
Abstract
A molecular dynamics (MD) simulation with atomistic details was performed to examine the partitioning and transport behavior of moderately cytotoxic ionic liquids (ILs), namely choline bis(2-ethylhexyl) phosphate (CBEH), choline bis(2,4,4-trimethylpentyl) phosphinate (CTMP) and choline O,O-diethyl dithiophosphate (CDEP) in a fully hydrated dipalmitoylphosphatidylcholine (DPPC) bilayer in the fluid phase at 323 K. The structure of ILs was so selected to understand if the role of dipole and dispersion forces in the ILs distribution in the membrane can be possible. Several analyses including mass density, electrostatic potential, order parameter, diffusion coefficients and hydrogen bond formation, was carried out to determine the precise location of the anionic species inside the membrane. Moreover, the potential of the mean force (PMF) method was used to calculate free energy profile for transferring anionic species from the DPPC membrane into the bulk water. While less cytotoxic DEP is located within the bulk water, more cytotoxic TMP and BEH ILs were found to remain in the membrane and the energy barrier for crossing through the bilayer center of BEH was higher. Various ILs have no significant effect on P–N vector. The thickness of lipid bilayer decreased in all systems comprising ILs, while area per lipid increased.
Introduction
Ionic liquids (ILs), salts with melting points below 100 °C, are versatile materials that have been attracting much attention as excellent alternatives to organic solvents in homogeneous and biphasic processes (Ohno, Citation2011; Welton, Citation1999; Xu & Angell, Citation2003). Solubility in both aqueous and organic media makes ILs a good candidate for pharmacological studies. Preliminary investigations on the applications of ILs in the pharmaceutical industry have been allocated for studying their antimicrobial activity and toxicity. Because of their nontoxic nature, they have been used widely in food products as non-nutritive sweeteners (Carter et al., Citation2004; Pernak et al., Citation2005). Current applications of ILs are focused on the formation of microemulsion droplets for transportation and release of drugs, and stabilizing of actives and additives in pharmaceutics (Moniruzzaman et al., Citation2010). Studying the behaviors of ILs in water environment is of particular importance due to the fact that many metabolic processes are feasible in aqueous media.Therefore, studying the behaviors of ILs in a water environment is of particular importance (Fujita et al., Citation2005; Xu & Angell, Citation2003). On the other hand, ILs have been probed as solvents/co-solvents in different biotechnology applications, including the formulation of biopharmaceuticals and media for biocatalysis (Fujita et al., Citation2006; Vrikkis et al., Citation2009; Weaver et al., Citation2010). This has led, in turn, to many questions concerning the behavior of ILs in either the human body or in the living ecosystem. The situation of ILs in these environments is dictated by the behavior of organic ions after dilution in an aqueous solution. Up to now, biological adaptability of ILs formulated in aqueous media has generally been probed in the context of high-throughput assays (pluripotent methods) which study the relative toxicity of the ILs toward specific species or cell types (Jastorff et al., Citation2005, Citation2003; Ohno, Citation2011; Petkovic et al., Citation2010; Stolte et al., Citation2006). These types of assays provide useful information as to how certain chemical structures affect toxicity trends, but cannot provide a detailed molecular description of the interaction between ILs and the cell membrane. In order to study the structure and stability of two model biomembranes, neutron reflectometry (NR) measurements were undertaken (Benedetto et al., Citation2014), it was found that the changes in the bilayer due to the Room-temperature ionic liquids (RTIL) sorption are irreversible and a measurable amount of IL remains in the lipid fraction. The determination of the microscopic mechanism of the RTILs cytotoxicity with the aim to design greener RTILs for industrial applications, is the first target of RTILs/biomembrane interaction studies (Benedetto, Citation2017). Considering of RTILs combinations with phospholipids, proteins and peptides, nucleic acids (mainly DNA) and with carbohydrates from simple sugars to large polysaccharides, studies have been conducted to identify the basic principles able to organize and rationalize the vast variety of properties and phenomena displayed by these systems (Benedetto & Ballone, Citation2015, Citation2016).
Today, over half of the drugs in the marketplace are sold as organic salts (Stoimenovski et al., Citation2010). Therefore, the transformation of a drug into a salt is a vital step in the drug development process (Hough & Rogers, Citation2007). In this regard, ILs can play a crucial role in the development of novel liquid salts on the basis of pharmacologically active ions. The other most important issue is the transformation of ILs anions through the cell membrane which has rarely been investigated (Gulati et al., Citation1998). Most of our knowledge about the interactions of ILs with membranes is obtained from the results of cytotoxicity studies. For example, a direct relationship between the alkyl side-chain length of alkylimidazolium salts and cellular toxicity has been found (Stolte et al., Citation2006). Furthermore, some experimental studies have been conducted to determine the toxicity of choline-based ILs (Weaver et al., Citation2013). The results revealed a range of toxicities from low (choline dihydrogen phosphate (CDHP)) to high (choline bis (2,4,4-trimethylpentyl) phosphinate (CTMP) and choline bis (2-ethylhexyl) phosphate (CBEH)) based on the anion effect ratio (Stolte et al., Citation2006; Weaver et al., Citation2010). It was determined that by increasing the alkyl chain length on anionic component of ILs, the cytotoxicity rises with respect to mammalian cells. This increase was assumed to be associated with an increment of lipophilicity (Weaver et al., Citation2010). Furthermore, in a study of the thermotropic behavior of the DPPC in the presence of diluted choline-based organic salts, a high relative toxicity along with most changes in the lipid phase transition behavior of lipid membrane was confirmed in the cases of CTMP and CBEH ILs (Weaver et al., Citation2013).
