Abstract
Anion exchanger 1 (AE1, or Band 3) is an integral membrane glycoprotein found in erythrocytes, responsible for the electroneutral exchange of chloride and bicarbonate ions across the plasma membrane. Southeast Asian ovalocytosis (SAO) results from a nine-amino acid deletion in the first transmembrane segment (TM) of the AE1 protein that abolishes its transport function. The effects of the SAO deletion on: (1) the efficiency of integration of TM1 into the membrane, and (2) the precise positioning of TM1 relative to the membrane were investigated using scanning N-glycosylation mutagenesis in a cell-free transcription/translation system and in transfected HEK293 cells. AE1 or SAO constructs containing either the endogenous N-glycosylation site at Asn642 in extracellular loop 4 (EC4) or single N-glycosylation sites engineered into an expanded extracellular loop 1 (EC1) were used. N-glycosylation efficiency of EC1 in the SAO construct was significantly lower than that of the AE1 construct, indicating that the SAO deletion impairs membrane integration of TM1 and the translocation of EC1 across the membrane. Scanning N-glycosylation mapping of EC1 in the cell-free system and in transfected cells showed that the C-terminus of both AE1 and SAO TM1 were at the same position relative to the membrane. Thus, the SAO deletion is likely to cause a pulling-in of the polar amino acid sequence immediately N-terminal to the deletion into the lipid bilayer, allowing SAO TM1 that was inserted to assume a transmembrane disposition.
| Abbreviations | ||
| AE | = | anion exchanger |
| EC | = | extracellular loop |
| ER | = | endoplasmic reticulum |
| HEK | = | human embryonic kidney |
| NBC | = | sodium-bicarbonate cotransporter |
| OST | = | oligosaccharyltransferase |
| SAO | = | Southeast Asian ovalocytosis |
| TM | = | transmembrane segment |
Introduction
Anion exchanger 1 (AE1) belongs to the SLC4A family of transporters, which also includes AE2, AE3, and the sodium-bicarbonate co-transporters (NBCs) (Alper et al. [Citation2002], Romero & Boron [Citation1999]). AE1 is expressed in the erythrocyte membrane, where it carries out the electroneutral exchange of bicarbonate and chloride ions, a process important for increasing the carbon dioxide-carrying capacity of blood. Human AE1 protein consists of 911 amino acids, which forms two domains (Lux et al. [Citation1989], Reithmeier et al. [Citation1996], Tanner [Citation1993], Tanner et al. [Citation1988]). The N-terminal cytosolic domain provides anchor sites between the red cell membrane and the underlying cytoskeleton and binds cytosolic proteins such as hemoglobin and glycolytic enzymes (Low [Citation1986]). The C-terminal membrane-embedded domain contains 12–14 transmembrane segments (TMs) and is responsible for the anion transport function (Grinstein et al. [Citation1978], Groves & Tanner [Citation1992], Lepke et al. [Citation1976]). N-glycosylation occurs endogenously at Asn642 in the fourth EC loop within the membrane domains (Drickamer [Citation1978], Lux et al. [Citation1989], Tanner et al. [Citation1988]). The short cytosolic C-terminal tail contains a binding site for carbonic anhydrase II, giving rise to a transport metabolon (Reithmeier [Citation2001], Sterling et al. [Citation2001], Vince et al. [Citation2000], Vince & Reithmeier [Citation1998], Vince & Reithmeier [Citation2000]). The protein exists as dimers and tetramers in the red cell membrane (Casey & Reithmeier [Citation1991], Jennings [Citation1984]). An N-terminally truncated isoform of AE1 is expressed in the α-intercalated cells of the kidney where it mediates bicarbonate transport into the blood (Kollert-Jons et al. [Citation1993]).
A number of mutations have been identified in the AE1 gene that result in a change in anion transport and/or red cell morphology (Bruce & Tanner [Citation1999], Tanner [Citation2002]). One of these mutations, Southeast Asian ovalocytosis (SAO), produces a protein present in the erythrocyte membrane that is inactive in anion transport (Schofield et al. [Citation1992]). The condition is characterized by the presence of ovalocytic red cells in blood film (Liu et al. [Citation1990]) that are rigid compared to normal red cells (Mohandas et al. [Citation1992]). The molecular defect is a 27-bp in-frame deletion in exon 11 of the AE1 gene, corresponding to a nine-amino acid deletion (Ala400–Ala408 inclusive) at the boundary of the cytosolic domain and TM1 of the protein (Jarolim et al. [Citation1991], Mohandas et al. [Citation1992], Schofield et al. [Citation1992], Takeshima et al. [Citation1994]). The deletion is linked to a common polymorphism (Lys56Glu) in the cytosolic domain (Memphis I). Interestingly, the SAO deletion is found in regions endemic to malaria, and has been suggested to confer resistance to cerebral malaria (Genton et al. [Citation1995]). However, homozygosity of the condition may be embryonic lethal (Liu et al. [Citation1994]), and only heterozygotes have been found.
The effect of the SAO deletion on the overall structure of AE1 has been studied. AE1 SAO does not bind anion transport inhibitors such as stilbene disulfonates (Sarabia et al. [Citation1993], Schofield et al. [Citation1992]). The mutant protein does not possess the extended polylactosaminyl N-glycan found on normal AE1, and a higher proportion of tetramers are found in SAO red cells compared to normal red cells (Sarabia et al. [Citation1993]). While circular dichroism shows that there is no significant change in the helical content of AE1 SAO compared to normal AE1 (Moriyama et al. [Citation1992], Sarabia et al. [Citation1993]), calorimetric data suggests that the membrane domain of AE1 SAO is denatured (Moriyama et al. [Citation1992]). Solution-state NMR experiments with synthetic peptides encompassing TM1 of AE1 (residues 389 to 430) in 1:1 (v/v) chloroform/methanol showed that the structure of TM1 is altered in AE1 SAO. Whereas the normal peptide assumes a structure with three helical regions, the SAO peptide contains a single stable helix due to the deletion of a turn at Pro403 (Chambers et al. [Citation1999]). The release kinetics of H2DIDS (4, 4′-diisothiocyano-2, 2′-dihydrostilbenedisulfonate) from normal AE1 in SAO red cell membranes is altered, indicating the conformation of the normal subunit is affected by the neighboring SAO subunit (Salhany & Schopfer [Citation1996]). Recent studies have shown that AE1 SAO has a dominant structural effect on the normal subunit in a hetero-oligomer, with altered patterns of proteolytic cleavage and chemical modification throughout the membrane domain (Kuma et al. [Citation2002]). These observations agree with the more than 50% reduction in sulfate transport activity in SAO red cells (Schofield et al. [Citation1992]) despite the fact that only about half of the AE1 proteins in SAO red cells are the mutant form (Sarabia et al. [Citation1993]). Cell-free translation and N-glycosylation assay have shown that TMs 3 and 4 in an SAO construct encompassing TMs 1 to 4 had the correct transmembrane orientation (Kanki et al. [Citation2003]). The hydrophobic length of TM1 is important for membrane integration, as inserting a trileucine sequence at the SAO deletion was sufficient to restore N-glycosylation of the following loop, and thus the membrane integration of TM1, to wild type level (Kanki et al. [Citation2003]).
