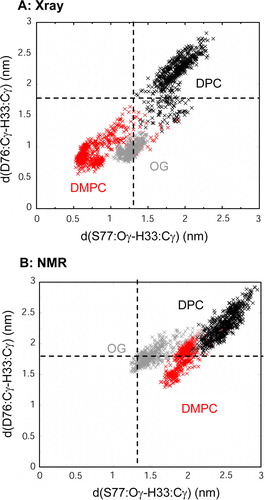
Scatter plot analysis, comparing the two micelle environments with the DMPC bilayer, suggests that the bilayer environment promotes smaller distances between the three active site residues than do the micelle environments. Sim1 (X-ray, DMPC) showed the closest approach of the three residues (such that S77:Oγ to H33:Cγ = ~0.6 nm; and D76:Cγ to H33:Cγ = ~0.8 nm; compared with 1.3 nm and 1.8 nm, respectively, for the X-ray structure). In Sim1 a water molecule was ‘captured’ early on in the simulation, forming bridging H-bonds between H33 and D76, and resulting in closure of loops L1 and L2 from ~4 ns onwards. It is clear that the DPC allows a greater distance between the residues. A comparison of the active site residues was also carried out for the various lipid species in bilayers (data not shown). The closest approaches were seen for saturated lipids i.e. DMPC and DPPC.
Discussion
A number of the results from the PagP simulations are of relevance to our understanding of the stability and function of this protein. Thus, we observe substantial flexibility of the L1 loop which may be correlated with both substrate access to the binding pocket and with formation of the active site from otherwise distant residues. The differences in number of protein contacts to detergent headgroups between the DPC and OG simulations may be related to the differences in the NMR structures observed between the two detergent micelle species Citation[14]. The interactions of the N-terminal α-helix seen in the simulations provide an alternative interpretation of the nature of the ‘clamp’ formed by the helix in folding studies of PagP Citation[19]. In general, the interactions of Trp, Tyr and Arg sidechains with lipid headgroups are in agreement with the emerging general picture of the role of such sidechains in locking membrane proteins into position in a lipid bilayer Citation[43], Citation[44]. The tilt of the β-barrel axis relative to the bilayer normal observed in the simulations correlates well with infrared spectroscopic studies of a number of OMPs in lipid bilayers Citation[11], Citation[45]. This tilting, as noted above, seems to play a role in helping lipid acyl chains to access the central binding pocket of PagP. The simulations provide direct evidence for multiple conformations of the loops containing the putative active site residues, with ‘closure’ of the active site loops observed in some bilayer simulations. This is of particular interest given the suggestion from NMR studies Citation[17] of two states of the L1 loop: an R state corresponding to an ensemble of conformers, and a catalytically active T state corresponding to a more defined configuration. It is tempting to identify the T state with the H33-S77 ‘closed’ state seen in, e.g., Sim1.
It is useful to appraise critically the MD simulation methodology used in this study. The simulations (15 ns) are relatively short. However, the total simulation time (135 ns) is reasonable, and by performing multiple simulations we were better able to assess the influence of starting structure and environment on the simulated behaviour of PagP. The major limitation is the restriction to simple lipids or detergents in the simulations. Whilst this reflects the conditions under which PagP has been studied in vitro, the lipid bilayer environments studied are considerably simpler than that present in the bacterial outer membrane in vivo. Recent developments in modelling bacterial lipopolysaccharide (LPS) Citation[46], Citation[47] may enable future simulations to include a more physiologically realistic environment.
In summary, these studies demonstrate the utility of a simulation approach to exploring structure/function relationships in individual outer membrane proteins. Future studies may extend this towards modelling of more complex assemblies of multiple OMPs and of LPS in order to more fully characterise the nature of the outer membrane as a whole.
Acknowledgements
This work was supported by grants from The Biotechnology and Biological Sciences Research Council (BBSRC). Our thanks to our colleagues for their interest in this work, especially Syma Khalid and Peter Bond. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol 2000; 37: 239–253
- Bishop RE. Structural biology of membrane-intrinsic β-barrel enzymes: sentinels of the bacterial outer membrane. Biochim Biophys Acta 2008; 1778: 1881–1896
- Kleinschmidt JH, Wiener MC, Tamm LK. Outer membrane protein A of E. coli folds into detergent micelles, but not in the presence of monomeric detergent. Prot Sci 1999; 8: 2065–2071
- Bulieris PV, Behrens S, Holst O, Kleinschmidt JH. Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J Biol Chem 2003; 278: 9092–9099
- Kleinschmidt JH. Folding kinetics of the outer membrane proteins OmpA and FomA into phospholipid bilayers. Chem Phys Lipids 2006; 141: 30–47
- Marsh D, Shanmugavadivu B, Kleinschmidt JH. Membrane elastic fluctuations and the insertion and tilt of beta-barrel proteins. Biophys J 2006; 91: 227–232
- Pocanschi CL, Patel GJ, Marsh D, Kleinschmidt JH. Curvature elasticity and refolding of OmpA in large unilamellar vesicles. Biophys J 2006; 91: L75–77