Molecular dynamics (MD) simulation is a powerful tool that can provide key complementary experimental information regarding the details of interactions between ILs molecules and bio-membrane (Yoo et al., Citation2016). However, MD simulations utilizing GROMACS package of the choline-based ILs (i.e. CTMP, CBEH and CDEP) in the lipid bilayer are not done. In this study, for the first time (in the range of empirically studied concentrations) the interaction between DPPC and choline-based ILs was investigated using MD simulation. To better understand the nature of specific interactions between the ILs and the lipid bilayer, we investigated in this work the effect of certain choline-based ILs and organic salts (CTMP, CDEP and CBEH) on the model lipid membrane systems prepared from the DPPC ().
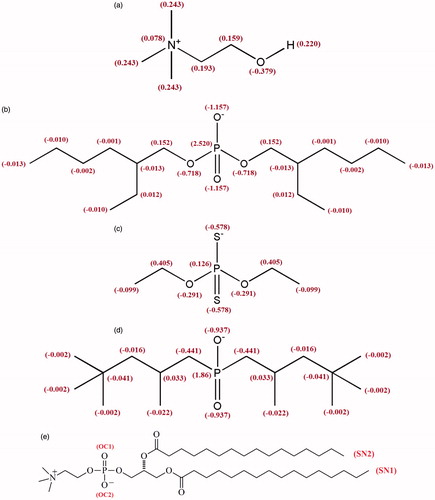
Figure 1. Molecular structures of Choline (a), BEH (b), DEP (c) and TMP (d) with partial atom charges along with the structure of DPPC lipid (e).
Methodology
Simulation set up
Simulations were carried out along four different simulation systems. Three of these systems comprised of 128 DPPC lipids (64 per leaflet) with one IL molecule in each leaflet (anion species and Choline), which were hydrated by 4000 water molecules. The fourth system comprised of only lipid and water molecules that was utilized as a reference system. Molecular structures of DPPC Lipid and ILs are shown in . For water molecules, the simple point charge model (SPC) was used (Berendsen et al., Citation1981). The InflateGRO methodology was used for embedding ILs in the lipids (Kandt et al., Citation2007). The topology and coordinate files of ILs based on the GROMOS 53A6 force field was received from ATB server (Canzar et al., Citation2013; Koziara et al., Citation2014).
Simulation details
In this study, two different types of molecular dynamics (MD) simulation were performed. The first study, which was run over four different systems was conducted to investigate the behavior of the ILs in the lipid bilayer. The second simulation was set up for PMF calculations in systems containing the 128 DPPC lipids and one of the CBEH, CDEP or CTMP molecules. The simulations were executed in the NPT ensemble using GROMACS version 4.5.5 package (Hess et al., Citation2008; KTH Royal Institute of Technology, Stockholm, Sweden). All simulations were carried out under pressure (1 bar) and temperature (323 K), and with periodic boundary conditions. Temperature was controlled by the Nose–Hoover thermostat with a coupling time of 0.5 ps (Boggara & Krishnamoorti, Citation2010). Constraining the bond lengths was carried out by using the LINCS algorithm (Hess et al., Citation1997). The pressure control was carried out by coupling the simulation cell to a Parrinello–Rahman barostat, with coupling time constant 2 ps. Semi-isotropic pressure coupling was applied with two degrees of freedom, one in XY direction and another in Z direction (Parrinello & Rahman, Citation1980). To integrate Newton’s equations of motion, the leap-frog algorithm was used with time step 2 fs (Hockney et al., Citation1974). Coulomb and Van der Waals interactions were cutoff at 1.2 nm, whereas for long-range electrostatics, the particle mesh Ewald treatment was applied (Essmann et al., Citation1995). Starting structure for DPPC lipid bilayer was taken from the Berger lipid (Berger et al., Citation1997) modified by Tieleman (Tieleman et al., Citation2006). In all systems, unfavorable atomic contacts were removed by steepest descent energy minimization (Snyman, Citation2005). At first, positions of ILs were restrained and equilibration conducted in the NVT ensemble for 2 ns and then followed by equilibration in NPT ensemble for 8 ns. After equilibration steps, all simulations were run for 125 ns from their starting conditions and coordinates of the atoms. The GridMAT-MD program (Allen et al., Citation2009) was used to measure the thickness of the DPPC membrane. The free energy profiles as the potential of mean force (PMF) for translocation of one anion species across the lipid bilayers were calculated using umbrella sampling (Torrie & Valleau, Citation1977) and the weighted histogram analysis method (WHAM) (Hub et al., Citation2010; Kumar et al., Citation1992). Statistical errors were estimated using Bayesian bootstrap analysis (N = 200). Starting configurations were obtained by positioning the center of mass of an anion species molecule in the bilayer center, and then pulling it into the bulk water along the z-axis using the umbrella method. Anion species, in the presence of choline, in order to provide electroneutrality, were pulled along the bilayer normal (the z-axis) by using a pulling force constant of 1000 kJ mol−1nm−² and pulling rate of 0.01 nmps−1. Pulling exerts a harmonic potential on the anionic species and moves the molecule center with a given pull rate. From the pulling simulation, the snapshots were obtained and among them 21 windows (∼0.17 nm apart) along the z-axis, ranging from the bilayer center (Z= ±0.0 nm) to the bulk water (Z= ± 3.5 nm), were selected. Each window was explored in a separate simulation (10 ns run time each) to avoid any ILs interactions. Visualizations for our systems were created with VMD (Humphrey et al., Citation1996). It should be noted that, we attempted to create similarities as much as possible between the systems selected in this study and those empirically investigated. We did not particularly take into account the presence of cholesterol in the membrane which showed to greatly affect the partitioning of solute into the center of the membrane (Wennberg et al., Citation2012).