Because of the misfolded state of AE1 SAO, crystallographic techniques may not be feasible for examining its structure. In this study, the effect of the SAO deletion on the integration and precise positioning of TM1 of AE1 in the membrane was examined by N-glycosylation techniques. Scanning N-glycosylation mutagenesis allows a determination of the position of a TM with respect to the membrane, since the active site of the oligosaccharyltransferase is fixed (Nilsson & von Heijne [Citation1993]) and the minimum glycosylation distance of a loop may be used as a ruler to infer the lumenal end position of the preceding or following TM (Popov et al. [Citation1997], Popov et al. [Citation1999]). Full-length AE1 constructs were used in order to more closely approximate the native protein. Results obtained from a cell-free translation system and transfected HEK293 cells showed that the SAO deletion significantly impairs ability of TM1 to act as a signal sequence and translocate EC1 across the ER membrane. The C-terminal position of TM1 relative to the membrane was however not altered by the SAO deletion, suggesting that the sequence N-terminal to the deletion may be able to compensate for the helical length of the segment in the SAO mutant.
Materials and methods
The following is a list of materials used and their suppliers: pcDNA3 (Invitrogen, San Diego, CA, USA); QuikChange™ site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA); Transformer™ site-directed mutagenesis kit (Clontech, Palo Alto, CA, USA); TNT® T7 Quick Coupled Transcription/Translation System, canine pancreatic microsomal membranes (Promega, Madison, WI, USA); Dulbecco's modified Eagle medium (DMEM), calf serum, penicillin and streptomycin (Gibco BRL, Burlington, ON, Canada); EasyTag™ L-[35S]-Methionine, EasyTag™ EXPRE35S35S protein labeling mix (Perkin Elmer Life Sciences, Mississauga, ON, Canada); concanavalin A (Sigma-Aldrich Canada Ltd., Oakville, ON, Canada); endoglycosidase H (New England Biolabs, Mississauga, ON, Canada); protein G-Sepharose (Amersham Pharmacia Biotech, Baie d'Urfé, QC, Canada); dropping bottle with 20–30 µm filter unit (Wheaton, Millville, NJ, USA); mutagenic primers (ACGT Corp., Toronto, ON, Canada).
Site-directed mutagenesis
The entire coding sequence for normal human AE1 was inserted into the XhoI and BamHI sites of pcDNA3 (Popov et al. [Citation1999]). The construction of AE1 SAO and the EC4 insertion constructs AE1 N642′D N642D, AE1 N642′ N642D and AE1 N642′D N642, (or AE1 1 − /4−, AE1 1 + /4− and AE1 1 − /4+, respectively) have been described previously (Popov et al. [Citation1997]; Popov et al. [Citation1999]; Popov et al. [Citation2000]). The corresponding SAO EC4 insertion constructs SAO 1 − /4−, SAO 1 + /4−, and SAO 1 − /4+ were created similarly by insertion of a 34-amino acid sequence derived from EC4 (residues 626 to 659) into the AE1 SAO template at an XbaI site in EC1 (as described in Popov et al. ([Citation1997]) and subsequent mutation (Asn→Asp) of the endogenous or novel N-glycosylation site. The scanning N-glycosylation mutants in EC1 were constructed using Transformer™ or QuikChange™ mutagenesis kits, using AE1 1 − /4− or SAO 1 − /4− as templates. Sequencing of the constructs was carried out by the ACGT Corp. See for an illustration of the insertion constructs and for a complete list of constructs.
Figure 1. Folding model of human AE1 and expansion of extracellular loop 1 by insertion. Human AE1 is predicted to span the lipid bilayer 12–14 times, with both N- and C-termini cytosolic, and contains an endogenous N-glycosylation site at Asn642. The insertion constructs used in this study were created by duplicating the EC4 segment from residue 626 to 659 and inserting it after residue 428 in EC1. The duplicated residues are denoted by “ ‘ ”. The SAO mutation involves deletion of residues 400 to 408 in TM1 as indicated in the diagram.
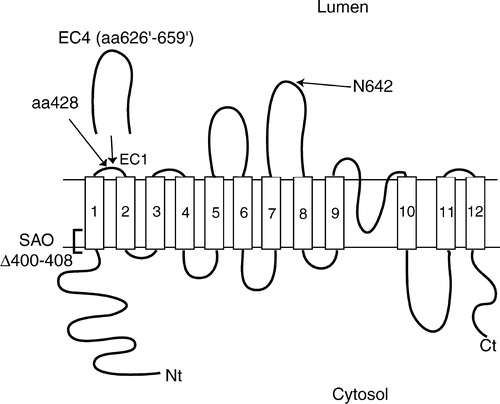
Table I. Constructs used in TM1 insertion study and scanning N-glycosylation mapping.
Cell-free transcription/translation of AE1 and SAO mutants
For each construct, 0.5 µg of plasmid DNA was transcribed and translated using TNT® T7 Quick Coupled transcription/translation system supplemented with canine pancreatic microsomes, in the presence of [35S]methionine to a total volume of 25 µL. The reactions were allowed to proceed for 1 h at 30°C. Samples were then extracted with 250 µL ice-cold 0.1 M Na2CO3, pH 11.5 and centrifuged at 16?000×g for 15 min to recover stripped microsomes. The translation products were solubilized in sample buffer and analyzed by concanavalin A lectin-shift SDS-PAGE in order to resolve N-glycosylated (high mannose) from unglycosylated protein (Popov et al. [Citation2000]), and autoradiography. Intensities of the bands were quantified using NIH Imager, and multiple exposures of the autoradiographs were obtained to ensure band intensities were within linear range of measurement. Percentage N-glycosylation was calculated as intensity of upper (glycosylated) band divided by total intensity of upper and lower (unglycosylated) bands. Due to varying quality of the microsomes, variation in N-glycosylation efficiencies was observed from experiment to experiment. Thus percentage N-glycosylation was converted to relative N-glycosylation efficiencies, with the efficiency of AE1 in each experiment set to one. Statistical significance of relative N-glycosylation efficiencies was analyzed using Student's t-test (p<0.05).