- Tamm LK, Hong H, Liang B. Folding and assembly of β-barrel membrane proteins. Biochim Biophys Acta 2004; 1666: 250–263
- Hong H, Szabo G, Tamm LK. Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nature Chem Biol 2006; 2: 627–635
- Hong HD, Park S, Jimenez RHF, Rinehart D, Tamm LK. Role of aromatic side chains in the folding and thermodynamic stability of integral membrane proteins. J Am Chem Soc 2007; 129: 8320–8327
- Anbazhagan V, Qu J, Kleinschmidt JH, Marsh D. Incorporation of outer membrane protein OmpG in lipid membranes: protein-lipid interactions and beta-barrel orientation. Biochem 2008; 47: 6189–6198
- Burgess NK, Dao TP, Stanley AM, Fleming KG. β-Barrel proteins that reside in the Escherichia coli outer membrane in vivo demonstrate varied folding behavior in vitro. J Biol Chem 2008; 283: 26748–26758
- Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Privé GG. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO J 2004; 23: 2931–2941
- Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Privé GG, Bishop RE, Kay LE. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Nat Acad Sci USA 2002; 99: 13560–13565
- Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998; 95: 189–198
- Kawasaki K, Ernst RK, Miller SI. 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J Biol Chem 2004; 279: 20044–20048
- Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc Natl Acad Sci USA 2004; 101: 9618–9623
- Bishop RE. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Molec Microbiol 2005; 57: 900–912
- Huysmans GHM, Radford SE, Brockwell DJ, Baldwin SA. The N-terminal helix is a post-assembly clamp in the bacterial outer membrane protein PagP. J Mol Biol 2007; 373: 529–540
- Ash WL, Zlomislic MR, Oloo EO, Tieleman DP. Computer simulations of membrane proteins. Biochim Biophys Acta 2004; 1666: 158–189
- Lindahl E, Sansom MSP. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol 2008; 18: 425–431
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol 1993; 234: 779–815
- Fiser A, Kinh Gian Do R, Sali A. Modeling of loops in protein structures. Prot Sci 2000; 9: 1753–1773
- Vriend G. WhatIf – a molecular modeling and drug design program. J Mol Graph 1990; 8: 52–56
- Roseman MA. Hydrophilicity of polar amino acid side-chains is markedly reduced by flanking peptide bonds. J Mol Biol 1988; 200: 513–522
- Faraldo-Gómez JD, Smith GR, Sansom MSP. Setup and optimisation of membrane protein simulations. Eur Biophys J 2002; 31: 217–227
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. Intermolecular forces, B Pullman. Reidel, Dordrecht 1981; 331–342
- Bond PJ, Sansom MSP. Membrane protein dynamics vs. environment: simulations of OmpA in a micelle and in a bilayer. J Mol Biol 2003; 329: 1035–1053
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Molec Model 2001; 7: 306–317
- van Gunsteren, WF, Kruger, P, Billeter, SR, Mark, AE, Eising, AA, Scott, WRP, Huneberger, PH, Tironi, IG. 1996. Biomolecular simulation: the GROMOS96 Manual and User Guide. Biomos & Hochschulverlag AG an der ETH Zurich, Groningen & Zurich.
- Darden T, York D, Pedersen L. Particle mesh Ewald – an N.log(N) method for Ewald sums in large systems. J Chem Phys 1993; 98: 10089–10092
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys 1984; 81: 3684–3690
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comp Chem 1997; 18: 1463–1472
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22: 2577–2637
- Humphrey W, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Molec Graph 1996; 14: 33–38
- Sayle RA, Milner-White EJ. RasMol: Biomolecular graphics for all. Trends Biochem Sci 1995; 20: 374–376
- Bond PJ, Sansom MSP. The simulation approach to bacterial outer membrane proteins. Mol Memb Biol 2004; 21: 151–162
- Cox K, Bond PJ, Grottesi A, Baaden M, Sansom MSP. Outer membrane proteins: comparing X-ray and NMR structures by MD simulations in lipid bilayers. Eur Biophys J 2007; 37: 131–141
- Khalid S, Bond PJ, Carpenter T, Sansom MSP. OmpA: gating and dynamics via molecular dynamics simulations. Biochim Biophys Acta 2008; 1778: 1871–1880
- Deol SS, Bond PJ, Domene C, Sansom MSP. Lipid-protein interactions of integral membrane proteins: a comparative simulation study. Biophys J 2004; 87: 3737–3749
- Fowler PW, Balali-Mood K, Deol S, Coveney PV, Sansom MSP. Monotopic enzymes and lipid bilayers: a comparative study. Biochem 2007; 46: 3108–3115
- Killian JA, von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem Sci 2000; 25: 429–434
- Granseth E, von Heijne G, Elofsson A. A study of the membrane-water interface region of membrane proteins. J Mol Biol 2005; 346: 377–385
- Scott KA, Bond PJ, Ivetac A, Chetwynd AP, Khalid S, Sansom MSP. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 2008; 16: 621–630
- Ramakrishnan M, Qu J, Pocanschi CL, Kleinschmidt JH, Marsh D. Orientation of β-barrel proteins OmpA and FhuA in lipid membranes. Chain length dependence from infrared dichroism. Biochem 2005; 44: 3515–3523
- Shroll RM, Straatsma TP. Molecular structure of the outer bacterial membrane of Pseudomonas aeruginosa via classical simulation. Biopolymers 2002; 65: 395–407
- Soares TA, Straatsma TP. Assessment of the convergence of molecular dynamics simulations of lipopolysaccharide membranes. Molec Simul 2008; 34: 295–307