Results and discussion
Mass density
Analysis of mass density distribution can be effectively used for obtaining the profile of ILs accumulation within the simulation box. As can be seen from , lipid and water mass density variations in different parts of the simulation box are similar to those reported in the literature (Gullapalli et al., Citation2008; Hughes et al., Citation2012). From , it can be seen that the maximum of the density of TMP and BEH is very close to each other and located at approximately 1.3 nm from the center of the bilayer corresponding to the upper portion of the lipid tail region. It seems that the maximum density profile of ILs is not deeply affected by either the P-O bonds in BEH and TMP or the configuration of hydrocarbon chains in DPPC. For DEP, the maximum occurs far from the center of the bilayer (around 1.6 nm), an indication of accumulation of molecules outside the membrane. Moreover, the density for DEP is zero in the middle of the bilayer, indicating that DEP molecules are not able to cross the bilayer on the time scale of simulation. Regarding the smaller contribution of dispersion forces in DPPC/IL interactions for DEP compared to TMP and BEH, these results are quite meaningful. Moreover, while the maximum position of the DPPC density for the CTMP and CBEH including systems is slightly shifted toward the middle of the bilayer, it has not been seriously affected by DEP. As can be seen from , the density of water is the same in all systems and the choline maximum density, which occurs around 1.5 nm from the center of the bilayer near the polar head group and is not presented here is the same for all simulated systems. In order to get an insight into the penetration of water molecules into the lipid bilayer, several radial distribution functions (RDF) were analyzed. The RDF of water oxygen atoms around both the phosphate oxygen atom and the nitrogen atom of the lipid headgroup were calculated in the presence of all three ILs as well as for the reference bilayer (Figure S1(a,b)). The RDFs schemes of the systems are nearly the same. In all of the RDFs, compared to the reference system, the ILs including systems have somewhat higher RDF values. The coordination numbers obtained from the appropriate integration of RDF up to the first minimum, are shown in . It can be concluded that water penetration into the headgroup is minor.
Figure 2. Mass density of different components for DPPC lipid bilayer system (a), different anionic species of ILs (b), and water (c).
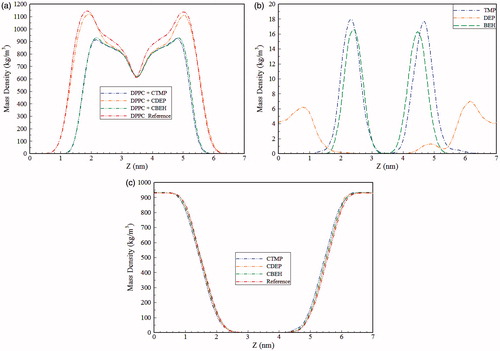
Table 1. Coordination numbers of water to DPPC atoms in the simulated systems.
Potential of mean force
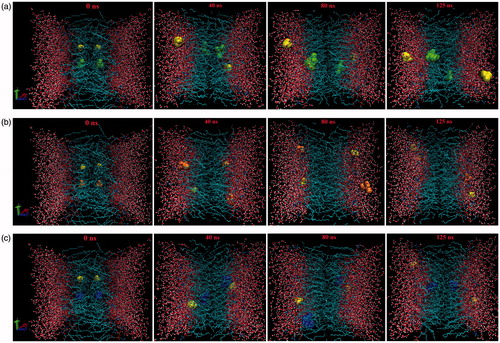
shows the PMFs obtained for the anionic species of ILs. As seen in this diagram, the free energies of TMP and BEH show a minimum in the hydrocarbon region. TMP shows a discernible minimum at 1.0 nm from the bilayer center, and the free energy minimum of BEH occurring in a flat area around 1.0–1.4 nm is more negative than that of TMP. These results are consistent with the results of maximum density explained in the previous section. As a consequence, in this section, the value of ΔG is negative for the TMP(∼ −30) and BEH(∼ −36), while for DEP(∼ +63) is positive. Hence, penetration into the membrane is easier for TMP and BEH than DEP. The authors suggest that, in agreement with experimental results, more cytotoxicity of these two ILs in comparison with CDEP is attributed to this behavior (Stolte et al., Citation2006; Weaver et al., Citation2013). The more negative free energy minimum of BEH can be attributed to the larger non-polar domains and less space crowding. These findings are supported by a final snapshot of ILs as shown in . Regarding the higher energy barrier in the bilayer center, passing across the membrane is somewhat more difficult for the BEH than TMP. The free energy profile of DEP is considerably different from that of BEH and TMP. As observed, the minimum free energy of DEP is found in a flat region around 2.25–3.50 nm from the bilayer center corresponding to the bulk water. Because of the larger contribution of polar/non-polar interactions, compared to BEH and TMP, it is more difficult for DEP to penetrate in the bilayer and as can be seen from the final snapshot in , it is located within the bulk water.