Transient transfection and expression of AE1 mutants. HEK293 cells were transfected using the DEAE-dextran method (Lopata et al. [Citation1984]) with 2 µg of plasmid DNA per well of a 6-well plate, or by the calcium phosphate method (Graham & van der Eb [Citation1973]) with 1 µg of plasmid DNA per well. Cells were grown in DMEM supplemented with 10% (v/v) calf serum, 0.5% penicillin and 0.5% streptomycin under air/CO2 (19:1) at 37°C (Popov et al. [Citation1999]). One day post-transfection cells were labeled with EasyTag™ EXPRE35S35S protein labeling mix for 2 h at 37°C, then washed and lyzed with RIPA buffer (1% deoxycholate, 1% Triton X-100, 0.1% SDS, 0.15 M NaCl, 10 mM Tris·HCl, pH7.5, 1 mM EDTA). Immunoprecipitation of AE1 was carried out using a rabbit polyclonal antibody directed against a synthetic peptide corresponding to the C-terminal 16 residues of human AE1 (Popov et al. [Citation1999]) and protein G-Sepharose. Endoglycosidase H digestion was carried out in control samples solubilized in sample buffer, at room temperature for 30 min. Samples were then analyzed by concanavalin A lectin-shift SDS-PAGE (Popov et al. [Citation2000]) and autoradiography. Intensities of the bands were quantified using NIH Imager. Percentage N-glycosylation was calculated as intensity of upper (glycosylated) band divided by total intensity of upper and lower (unglycosylated) bands. Relative N-glycosylation efficiencies were calculated and statistical significance was analyzed using Student's t-test (p<0.05).
Results
SAO deletion significantly impairs TM1 integration in the full-length protein
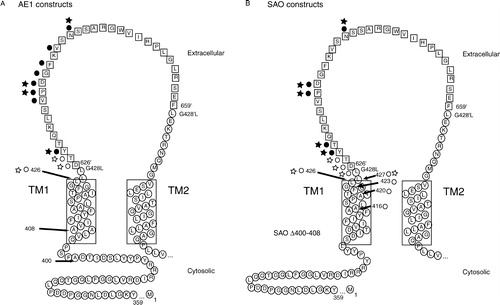
N-glycosylation is useful for identifying lumenally or extracellularly exposed regions of membrane proteins, since the modification requires accessibility to the active site of the oligosaccharyltransferase (OST), which is located in the ER lumen. The technique has been used to investigate the topology of many membrane proteins (reviewed in van Geest & Lolkema [Citation2000]), including AE1 (Popov et al. [Citation1999], Popov et al. [Citation1997]), the sodium/glucose cotransporter SGLT1 (Turk et al. [Citation1996]), glucose transporter Glut1 (Hresko et al. [Citation1994]), glucose-6-phosphatase (Ukaji et al. [Citation2002]), and the secretory Na+-K+-2Cl− cotransporter (Gerelsaikhan & Turner [Citation2000]). In order to assess the effect of the SAO deletion on TM1 integration into lipid bilayer and EC1 translocation into the ER lumen, the N-glycosylation efficiency of EC1 in a cell-free translation system was monitored. Since EC1 in native AE1 is only about 10 amino acids long and too short to be N-glycosylated (Landolt-Marticorena & Reithmeier [Citation1994], Popov et al. [Citation1997]), it was necessary to extend the loop. A 34-residue segment, consisting of residues 626 to 659, of EC4 of AE1 was duplicated and inserted after position 428 of EC1 to create the insertion constructs () (Popov et al. [Citation1997]). The inserted residues are denoted by the ‘ ′ ’ symbol. To mutate one or both N-glycosylation acceptor sites present, the Asn residue at positions 642′ (EC1) and/or 642 (EC4) was substituted with Asp by site-directed mutagenesis. Constructs with no N-glycosylation acceptor site at either loop (1 − /4 − ) or with a single N-glycosylation site at either EC1 (1 + /4 − ) or EC4 (1 − /4 + ) were created ().
The various constructs were transcribed and translated using a rabbit reticulocyte lysate supplemented with canine pancreatic microsomes in the presence of [35S]methionine. The microsomal membranes were then alkaline-stripped to remove any non-integrated proteins, solubilized with sample buffer and analyzed by lectin-shift SDS-PAGE (). The presence of a layer of concanavalin A in the upper separating layer of the polyacrylamide gel allowed better separation of the high mannose-glycosylated from the unglycosylated polypeptides (Popov et al. [Citation1999], Popov et al. [Citation2000]). The upper band represents N-glycosylated AE1, which disappears upon inclusion of a competitive tripeptide glycosylation inhibitor (Asn-Tyr-Thr) in the transcription/translation mix (Popov et al. [Citation1997]; Popov et al. [Citation1999]; Popov et al. [Citation2000], and data not shown). The lower band is unglycosylated AE1. The N-glycosylation efficiency of AE1 at N642 in the cell-free translation system averaged 44% and was normalized to one to account for the variability observed with different batches of microsomes. The N-glycosylation efficiency of AE1 SAO (39%) at the endogenous site (Asn642) was not significantly different from of AE1 (). Thus, the SAO deletion in TM1 does not affect the N-glycosylation efficiency of the N-glycosylation site located downstream in EC4. The 1 − /4− constructs, with loop insertion in EC1 but no N-glycosylation acceptor site at either EC1 or EC4, were not N-glycosylated as expected (). The N-glycosylation efficiency of AE1 1 − /4+ (38%) at Asn642 is not statistically different from AE1 (44%), indicating that the presence of the EC1 insertion does not alter the topology of EC4. The N-glycosylation of SAO 1 − /4+, with the insertion in EC1 and the endogenous N-glycosylation site in EC4, was slightly lower (29%) but was not statistically different from AE1 1 − /4+ (38%) (). Taken together, these results indicate that EC4 could translocate into the ER lumen despite the SAO deletion, suggesting that the topology of TM7–8 was not altered. In contrast, SAO 1 + /4−, with an N-glycosylation acceptor site in the expanded EC1 but not in EC4, had an N-glycosylation efficiency at about half that of AE1 1 + /4− (17% versus 36%), which was a statistically significant difference (). This indicates that in the context of the full-length protein, the SAO deletion exerted a deleterious effect on membrane integration of TM1 and translocation of EC1.
Figure 2. Membrane integration of TM1 in full-length AE1 and SAO constructs in cell-free translation system. (A) Plasmid DNA of various AE1 and SAO constructs were transcribed and translated in a cell-free system of rabbit reticulocyte lysate supplemented with canine pancreatic microsomes, in the presence of [35S]methionine. The microsomal membranes were then extracted with 0.1 M Na2CO3, pH 11.5 to remove non-integrated proteins. The samples were solubilized in sample buffer and analyzed by lectin-shift SDS-PAGE and autoradiography. Closed circle indicates position of N-glycosylated AE1 and open circle indicates position of unglycosylated AE1. Percentage N-glycosylation of each construct is shown underneath each lane (n=5). (B) Relative N-glycosylation efficiencies were calculated by dividing the intensity of the upper (glycosylated) band by the total intensity of the upper and lower (unglycosylated) bands. Shown are the results of five experiments (mean±SD), with the N-glycosylation efficiency of AE1 set to one in each experiment.