Figure 3. Free energy profiles for anionic species of ILs along the bilayer z-axis.
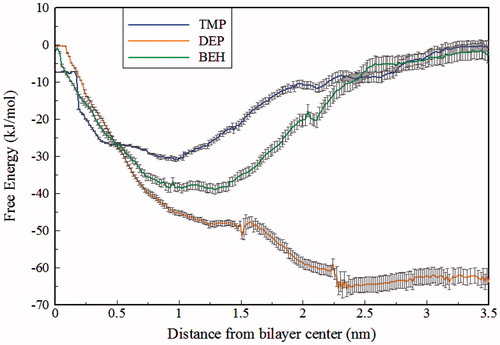
Figure 4. Snapshots of the final configuration of the DPPC lipids and water molecules and ILs, BEH (Green) (a), DEP (Orange) (b), TPM (Blue) (c). In all systems choline is as yellow.
Electrostatic potential
The electrostatic potential (EP) across the membrane is one of the most important properties of lipid bilayers that can affect the mechanism of a voltage-gated ion channel. One reason for the appearance of this potential is the orientation of water dipoles and lipid headgroup dipoles at the water-membrane interface (Mojumdar & Lyubartsev, Citation2010; Yousefpour et al., Citation2013). The EP can be influenced by the presence of ions and other charged components (Högberg & Lyubartsev, Citation2008). In , the total EP is presented for all four simulations. The values of the potential in the middle of the membrane and the maximum values of the potential are provided in . For the reference system, a positive dipole potential of around 600 mV in the center of bilayer was seen that is in agreement with the results of previous simulations conducted by GROMOS force field (Azizi & Koli, Citation2016; Patra et al., Citation2006; Wohlert & Edholm, Citation2004). Accurate determination of the EP is difficult and multiple sources have reported different values (Cevc, Citation1990; Clarke, Citation2001). A value of 510 mV has been reported for diphytanoylphosphatidylcholine (ester-DPhPC) membrane (Wang et al., Citation2006), that is close to our result in this work for the reference system. As seen in , in headgroup area, by adding 2 units of CTMP, CDEP and CBEH to the DPPC membrane causes approximately 90, 60 and 50 mV decreases in the EP compared to the reference system, respectively. Additional oxygen atoms in BEH and the presence of two more sulfur atoms in DEP structure compared to TMP leads to spatial crowding (near the headgroup) and interactions between the ionic head group are limited. Hence, interactions with polar headgroup and reduction of EP in the headgroup area for CTMP are more effective than CBEH and CDEP. In the hydrocarbon region, the decrease of EP for CTMP and CDEP is 80 and 40 mV relative to the reference system, respectively. However, the EP in the hydrocarbon region is not affected by CBEH.
Figure 5. Electrostatic potential of the simulated systems.
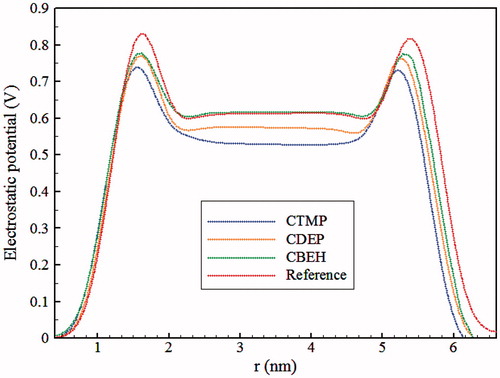
Table 2. The values of area per lipid (APL), thickness of bilayer (DB), lipid lateral diffusion coefficient (DL), anion species lateral diffusion coefficient (DA), electrostatic potential in the middle of membrane (ωMid), maximum electrostatic potential (ωMax) and DPPC head group P–N tilt angle (αP–N) of the simulated bilayer systems.