![Figure 2. Membrane integration of TM1 in full-length AE1 and SAO constructs in cell-free translation system. (A) Plasmid DNA of various AE1 and SAO constructs were transcribed and translated in a cell-free system of rabbit reticulocyte lysate supplemented with canine pancreatic microsomes, in the presence of [35S]methionine. The microsomal membranes were then extracted with 0.1 M Na2CO3, pH 11.5 to remove non-integrated proteins. The samples were solubilized in sample buffer and analyzed by lectin-shift SDS-PAGE and autoradiography. Closed circle indicates position of N-glycosylated AE1 and open circle indicates position of unglycosylated AE1. Percentage N-glycosylation of each construct is shown underneath each lane (n=5). (B) Relative N-glycosylation efficiencies were calculated by dividing the intensity of the upper (glycosylated) band by the total intensity of the upper and lower (unglycosylated) bands. Shown are the results of five experiments (mean±SD), with the N-glycosylation efficiency of AE1 set to one in each experiment.](/cms/asset/3218de57-fb5f-4ad2-a7a0-82ce257adb67/imbc_a_109294_uf0002_b.jpg)
Scanning N-glycosylation mapping of C-terminal end of TM1 in AE1 and SAO constructs in cell-free translation system
Because the SAO deletion involves residues in TM1, the exact disposition of TM1 in the membrane may be altered. To investigate the effect of the deletion on the position of TM1 in the membrane, scanning N-glycosylation mapping technique was used to determine the position of C-terminal ends of TM1 in both normal and SAO proteins relative to the membrane. The active site of OST has been determined to be about 30–40Å from the membrane surface of the ER lumen (Nilsson & von Heijne [Citation1993]). Thus, the minimum distance requirement for N-glycosylation is useful for determining the positioning of a TM segment in the lipid bilayer, and has been used to map the lumenal ends of TM7 and TM8 in AE1 (Popov et al. [Citation1997], Popov et al. [Citation1999]). An acceptor has to be at least 12 amino acids away from the preceding TM and 14 amino acids away from the following TM to be N-glycosylated (Nilsson & von Heijne [Citation1993]), which has been referred to as the ‘12 + 14 rule’ (Popov et al. [Citation1997]).
Here we employed scanning N-glycosylation mutagenesis to map the lumenal end of TM1 in normal and SAO proteins. As explained above, insertion constructs were required in this experiment since endogenous EC1 was too short to be N-glycosylated (; ) (Landolt-Marticorena & Reithmeier [Citation1994], Popov et al. [Citation1997]). Each construct contained a single consensus N-glycosylation site (Asn-Xaa-Ser/Thr) at various positions along the expanded EC1 with the N-glycosylation acceptor site at EC4 mutated (). When expressed in the cell-free translation system, a sharp boundary for efficient N-glycosylation was observed (). Positions close to the end of TM1 were not N-glycosylated. Several positions in the SAO constructs, for example N626′ and N627′, had a faint glycosylated band on the autoradiographs, visible to the unaided eye. These were difficult to quantify accurately relative to the high intensity of the lower unglycosylated band, and did not register on densitometry. We estimate the glycosylated bands represent less than 5% of the total signal. The first position that could be efficiently N-glycosylated was N628′ in both normal () and SAO () proteins, the third residue in the inserted segment. This indicates a similar positioning of the C-terminal end of TM1 in the membrane in both normal and SAO proteins. Consistent with the membrane insertion experiment (), all the AE1 constructs have higher N-glycosylation efficiencies than SAO constructs, again showing that the SAO deletion results in less efficient integration of TM1.
Figure 3. Scanning N-glycosylation mapping of TM1 in AE1 and SAO constructs in cell-free translation system. Insertion constructs with N-glycosylation acceptor sites at various positions along EC1 were subjected to cell-free translation in the presence of microsomal membranes and [35S]methionine. The samples were alkaline extracted, solubilized in sample buffer, and analyzed by lectin-shift SDS-PAGE. Representative autoradiographs are shown. The positions of glycosylated and unglycosylated proteins are indicated by closed and open circles, respectively. (A) AE1 constructs; (B) SAO constructs. Percentage N-glycosylation of each construct is shown underneath each lane (n=4 where standard deviation is included). N-glycosylation efficiencies were calculated by dividing the intensity of the upper (glycosylated) band by the total intensity of upper and lower (unglycosylated) bands.
![Figure 3. Scanning N-glycosylation mapping of TM1 in AE1 and SAO constructs in cell-free translation system. Insertion constructs with N-glycosylation acceptor sites at various positions along EC1 were subjected to cell-free translation in the presence of microsomal membranes and [35S]methionine. The samples were alkaline extracted, solubilized in sample buffer, and analyzed by lectin-shift SDS-PAGE. Representative autoradiographs are shown. The positions of glycosylated and unglycosylated proteins are indicated by closed and open circles, respectively. (A) AE1 constructs; (B) SAO constructs. Percentage N-glycosylation of each construct is shown underneath each lane (n=4 where standard deviation is included). N-glycosylation efficiencies were calculated by dividing the intensity of the upper (glycosylated) band by the total intensity of upper and lower (unglycosylated) bands.](/cms/asset/78164d54-ff0a-45b6-b6fb-b690efed33b0/imbc_a_109294_uf0003_b.jpg)
Scanning N-glycosylation mapping in transfected HEK 293 cells
The same scanning N-glycosylation constructs were expressed by transient transfection in HEK293 cells as a comparison to results obtained using the cell-free system. Twenty-four hours post-transfection the cells were labeled with [35S]methionine for two hours, solubilized, and AE1 and SAO proteins were immunoprecipitated by an anti-AE1 Ct antibody. The samples were analyzed by lectin-shift SDS-PAGE. A representative autoradiograph is shown in . The lower bands consist of unglycosylated polypeptides, likely due to the overexpression of AE1 in transiently transfected HEK cells and inefficient N-glycosylation. The upper bands represent N-glycosylated polypeptides that could be shifted by endoglycosidase H treatment (, top), indicating a high-mannose oligosaccharide structure on the endogenous acceptor site on AE1 in transfected HEK cells, consistent with previous observations (Li et al. [Citation2000]). The upper band of AE1 1 + /4− was also susceptible to endoglycosidase H digestion (, top), confirming the high-mannose N-glycan structure present on the expanded EC1 (Popov et al. [Citation1999]). AE1 and AE1 SAO (no loop expansion) were glycosylated at 57% and 48% respectively (n=5). Although AE1 SAO is less efficiently glycosylated than AE1, the observed difference was not statistically significant. The glycosylation efficiency of insertion mutant SAO 1 − /4+ (39%) was also lower than AE1 1 − /4+ (56%) (n=4). Despite the differences in glycosylation level, these results indicate that in cells, EC4 of both proteins were translocated into the ER lumen. 1 − /4− constructs were not N-glycosylated, as expected. Both 1 + /4− constructs were N-glycosylated, indicating that, as in the cell-free system, TM1 in both normal and SAO context was able to integrate into the lipid bilayer. The N-glycosylation efficiency of SAO 1 + /4− (22%) was significantly lower than AE1 1 + /4− (45%) (n=8). This agrees with the results from the cell-free system that membrane integration of TM1 in transfected cells is also impaired in the SAO context. The residues close to the end of TM1 were not efficiently N-glycosylated (less than 10%), up to position N627′. The first position to be efficiently N-glycosylated was N628′ in both normal and SAO constructs, exactly the same as the findings from the cell-free system. This indicates that in both cell-free system and transfected HEK293 cells, the dispositions of normal and SAO TM1 in the membrane are identical.
Figure 4. Scanning N-glycosylation mapping of TM1 in AE1 and SAO constructs in transiently transfected HEK293 cells. HEK293 cells were transiently transfected with various AE1 and SAO constructs. Twenty-four hours post-transfection, cells were labeled with [35S]methionine for 2 h at 37°C, then lyzed in RIPA buffer. AE1 and SAO constructs were immunoprecipitated by an anti-C-terminal antibody. The samples were solubilized in sample buffer and analyzed by lectin-shift SDS-PAGE. Representative autoradiographs are shown. (A) AE1 constructs; (B) SAO constructs. C, undigested control; H, endoglycosidase H-treated sample. Closed and open circles indicate positions of glycosylated and unglycosylated proteins, respectively. Percentage N-glycosylation of each construct is shown underneath each lane (n=3–8 where standard deviation is included).
![Figure 4. Scanning N-glycosylation mapping of TM1 in AE1 and SAO constructs in transiently transfected HEK293 cells. HEK293 cells were transiently transfected with various AE1 and SAO constructs. Twenty-four hours post-transfection, cells were labeled with [35S]methionine for 2 h at 37°C, then lyzed in RIPA buffer. AE1 and SAO constructs were immunoprecipitated by an anti-C-terminal antibody. The samples were solubilized in sample buffer and analyzed by lectin-shift SDS-PAGE. Representative autoradiographs are shown. (A) AE1 constructs; (B) SAO constructs. C, undigested control; H, endoglycosidase H-treated sample. Closed and open circles indicate positions of glycosylated and unglycosylated proteins, respectively. Percentage N-glycosylation of each construct is shown underneath each lane (n=3–8 where standard deviation is included).](/cms/asset/9f2ca3bc-40ce-43db-80b3-6d2de0ab85d9/imbc_a_109294_uf0004_b.jpg)
Discussion
The effect of the nine-amino acid SAO deletion on membrane integration and topology of TM1 in human AE1 has been investigated by N-glycosylation in both cell-free system and transfected cells. Our investigation showed that the presence of the deletion results in an impairment of TM1 integration in the full-length protein. However, the relative position of the C-terminal end of TM1 with respect to the membrane bilayer is not altered due to the SAO deletion, in the case where TM1 manages to integrate.
Using a truncated construct consisting of TM1–4 and a glycosylation site in EC2, Kanki et al. ([Citation2003]), had shown that the SAO deletion does not affect the topology of TM3–4. Our work using the full-length AE1 protein extends the same observation to regions further downstream, showing that the SAO deletion does not grossly alter the topology of TM7–8. Comparison of the relative N-glycosylation efficiencies between AE1 and AE1 SAO constructs in the cell-free system indicates that translocation of EC4 into the microsomal lumen was not significantly affected by the SAO deletion. A similar observation is made in transfected HEK293 cells, with 57% and 48% of AE1 and AE1 SAO glycosylated, respectively. Our results suggest that EC4 of AE1 and AE1 SAO are accessible to the OST active site to a similar extent.
Our study also shows that SAO TM1 integrates into the membrane much less efficiently than AE1 TM1, as indicated by the reduction in EC1 N-glycosylation. Similar results were obtained by Chambers et al. ([Citation1999]) and Kanki et al. ([Citation2003]), who based their studies on various truncated constructs of the membrane domain. We note that the translocation efficiencies of SAO TM1 seem to vary with the sizes of the constructs. The glycosylation efficiency of our SAO 1 + /4− construct was approximately half of that of AE1 1 + /4− in both cell-free system () and in cells (). However, an SAO construct consisting of TM1–4 with a similar EC1 loop insertion in the study of Kanki et al. ([Citation2003]), only achieved 19% of that of the AE1 construct. The difference between our results and the results from Kanki et al. ([Citation2003]), could be due to the use of full-length versus truncated constructs.
The work of Nilsson and von Heijne ([Citation1993]) on E. coli leader peptidase expressed in a cell-free transcription/translation system had shown that there is a sharp threshold for N-glycosylation. They determined that only positions in a lumenal loop that are 12–14 residues away from the preceding and following TMs can be N-glycosylated, and estimated from these results that the active site of the oligosaccharyltransferase is 30–40 Å from the membrane (Nilsson & von Heijne [Citation1993]). The ‘12 + 14 rule’ used in previous studies in our lab to map the lumenal ends of TMs in AE1 by scanning N-glycosylation mutagenesis (Popov et al. [Citation1997], Popov et al. [Citation1999]) was based on these results. Although slight variations were seen, in all cases there was a sharp N-glycosylation boundary and reasonable agreement with the ‘12 + 14 rule’. AE1 TMs 7 and 8 () are clearly defined by a hydrophobic stretch of about 20 amino acids each, from Val604–Ile624 and Met663–Gln683, respectively (Reithmeier [Citation1996]). In scanning N-glycosylation of EC4 expressed in a cell-free system, it was found that the first positions to be glycosylated, counting from the lumenal end of TMs 7 and 8, were 637 and 648, respectively (Popov et al. [Citation1997]). These translate to 13 and 15 amino acids away from TMs 7 and 8, respectively, agreeing well with the ‘12 + 14 rule’. In vivo work shows a continuous stretch of N-glycosylation from positions 635 to 648 (Popov et al. [Citation1999]), 11 and 15 residues away from TMs 7 and 8, respectively. The difference observed between the in vitro and in vivo systems may reflect the differing efficiencies of the oligosaccharyltransferase. Scanning cysteine mutagenesis on AE1 (Tang et al. [Citation1998]) revealed a stretch of amino acids, from residues 664 to 683, in a hydrophobic environment. Using position 664 as the lumenal end of TM8 as suggested by these results (Tang et al. [Citation1998]), the first position to be glycosylated (648) (Popov et al. [Citation1999]) is 16 amino acids away from TM8. Scanning N-glycosylation mutagenesis of the region between TMs 9 and 10 () in vitro shows efficient glycosylation 13 and 15 amino acids away from the ends of the TMs (Popov et al. [Citation1997]). These results show that the ‘12 + 14 rule’ is useful for estimating the lumenal ends of TMs, with consideration of its limitation. Subsequent studies on constructs of E. coli leader peptidase (Nilsson et al. [Citation1994], Nilsson et al. [Citation1998]) indicated that the minimum N-glycosylation distance is dependent on the length of the TM preceding or following the acceptor site. Although the active site of the OST is at a fixed distance from the membrane surface (Nilsson & von Heijne [Citation1993]), depending on the length of the TM segment, the end residue of the helix may be various distances away from the membrane-water interface and thus, from the OST active site (Nilsson et al. [Citation1998]). This may account for the small differences seen between the mapping results for TMs 7–8 and TMs 9–10 (Popov et al. [Citation1997], Popov et al. [Citation1999]) and the ‘12 + 14 rule’. In our human AE1 folding model (Popov et al. [Citation1999]), TM1 is predicted to include residues Gln404 to Leu427, according to hydropathy analysis (Reithmeier [Citation1996]). A proline residue at 403 delineates the cytosolic end of TM1, a prediction supported by a solution NMR structure which shows a sharp bend separating the cytosolic amphipathic helix (Pro391–Ala401) and the transmembrane region (Pro403–Ala416 and Ser418–Phe423) (Chambers et al. [Citation1999]). We note that applying the ‘12 + 14 rule’ literally on our current results ( and ) would put the lumenal end of TM1 at Pro419 (). Depending on the length and tilt of TM1, however, different ending residues may be predicted. Nilsson et al. ([Citation1998]) found a roughly linear relationship between the length of the hydrophobic TM (n, number of leucine residues) and the minimum glycosylation distance, which ranges from 12.5 residues to 9.5 residues for 12 < n<23. Minimum glycosylation distances are estimated at 12.5 for n ≤ 12 and 9.5 for n ≥ 23 (Nilsson et al. [Citation1998]). Assuming an average hydrophobic length of 20 amino acids for TM1 would give a minimum glycosylation distance of 11 amino acids, and a glycosylation threshold at N628′ would place the lumenal end of TM1 at Ala 420. Assuming TM1 consists of the hydrophobic stretch of 24 amino acids (Gln404–Leu427) predicted by hydropathy analysis (Reithmeier [Citation1996]), with a minimum glycosylation distance of 9.5 (Nilsson et al. [Citation1998]) would place the glycosylation threshold at N633′/N634′, contradictory to our results ( and ). We cannot rule out the possibility that expanding EC1 by insertion may affect the positioning of TM1. Despite this caveat, scanning N-glycosylation mapping has shown that the lumenal ends of AE1 TM1 and SAO TM1 are very similar. Determining the precise placement of TM1 with respect to the membrane will require a high-resolution structure.
Figure 5. Summary of scanning N-glycosylation results. Folding models of TM1 to TM2 of (A) AE1 and (B) SAO EC1 insertion constructs. Inserted residues of EC4 are represented by squares. Results from N-glycosylation mapping are summarized using symbols of circles and stars, representing cell-free and transfected cells results, respectively. Open symbols indicate absence of N-glycosylation at the position; filled symbols indicate the position could be N-glycosylated.
As our results showed that the positioning of the lumenal end of SAO TM1 remains unaltered, the cytosolic sequence immediately N-terminal to the 9-aa deletion may be pulled into the lipid bilayer to compensate for the shortened helix. Assuming again that a minimum of 20 amino acids are required to span the hydrophobic core of the lipid bilayer in an α-helical conformation, the sequence Ser395-Asp399 could become the N-terminal ∼1.5 turns of TM1 in the SAO protein in our folding model (Popov et al. [Citation1999]). Compared to the deleted membrane-embedded sequence (AFSPQ), Ser395–Asp399 (SDITD) is less hydrophobic. The Kyte-Doolittle hydropathy score (Kyte & Doolittle [Citation1982]) for the 24-residue wild type TM1 (Gln404–Leu427; ) is 1.9 and for SAO TM1 (Ser395–Asp399, ΔAla400–Ala408, Val409–Leu427; ) is 1.4. Thus, SAO TM1 may not be as stably integrated in the membrane, when compared to normal TM1. This accounts for the decreased EC1 N-glycosylation efficiency in SAO 1 + /4− (). Indeed, Kanki et al. ([Citation2003]) found that lengthening the hydrophobic segment in SAO TM1 by a trileucine sequence restored efficient membrane integration of the TM (Kyte-Doolittle score = 2.2 assuming a 22-residue TM sequence of Leu3+Val409–Leu427). As well, very low levels of N-glycosylation (less than 10%) were detected in EC1 positions (N428, N626′, and N627′) preceding the threshold of N628′ in the SAO construct in several experiments in cell-free system and in transfected cells ( and ). This observation suggests that there could be a ‘slippage’ of SAO TM1 in the translocon, where a portion of the TM helix would extend beyond the membrane surface, at least during N-glycosylation, a co-translational process that occurs before the TM moves out of the translocon (Kiely et al. [Citation1976], Lingappa et al. [Citation1978]), since OST is associated with the translocon (Scheper et al. [Citation2003]). The majority of the SAO proteins with a membrane-integrated TM1, however, had a threshold at N628′. Membrane integration of TM2 is influenced by TM1. In the absence of a closely spaced TM1, TM2 integrates into the bilayer inefficiently (Groves & Tanner [Citation1999], Ota et al. [Citation1998], Ota et al. [Citation2000]). The hydrophobic length of TM1 is also critical for membrane integration of TM2, suggesting that the two TMs may interact (Ota et al. [Citation2000]). The failure of TM1 to integrate into the membrane would result in the scenario where both TM1 and TM 2 become exposed to the cytosol (, bottom). Even in the case where SAO TM1 integrates into the lipid bilayer, as indicated by the successful lumenal translocation and N-glycosylation of EC1 in our experiments, the shortened hydrophobic length of SAO TM1 would not be sufficient to support proper integration of TM2, or interaction of TM1 with TM2, and may result in the extrusion of TM2 into the lumenal space (, middle). In either case, the misfolding of TM1 and 2 causes widespread alteration in the folding of the SAO protein, affecting transport (Groves et al. [Citation1993], Schofield et al. [Citation1992]), inhibitor binding (Sarabia et al. [Citation1993], Schofield et al. [Citation1992]), and N-glycan processing (Sarabia et al. [Citation1993]).
Figure 6. Proposed membrane topologies of AE1 SAO. (A) Folding model of normal human AE1 with 12 transmembrane segments in the ER membrane. The endogenous N-glycosylation site is N642. EC1, extracellular loop 1. (B) Possible topologies of AE1 SAO. Top, Decreased hydrophobic length of SAO TM1 results in weakened interaction with TM2. Middle, Weakened interaction with TM2 may cause TMs 2 and 3 to translocate into the ER lumen. Bottom, SAO TM1 may fail to integrate into the membrane, leaving both TMs 1 and 2 on the cytosolic side of the membrane. The dark area in TM1 represents residues N-terminal to the SAO deletion, which may be drawn in to form part of TM1 in AE1 SAO.