Lipid disorder quantities
The average area per lipid (APL) is one of the most fundamental characteristics of lipid bilayers, as many other membrane properties like membrane thickness, elasticity, lateral diffusion and order parameters are related to it. The APL was calculated in order to ensure the equilibration of the simulated systems. In this work, the average APL was calculated by dividing the result of multiplying the XY dimensions of the simulation box by the number of lipid molecules present in one leaflet of the bilayer. Figure S2 shows the APL for the fully equilibrated lipid systems and the average APL for different systems is shown in . As seen, the average value of APL for pure DPPC system is equivalent to 61.04 Å2 that is within the range of previously reported results (Berger et al., Citation1997; Marrink et al., Citation2001). It is also in good agreement with experimental amount of 62 Å2 at 323 K and 1 bar (Nagle et al., Citation1996). From the results of , it can be seen that membrane in the presence of ILs molecules has somewhat larger APL than pure DPPC system. As mentioned in Section 3.1, penetration of water after ILs insertion in headgroups in all of the systems is negligible. Hence, the differences in the APL of different systems may be caused by the positioning of ILs molecules in the membrane. shows that the increase in the APL for CTMP and CBEH is almost identical and larger than that of CDEP. This is consistent with the results of profiles of maximum density and potential of mean force, indicating that DEP does not penetrate the membrane and is located within the bulk water. To complement the average APL measurements, we computed the area probability distribution. As shown in Figure S3, by adding the ILs to DPPC bilayer, distributions of APL reaches higher amounts and for CTMP and CBEH including systems are approximately the same. The bilayer thickness (DB) of 3.87 nm for the reference system is in excellent agreement with the experimental value of 3.9 nm (Kučerka et al., Citation2011; Nagle et al., Citation1996). According to , by adding two molecules of every type of ILs, DB was decreased. For CTMP and CDEP systems, the decrements are quite noticeable (3.75 and 3.77 nm, respectively). It seems that the reduction of the EP in the hydrocarbon area, as discussed in the previous section, increases the attraction between adjacent DPPC units. Moreover, it has been well established that as the size of the system increases, DB values suffer from a smaller statistical error (Benedetto et al., Citation2015). The structural-transition during the simulation can be verified by order parameter analysis (Wohlert & Edholm, Citation2006). The order parameters related to CH bonds can be measured in NMR experiments and is defined by the following equation (Gullapalli et al., Citation2008):
(1)
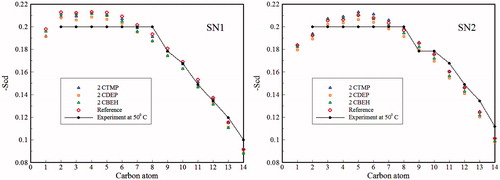
where θ is the angle between the molecular vector and Z-axis which is assumed to be parallel to the bilayer normal. The angular brackets denote an ensemble time average. For the reference bilayer, the observed order parameters agree well with previous simulations (Azizi & Koli, Citation2016; Berger et al., Citation1997) and experimental data (Petrache et al., Citation2000). As seen in , incorporation of ILs leads to small but noticeable effects on –Scd of both acyl chains. The overall effect is that –Scd of both acyl chains show the largest and the smallest amount of reduction for the CDEP and the CTMP containing systems, respectively. It seems that these differences are mainly originated from the two large sulfur atoms in the DEP structure. The tilt angle of the lipid molecules with respect to the bilayer normal can provide valuable information about the effects of ILs interpolation on the orientation of lipid chains. Figure S4 shows the distribution of the head group P–N vector of DPPC lipids relative to the bilayer normal at different kinds of ILs as well as for the reference system. An angle of 0° corresponds to the vector aligned parallel to the bilayer normal, while an angle of 90° corresponds to the vector perpendicular to the bilayer normal. The average angles between the P–N vector and the bilayer normal are given in . With regard to the data reported in and Figure S4, the presence of ILs has no significant effect on the orientation of the P–N vector in the bilayer. It seems that although ILs are ionic due to the presence of both anion and cation species, P–N vector was not affected by these kinds of molecules and average of the P–N angle is almost the same (∼90) as that of the reference system. Similar behavior of the P–N vector was also observed in the previous simulation for the reference system (Ulmschneider & Ulmschneider, Citation2009).
Figure 6. The deuterium order parameters for different carbon atoms in the SN-1 and SN-2 chains in presence three kinds of ILs.
Hydrogen bonds
All three types of ILs molecules are capable of establishing hydrogen bonds with water and different polar head group atoms of lipid and can also compete with lipids for hydrogen bonding with water. Hence, several types of hydrogen bonding was investigated and collected in . In this study, hydrogen bonds are determined by a maximum distance of 3.5 Å between acceptor and hydrogen and also the maximum angle of 30° between acceptor-hydrogen and hydrogen-donor vectors (van der Spoel et al., Citation2006). As shown in , the number of hydrogen bonds between DPPC and water, in the all of the ILs containing systems, are slightly more than the reference system. This result is in agreement with partial penetration of water into the membrane, which is discussed in Section 3.1. For all systems that included ILs, the formation of hydrogen bonds between Choline and DPPC are approximately the same, due to the location of Choline in all systems that is out of bilayer inside the water, as discussed in Section 3.1. Hydrogen bonding between anion species and water is notable in all ILs containing systems. BEH has four oxygen atoms in its structure, and TMP has two oxygen atoms, hence number of hydrogen bonds between water and BEH is double that of TMP. DEP has two oxygen atoms and two sulfur atoms. The number of hydrogen bonds between the oxygen of DEP and water was calculated to be 1.436, and is in agreement with that which is found in the experiment (Biswal, Citation2015). This observation indicates that the number of hydrogen bonds between sulfur atoms of DEP and water is negligible. Hence, due to the limited ability of sulfur atoms in the formation of hydrogen bonding and because of the presence of two oxygen atoms in two linkages within the DEP, less hydrogen bonding with water molecules is expected compared to TMP and BEH. The same observation for hydrogen bonding between anion species and Choline can be discussed in a similar way. It is interesting to note that while BEH and TMP remain within the membrane, DEP leaves this area and enters the aqueous environment. It seems that these effects are associated with the chemical structure and distribution of electrostatic charges within ILs and phospholipid units. Evaluations of e) show that, the hydrocarbon chains of phospholipid units, BEH and TMP that forms the main part of these molecules, is susceptible to dispersion interactions. Accordingly, the major contribution of intermolecular forces between phospholipids and units of BEH or TMP should be accounted for dispersion forces, and electrostatic interactions are negligible. The situation is different for DEP. Because of short hydrocarbon chains and a relatively significant electrical charge on the S atoms, this ion prefers to interact with its environment via electrostatic forces. Therefore, DEP prefers to be exposed to environments with high dielectric constant instead of staying amidst the hydrocarbon chains of phospholipids.