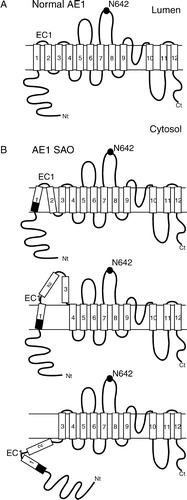
Our work using the scanning N-glycosylation technique has provided a clue on how the SAO deletion may affect the topology of TM1 and the surrounding sequences (). The N-glycosylation efficiency of SAO 1 + /4− is lower than that of AE1 1 + /4−, however SAO TM1 is able to support translocation of EC1 into the lumen to some extent, with the C-terminus positioning similar to that of normal TM1 (, top). However, even though the compensated helical length may allow SAO TM1 to integrate into the membrane, the shorter hydrophobic length of SAO TM1 is likely to cause an altered interaction with other TMs (, top). This may in turn result in extrusion of TMs 2 and 3, which have poor topogenic activity (Ota et al. [Citation1998], Ota et al. [Citation2000]), into the ER lumen (, middle). Unglycosylated proteins represent two possibilities: (1) SAO TM1 was integrated into membrane but EC1 was not N-glycosylated, or (2) TM1 was not integrated, leaving EC1 on the cytosolic side of the membrane. The first possibility is unlikely since the position of SAO TM1 was the same as normal TM1. In the second scenario, neither TM1 nor TM2 were inserted into the membrane and both were cytosolic (, bottom). Further experiments will help elucidate the effect of the SAO deletion on TMs 2 and 3.
This paper was first published online on prEview on 21 April 2005.
We would like to thank Jing Li and Mae Chiang for their assistance in site-directed mutagenesis.
Related Research Data
References
- Alper SL, Darman RB, Chernova MN, Dahl NK. The AE gene family of Cl/HCO3-exchangers. J Nephrol 2002; 15(Suppl 5)S41–53
- Bruce LJ, Tanner MJA. Erythroid band 3 variants and disease. Baillieres Best Pract Res Clin Haematol 1999; 12: 637–654
- Casey JR, Reithmeier RAF. Analysis of the oligomeric state of Band 3, the anion transport protein of the human erythrocyte membrane, by size exclusion high performance liquid chromatography. Oligomeric stability and origin of heterogeneity. J Biol Chem 1991; 266: 15726–15737
- Chambers EJ, Bloomberg GB, Ring SM, Tanner MJA. Structural studies on the effects of the deletion in the red cell anion exchanger (band 3, AE1) associated with South East Asian ovalocytosis. J Mol Biol 1999; 285: 1289–1307
- Drickamer LK. Orientation of the band 3 polypeptide from human erythrocyte membranes. Identification of NH2-terminal sequence and site of carbohydrate attachment. J Biol Chem 1978; 253: 7242–7248
- Genton B, al-Yaman F, Mgone CS, Alexander N, Paniu MM, Alpers MP, Mokela D. Ovalocytosis and cerebral malaria. Nature 1995; 378: 564–565
- Gerelsaikhan T, Turner RJ. Transmembrane topology of the secretory Na + -K + -2Cl- cotransporter NKCC1 studied by in vitro translation. J Biol Chem 2000; 275: 40471–40477
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973; 52: 456–467
- Grinstein S, Ship S, Rothstein A. Anion transport in relation to proteolytic dissection of band 3 protein. Biochim Biophys Acta 1978; 507: 294–304
- Groves JD, Ring SM, Schofield AE, Tanner MJA. The expression of the abnormal human red cell anion transporter from South-East Asian ovalocytes (band 3 SAO) in Xenopus oocytes. FEBS Lett 1993; 330: 186–190
- Groves JD, Tanner MJA. Glycophorin A facilitates the expression of human band 3-mediated anion transport in Xenopus oocytes. J Biol Chem 1992; 267: 22163–22170
- Groves JD, Tanner MJA. Topology studies with biosynthetic fragments identify interacting transmembrane regions of the human red-cell anion exchanger (band 3; AE1). Biochem J 1999; 344: 687–697
- Hresko RC, Kruse M, Strube M, Mueckler M. Topology of the Glut 1 glucose transporter deduced from glycosylation scanning mutagenesis. J Biol Chem 1994; 269: 20482–20488
- Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, Rubin HL, Zhai S, Sahr KE, Liu SC. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci USA 1991; 88: 11022–11026
- Jennings ML. Oligomeric structure and the anion transport function of human erythrocyte band 3 protein. J Membr Biol 1984; 80: 105–117
- Kanki T, Young MT, Sakaguchi M, Hamasaki N, Tanner MJA. The N-terminal region of the transmembrane domain of human erythrocyte band 3. Residues critical for membrane insertion and transport activity. J Biol Chem 2003; 278: 5564–5573
- Kiely ML, McKnight GS, Schimke RT. Studies on the attachment of carbohydrate to ovalbumin nascent chains in hen oviduct. J Biol Chem 1976; 251: 5490–5495
- Kollert-Jons A, Wagner S, Hubner S, Appelhans H, Drenckhahn D. Anion exchanger 1 in human kidney and oncocytoma differs from erythroid AE1 in its NH2 terminus. Am J Physiol 1993; 265: F813–821
- Kuma H, Abe Y, Askin D, Bruce LJ, Hamasaki T, Tanner MJA, Hamasaki N. Molecular basis and functional consequences of the dominant effects of the mutant band 3 on the structure of normal band 3 in southeast asian ovalocytosis. Biochemistry 2002; 41: 3311–3320
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol 1982; 157: 105–132
- Landolt-Marticorena C, Reithmeier RAF. Asparagine-linked oligosaccharides are localized to single extracytosolic segments in multi-span membrane glycoproteins. Biochem J 1994; 302: 253–260
- Lepke S, Fasold H, Pring M, Passow H. A study of the relationship between inhibition of anion exchange and binding to the red blood cell membrane of 4,4′-diisothiocyano stilbene-2,2′-disulfonic acid (DIDS) and its dihydro derivative (H2DIDS). J Membr Biol 1976; 29: 147–177
- Li J, Quilty J, Popov M, Reithmeier RAF. Processing of N-linked oligosaccharide depends on its location in the anion exchanger, AE1, membrane glycoprotein. Biochem J 2000; 349: 51–57
- Lingappa VR, Lingappa JR, Prasad R, Ebner KE, Blobel G. Coupled cell-free synthesis, segregation, and core glycosylation of a secretory protein. Proc Natl Acad Sci USA 1978; 75: 2338–2342
- Liu SC, Jarolim P, Rubin HL, Palek J, Amato D, Hassan K, Zaik M, Sapak P. The homozygous state for the band 3 protein mutation in Southeast Asian Ovalocytosis may be lethal. Blood 1994; 84: 3590–3591