Table 3. Average number of different hydrogen bonds in the simulated systems.
Lateral diffusion
It has been confirmed that many cell signaling processes of the membrane, are controlled by lipid molecules diffusion, an indicator of lipid viscosity (Gullapalli et al., Citation2008). Therefore, calculations of the lateral diffusion coefficients for all ILs as well as lipids in all simulated systems were carried out. The lateral diffusion constant can be obtained from the Einstein relation (Mosaddeghi et al., Citation2012):
(2)
where
is a two-dimensional vector in the membrane plane defined by the center of mass of a molecule, 〈….〉 indicates averaging over all possible initial times t0 and molecules of the given type, and D stands for the lateral diffusion coefficient. The brackets in EquationEquation (2)
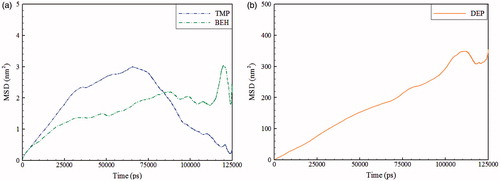
(2) define the time dependence of the lateral mean square displacement, which is shown in . shows the results obtained from the slope of the curves shown in . The selected time interval was 5–30 ns, where the MSD curves have the least fluctuations. It seems that the lateral diffusion coefficient of lipids, DL, are less affected by the presence of CDEP which is placed outside the membrane. CBEH and CTMP show opposite and quite different behaviors from that of CDEP. While the CBEH containing system shows an increment, in the CTMP containing system, DL is decreased compared to the reference system. Hydrocarbon portion of TMP has more branches than BEH, hence, TMP branches are wrapped within lipid chains and makes lipid movements more difficult. However, it seems that because of the strong interactions of BEH with head groups, due to the additional oxygen and less hindering of hydrocarbon portion, DL is increased compared to the reference system. As seen in , movement of BEH is less than TMP. This observation can be explained by additional oxygen in the BEH structure which leads to less mobility. However, DEP is located out of the bilayer (at the water) and, therefore, most movements are shown.
Figure 7. The mean square displacement of the anion species of ILs in the simulated systems, BEH and TMP (a), and DEP (b).
Conclusion
In order to compare the effects of different kinds of cytotoxic ILs on DPPC bilayer, a MD simulation study was conducted. Several analyses were performed to study the precise location of the anionic species inside the membrane and how it alters the structural and dynamic properties of the membrane. Both the profile of maximum density and potential of mean force of ILs in DPPC indicates that while TMP and BEH can remain in the middle of the bilayer lipid chains, DEP was not able to remain in the membrane and was located within the bulk water. Based on the findings, it seems that a direct relationship might be established between the dispersion/polar force ratio and the extent of penetration of anionic species in the membrane. The density of water molecules is zero in the middle of the bilayer, indicating that they are not able to cross the bilayer on the time scale of simulation. Regarding the higher energy barrier in the bilayer center, passing across the membrane is somewhat more difficult for the BEH than TMP. Analysis of the radial distribution function indicates that water penetration into the headgroup of DPPC is minor. In all of the ILs containing systems, the electrostatic potential in the headgroup, compared to the reference system, was decreased, while in the middle of the membrane it was only decreased for DEP and TMP. Compared with the reference system, in all of the ILs containing systems, distribution of APL goes toward higher values indicating a relationship between this property and cytotoxicity. The increase in the APL for CTMP and CBEH is almost identical and larger than that of CDEP. For all three Ils, the membrane thickness decreased compared to the reference system. Therefore, some relationships between cytotoxicity of ILs and reduction of the membrane thickness is expected. It seems that large sulfur atoms are responsible for the largest reduction in –SCD of the DEP-containing system. DL does not reveal a clearly identifiable trend, since it increases upon addition of CBEH and decreases in the CTMP case. A similar situation has been observed for other ILs containing syatems (Benedetto et al., Citation2015). Because of its special location in the bulk water, the lateral diffusion coefficient of lipids, DL, was less affected by the presences of CDEP than the other two ILs.
IMBC_1384859_Supplementary_Material.pdf
Download PDF (155.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Allen WJ, Lemkul JA, Bevan DR. 2009. GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J Comput Chem 30:1952–1958.
- Azizi K, Koli MG. 2016. Molecular dynamics simulations of Oxprenolol and Propranolol in a DPPC lipid bilayer. J Mol Graph Model 64:153–164.
- Benedetto A. 2017. Room-temperature ionic liquids meet bio-membranes: the state-of-the-art. Biophys Rev 1–12.
- Benedetto A, Ballone P. 2015. Room temperature ionic liquids meet biomolecules: a microscopic view of structure and dynamics. ACS Sustain Chem Eng 4:392–412.
- Benedetto A, Ballone P. 2016. Room temperature ionic liquids interacting with bio-molecules: an overview of experimental and computational studies. Philos Mag 96:870–894.
- Benedetto A, Bingham RJ, Ballone P. 2015. Structure and dynamics of POPC bilayers in water solutions of room temperature ionic liquids. J Chem Phys 142:124706.
- Benedetto A, Heinrich F, Gonzalez MA, Fragneto G, Watkins E, Ballone P. 2014. Structure and stability of phospholipid bilayers hydrated by a room-temperature ionic liquid/water solution: a neutron reflectometry study. J Phys Chem B 118:12192–12206.
- Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. 1981. Interaction models for water in relation to protein hydration. In Intermolecular forces. Berlin: Springer, 331–342.
- Berger O, Edholm O, Jähnig F. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J 72:2002.
- Biswal HS. 2015. Hydrogen bonds involving sulfur: new insights from ab initio calculations and gas phase laser spectroscopy. In Noncovalent forces. Berlin: Springer, 15–45.
- Boggara MB, Krishnamoorti R. 2010. Partitioning of nonsteroidal antiinflammatory drugs in lipid membranes: a molecular dynamics simulation study. Biophys J 98:586–595.
- Canzar S, El-Kebir M, Pool R, Elbassioni K, Malde AK, Mark AE, et al. 2013. Charge group partitioning in biomolecular simulation. J Comput Biol 20:188–198.
- Carter EB, Culver SL, Fox PA, Goode RD, Ntai I, Tickell MD, et al. 2004. Sweet success: ionic liquids derived from non-nutritive sweeteners. Chem Commun 6:630–631.
- Cevc G. 1990. Membrane electrostatics. Biochim Biophys Acta 1031:311–382.
- Clarke RJ. 2001. The dipole potential of phospholipid membranes and methods for its detection. Adv Colloid Interface Sci 89:263–281.
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. 1995. A smooth particle mesh ewald method. J Chem Phys 103:8577–8593.
- Fujita K, Forsyth M, MacFarlane DR, Reid RW, Elliott GD. 2006. Unexpected improvement in stability and utility of cytochrome c by solution in biocompatible ionic liquids. Biotechnol Bioeng 94:1209–1213.
- Fujita K, MacFarlane DR, Forsyth M. 2005. Protein solubilising and stabilising ionic liquids. Chem Commun 38:4804–4806.
- Gulati M, Grover M, Singh S, Singh M. 1998. Lipophilic drug derivatives in liposomes. Int J Pharm 165:129–168.
- Gullapalli RR, Demirel MC, Butler PJ. 2008. Molecular dynamics simulations of DiI-C18(3) in a DPPC lipid bilayer. Phys Chem Chem Phys 10:3548–3560.
- Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. 1997. LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18:1463–1472.
- Hess B, Kutzner C, Van Der Spoel D, Lindahl E. 2008. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447.
- Hockney RW, Goel SP, Eastwood JW. 1974. Quiet high-resolution computer models of a plasma. J Comput Phys 14:148–158.
- Högberg CJ, Lyubartsev AP. 2008. Effect of local anesthetic lidocaine on electrostatic properties of a lipid bilayer. Biophys J 94:525–531.
- Hough WL, Rogers RD. 2007. Ionic liquids then and now: from solvents to materials to active pharmaceutical ingredients. Bull Chem Soc Jpn 80:2262–2269.
- Hub JS, De Groot BL, Van Der Spoel D. 2010. G_wham—A free weighted histogram analysis implementation including robust error and autocorrelation estimates. J Chem Theory Comput 6:3713–3720.
- Hughes ZE, Mark AE, Mancera RL. 2012. Molecular dynamics simulations of the interactions of DMSO with DPPC and DOPC phospholipid membranes. J Phys Chem B 116:11911–11923.
- Humphrey W, Dalke A, Schulten K. 1996. “VMD–visual molecular dynamics”. J Mol Graph 14:33–38.
- Jastorff B, Mölter K, Behrend P, Bottin-Weber U, Filser J, Heimers A, et al. 2005. Progress in evaluation of risk potential of ionic liquids-basis for an eco-design of sustainable products. Green Chem 7:362–372.
- Jastorff B, Störmann R, Ranke J, Mölter K, Stock F, Oberheitmann B, et al. 2003. How hazardous are ionic liquids? Structure–activity relationships and biological testing as important elements for sustainability evaluation. Green Chem 5:136–142.
- Kandt C, Ash WL, Tieleman DP. 2007. Setting up and running molecular dynamics simulations of membrane proteins. Methods 41:475–488.
- Koziara KB, Stroet M, Malde AK, Mark AE. 2014. Testing and validation of the Automated Topology Builder (ATB) version 2.0: prediction of hydration free enthalpies. J Comput Aided Mol Des 28:221–233.
- Kučerka N, Nieh M-P, Katsaras J. 2011. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochim Biophys Acta Biomembr 1808:2761–2771.
- Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. J Comput Chem 13:1011–1021.
- Marrink SJ, Lindahl E, Edholm O, Mark AE. 2001. Simulation of the spontaneous aggregation of phospholipids into bilayers. J Am Chem Soc 123:8638–8639.
- Mojumdar EH, Lyubartsev AP. 2010. Molecular dynamics simulations of local anesthetic articaine in a lipid bilayer. Biophys Chem 153:27–35.