- Liu SC, Zhai S, Palek J, Golan DE, Amato D, Hassan K, Nurse GT, Babona D, Coetzer T, Jarolim P, et al. Molecular defect of the band 3 protein in southeast Asian ovalocytosis. N Engl J Med 1990; 323: 1530–1538
- Lopata MA, Cleveland DW, Sollner-Webb B. High level transient expression of a chloramphenicol acetyl transferase gene by DEAE-dextran mediated DNA transfection coupled with a dimethyl sulfoxide or glycerol shock treatment. Nucleic Acids Res 1984; 12: 5707–5717
- Low PS. Structure and function of the cytoplasmic domain of band 3: Center of erythrocyte membrane-peripheral protein interactions. Biochim Biophys Acta 1986; 864: 145–167
- Lux SE, John KM, Kopito RR, Lodish HF. Cloning and characterization of band 3, the human erythrocyte anion-exchange protein (AE1). Proc Natl Acad Sci USA 1989; 86: 9089–9093
- Mohandas N, Winardi R, Knowles D, Leung A, Parra M, George E, Conboy J, Chasis J. Molecular basis for membrane rigidity of hereditary ovalocytosis. A novel mechanism involving the cytoplasmic domain of band 3. J Clin Invest 1992; 89: 686–692
- Moriyama R, Ideguchi H, Lombardo CR, Van Dort HM, Low PS. Structural and functional characterization of band 3 from Southeast Asian ovalocytes. J Biol Chem 1992; 267: 25792–25797
- Nilsson I, Saaf A, Whitley P, Gafvelin G, Waller C, von Heijne G. Proline-induced disruption of a transmembrane alpha-helix in its natural environment. J Mol Biol 1998; 284: 1165–1175
- Nilsson I, Whitley P, von Heijne G. The COOH-terminal ends of internal signal and signal-anchor sequences are positioned differently in the ER translocase. J Cell Biol 1994; 126: 1127–1132
- Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem 1993; 268: 5798–5801
- Ota K, Sakaguchi M, Hamasaki N, Mihara K. Assessment of topogenic functions of anticipated transmembrane segments of human band 3. J Biol Chem 1998; 273: 28286–28291
- Ota K, Sakaguchi M, Hamasaki N, Mihara K. Membrane integration of the second transmembrane segment of band 3 requires a closely apposed preceding signal-anchor sequence. J Biol Chem 2000; 275: 29743–29748
- Popov M, Li J, Reithmeier RAF. Transmembrane folding of the human erythrocyte anion exchanger (AE1, Band 3) determined by scanning and insertional N-glycosylation mutagenesis. Biochem J 1999; 339: 269–279
- Popov M, Li J, Reithmeier RAF. Resolution of glycoproteins by a lectin gel-shift assay. Anal Biochem 2000; 279: 90–95
- Popov M, Tam LY, Li J, Reithmeier RAF. Mapping the ends of transmembrane segments in a polytopic membrane protein. Scanning N-glycosylation mutagenesis of extracytosolic loops in the anion exchanger, band 3. J Biol Chem 1997; 272: 18325–18332
- Reithmeier RAF. A membrane metabolon linking carbonic anhydrase with chloride/bicarbonate anion exchangers. Blood Cells Mol Dis 2001; 27: 85–89
- Reithmeier RAF, Chan S. L, Popov M. Structure of the erythrocyte band 3 anion exchanger. Transport processes in eukaryotic and prokaryotic organisms, WN Konings, HR Kaback, JS Lolkema. Elsevier Science, Amsterdam 1996; 281–309
- Romero MF, Boron WF. Electrogenic Na + /HCO3-cotransporters: Cloning and physiology. Annu Rev Physiol 1999; 61: 699–723
- Salhany JM, Schopfer LM. Interactions between mutant and wild-type band 3 subunits in hereditary Southeast Asian ovalocytic red blood cell membranes. Biochemistry 1996; 35: 251–257
- Sarabia VE, Casey JR, Reithmeier RAF. Molecular characterization of the band 3 protein from Southeast Asian ovalocytes. J Biol Chem 1993; 268: 10676–10680
- Scheper W, Thaminy S, Kais S, Stagljar I, Romisch K. Coordination of N-glycosylation and protein translocation across the endoplasmic reticulum membrane by Sss1 protein. J Biol Chem 2003; 278: 37998–38003
- Schofield AE, Reardon DM, Tanner MJA. Defective anion transport activity of the abnormal band 3 in hereditary ovalocytic red blood cells. Nature 1992; 355: 836–838
- Sterling D, Reithmeier RAF, Casey JR. A transport metabolon. Functional interaction of carbonic anhydrase II and chloride/bicarbonate exchangers. J Biol Chem 2001; 276: 47886–47894
- Takeshima Y, Sofro AS, Suryantoro P, Narita N, Matsuo M. Twenty seven nucleotide deletion within exon 11 of the erythrocyte band 3 gene in Indonesian ovalocytosis. Jpn J Hum Genet 1994; 39: 181–185
- Tang XB, Fujinaga J, Kopito R, Casey JR. Topology of the region surrounding Glu681 of human AE1 protein, the erythrocyte anion exchanger. J Biol Chem 1998; 273: 22545–22553
- Tanner MJA. Molecular and cellular biology of the erythrocyte anion exchanger (AE1). Semin Hematol 1993; 30: 34–57
- Tanner MJA. Band 3 anion exchanger and its involvement in erythrocyte and kidney disorders. Curr Opin Hematol 2002; 9: 133–139
- Tanner MJA, Martin PG, High S. The complete amino acid sequence of the human erythrocyte membrane anion-transport protein deduced from the cDNA sequence. Biochem J 1988; 256: 703–712
- Turk E, Kerner CJ, Lostao MP, Wright EM. Membrane topology of the human Na + /glucose cotransporter SGLT1. J Biol Chem 1996; 271: 1925–1934
- Ukaji K, Ariyoshi N, Sakaguchi M, Hamasaki N, Mihara K. Membrane topogenesis of the three amino-terminal transmembrane segments of glucose-6-phosphatase on endoplasmic reticulum. Biochem Biophys Res Commun 2002; 292: 153–160
- van Geest M, Lolkema JS. Membrane topology and insertion of membrane proteins: Search for topogenic signals. Microbiol Mol Biol Rev 2000; 64: 13–33
- Vince JW, Carlsson U, Reithmeier RAF. Localization of the Cl − /HCO3-anion exchanger binding site to the amino-terminal region of carbonic anhydrase II. Biochemistry 2000; 39: 13344–13349
- Vince JW, Reithmeier RAF. Carbonic anhydrase II binds to the carboxyl terminus of human band 3, the erythrocyte C1 − /HCO3-exchanger. J Biol Chem 1998; 273: 28430–28437
- Vince JW, Reithmeier RAF. Identification of the carbonic anhydrase II binding site in the Cl(−)/HCO(3)(−) anion exchanger AE1. Biochemistry 2000; 39: 5527–5533