- Moniruzzaman M, Kamiya N, Goto M. 2010. Ionic liquid based microemulsion with pharmaceutically accepted components: formulation and potential applications. J Colloid Interface Sci 352:136–142.
- Mosaddeghi H, Alavi S, Kowsari MH, Najafi B. 2012. Simulations of structural and dynamic anisotropy in nano-confined water between parallel graphite plates. J Chem Phys 137:184703.
- Nagle JF, Zhang R, Tristram-Nagle S, Sun W, Petrache HI, Suter RM. 1996. X-ray structure determination of fully hydrated Lα, phase dipalmitoylphosphatidylcholine bilayers. Biophys J 70:1419.
- Ohno H. 2011. Electrochemical aspects of ionic liquids. Hoboken (NJ): John Wiley & Sons.
- Parrinello M, Rahman A. 1980. Crystal structure and pair potentials: a molecular-dynamics study. Phys Rev Lett 45:1196.
- Patra M, Salonen E, Terama E, Vattulainen I, Faller R, Lee BW, et al. 2006. Under the Influence of Alcohol: the effect of ethanol and methanol on lipid bilayers. Biophys J 90:1121–1135.
- Pernak J, Stefaniak F, Węglewski J. 2005. Phosphonium acesulfamate based ionic liquids. European J Org Chem 2005:650–652.
- Petkovic M, Ferguson JL, Gunaratne HQN, Ferreira R, Leitao MC, Seddon KR, et al. 2010. Novel biocompatible cholinium-based ionic liquids—toxicity and biodegradability. Green Chem 12:643–649.
- Petrache HI, Dodd SW, Brown MF. 2000. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys J 79:3172–3192.
- Snyman J. 2005. Practical mathematical optimization: an introduction to basic optimization theory and classical and new gradient-based algorithms. Berlin: Springer Science & Business Media.
- Stoimenovski J, MacFarlane DR, Bica K, Rogers RD. 2010. Crystalline vs. ionic liquid salt forms of active pharmaceutical ingredients: a position paper. Pharm Res 27:521–526.
- Stolte S, Arning J, Bottin-Weber U, Matzke M, Stock F, Thiele K, et al. 2006. Anion effects on cytotoxicity of ionic liquids. Green Chem 8:621–629.
- Tieleman DP, MacCallum JL, Ash WL, Kandt C, Xu Z, Monticelli L. 2006. Membrane protein simulations with a united-atom lipid and all-atom protein model: lipid-protein interactions, side chain transfer free energies and model proteins. J Phys Condens Matter 18:S1221.
- Torrie GM, Valleau JP. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation – Umbrella sampling. J Comput Phys 23:187–199.
- Ulmschneider JP, Ulmschneider MB. 2009. United atom lipid parameters for combination with the optimized potentials for liquid simulations all-atom force field. J Chem Theory Comput 5:1803–1813.
- van der Spoel D, van Maaren PJ, Larsson P, Timneanu N. 2006. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J Phys Chem B 110:4393–4398.
- Vrikkis RM, Fraser KJ, Fujita K, MacFarlane DR, Elliott GD. 2009. Biocompatible ionic liquids: a new approach for stabilizing proteins in liquid formulation. J Biomech Eng 131:74514.
- Wang L, Bose PS, Sigworth FJ. 2006. Using cryo-EM to measure the dipole potential of a lipid membrane. Proc Natl Acad Sci USA 103:18528–18533.
- Weaver KD, Kim HJ, Sun J, MacFarlane DR, Elliott GD. 2010. Cyto-toxicity and biocompatibility of a family of choline phosphate ionic liquids designed for pharmaceutical applications. Green Chem 12:507–513.
- Weaver KD, Van Vorst MP, Vijayaraghavan R, MacFarlane DR, Elliott GD. 2013. Interaction of choline salts with artificial biological membranes: DSC studies elucidating cellular interactions. Biochim Biophys Acta Biomember 1828:1856–1862.
- Welton T. 1999. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem Rev 99:2071–2084.
- Wennberg CL, Van Der Spoel D, Hub JS. 2012. Large influence of cholesterol on solute partitioning into lipid membranes. J Am Chem Soc 134:5351–5361.
- Wohlert J, Edholm O. 2004. The range and shielding of dipole-dipole interactions in phospholipid bilayers. Biophys J 87:2433–2445.
- Wohlert J, Edholm O. 2006. Dynamics in atomistic simulations of phospholipid membranes: nuclear magnetic resonance relaxation rates and lateral diffusion. J Chem Phys 125:204703.
- Xu W, Angell CA. 2003. Solvent-free electrolytes with aqueous solution-like conductivities. Science 302:422–425.
- Yoo B, Jing B, Jones SE, Lamberti GA, Zhu Y, Shah JK, Maginn EJ. 2016. Molecular mechanisms of ionic liquid cytotoxicity probed by an integrated experimental and computational approach. Sci Rep 6:19889.
- Yousefpour A, Amjad Iranagh S, Nademi Y, Modarress H. 2013. Molecular dynamics simulation of nonsteroidal antiinflammatory drugs, naproxen and relafen, in a lipid bilayer membrane. Int J Quantum Chem 113:1919–1930.