Abstract
Haemolysis is usually episodic in glucose-6-phosphate dehydrogenase (G6PD) deficiency, often triggered by a period of oxidative stress. In the present work, we investigate a possible biochemical mechanism underlying the enhanced susceptibility of G6PD deficient red blood cells (RBC) to oxidative stress. We analysed eight male subjects with Mediterranean glucose-6P-dehydrogenase deficiency (G6PDd), class II, for their ability in phosphorylating erythrocyte membrane band 3 following oxidative and osmotic stress. Our findings show that this sensitivity is connected to an early membrane band 3 Tyr-phosphorylation in the presence of diamide. However, since both Syk, and Lyn kinases, and SHP-2 phosphatase, mostly implicated in the band 3 P-Tyr level regulation, are alike in content and activity in normal and patient erythrocytes, an alteration in the membrane organization is likely the cause of the anomalous response to the oxidant. We report, in fact, that hypertonic-induced morphological change in G6PDd erythrocyte induces a higher membrane band 3 Tyr-phosphorylation, suggesting a pre-existing membrane alteration, likely due to the chronic lowering of the redox systems in patients. We also report that 1-chloro-2,4-dinitrobenzene-pre-treatment of normal red cells can alter the normal protein–protein and protein–membrane interaction under hypertonic rather than oxidative stress, thus partially resembling the response in patients, and that RBC may utilize a wider range of redox defence, under oxidative conditions, including, but not exclusively, NADPH and glutathione. On the whole, these results would encourage a different approach to the evaluation of the effects of pharmacological administration to patients, giving more attention to the possible drug-induced membrane alteration evidenced by the abnormal band 3 Tyr-phosphorylation.
Introduction
Glucose-6-phosphate Dehydrogenase deficiency (G6PDd) is a common chronic metabolic disorder inherited as an X-linked genetic trait. Glucose-6-phosphate dehydrogenase (G6PD) catalyses the first step of the hexose monophosphate (HMP) shunt. This pathway involves the conversion of glucose into pentose phosphate sugars and also provides reducing power in the form of NADPH. The reduced coenzyme that is produced is used in the reduction of toxic peroxides (R–O–OH) via glutathione (Lee et al. [Citation1993]). In the absence of intracellular organelles, the red blood cell (RBC) capacity of reducing oxidized proteins depends solely on the penthose pathway for the generation of NADPH. G6PD deficiency is mainly asymptomatic but deficient subjects are at risk of severe acute haemolytic anaemia crises following the ingestion of particular drugs, during some infections and, notably, after eating fava beans (favism). Haemolysis of G6PDd RBC results from an increased susceptibility to oxidative stress, as subjects are unable to reduce NADP+ to NADPH at a normal rate. The major function of NADPH produced in RBC is to maintain glutathione in its reduced form (GSH). The latter compound plays a vital role in detoxification by reacting with hydrogen peroxide and organic peroxidases, and by maintaining the cysteine residues of haemoglobin and other red blood cell proteins in the reduced state (Jollow & McMillan [Citation2001]). Oxidative treatment of normal human erythrocytes with pervanadate and diamide acting as PTPase inhibitors induces an increase in protein Tyr-phosphorylation (Harrison et al. [Citation1994]; Zipser et al. [Citation1997]; Brunati et al. [Citation2000]) mostly involving four tyrosine residues of band 3 protein (Yannoukakos et al. [Citation1991]; Brunati et al. [Citation2000]) in a two step mechanism. Syk tyrosine kinase, belonging to the Syk family, triggers the so called ‘primary phosphorylation’ of Tyr 8 and 21, which, once phosphorylated, acts as docking sites for the Lyn tyrosine kinase, belonging to the Src family, which catalyses the ‘secondary phosphorylation’ of the Tyr 359 and 904 (Brunati et al. [Citation2000]). P-Tyr-359 is, therefore, recognized by SH2-domain of SHP-2, a protein tyrosine phosphatase which, when recruited to band 3, dephosphorylates Tyr-residues 8, 21 and 904, and thus contributes to the restoration of the normal erythrocyte band 3 Tyr-phosphorylation level (Bordin et al. [Citation2002]). Alteration in P-Tyr-protein content has been demonstrated to affect the cell's physiological status. From the regulation of glycolysis, through the release of glycolitic enzymes from the membrane band 3 following phosphorylation of its cytoplasmic domain (Low et al. [Citation1987]; Harrison et al. [Citation1991]; Low et al. [Citation1993]), to the alteration of RBC shape (Bordin et al. [Citation1995]) and volume (Musch et al. [Citation1991]), and membrane transport (Musch et al. [Citation1991]; De Franceschi et al. [Citation1997]), band 3 Tyr-phosphorylation is thought to be involved in regulating cell functions.
The aim of this work was to compare the behaviour of normal and G6PD deficient erythrocytes under oxidative or hypertonic stress, both known to induce Tyr-phosphorylation principally of band 3 (Harrison et al. [Citation1994]; Zipser et al. [Citation1997]; Minetti et al. [Citation1998]; Brunati et al. [Citation2000]). Our results show a higher sensitivity of G6PD deficient erythrocytes not only, as expected, towards oxidative stress, but also to hyperosmotic conditions, evidenced by a higher band 3 Tyr-phosphorylation rate. No alterations were observed in protein kinase and phosphatase content, or in total protein content. This work also reports that 1-chloro-2,4-dinitrobenzene pre-treatment of normal erythrocytes induces band 3 Tyr-phosphorylation in response to hypertonic stress, similar to that evidenced in patients, while when cells are exposed to oxidative stress neither GSH nor NADPH alone account for the alteration found in G6PD deficiency.
Materials and methods
Materials
Epiandrosterone, 1-chloro-2,4-dinitrobenzene (CDNB), anti-P-Tyr and anti-Syk monoclonal antibodies were purchased from ICN Biotecnology (Irvine CA) and Upstate (Lake Placid, NY), respectively. Rabbit anti-SHP-2 (C-18) polyclonal and mouse anti-Lyn monoclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Protease inhibitor cocktail was obtained from Calbiochem (Darmstadt, Germany). γ[32P] adenosine 5′-triphosphate (ATP) was purchased from Amersham Pharmacia Biotech (Little Chalfont, UK).
Methods
Isolation of human erythrocytes
Human erythrocytes were prepared, as previously described (Brunati et al. [Citation2000]), from fresh blood collected from healthy people and from eight patients with Mediterranean G6PDd, class II according to the WHO directives (Betke et al. [Citation1967]). Patients were not assuming any medications and did not show oxidative stress, and had normal bilirubin levels (0.2–1.2 mg/dL) (performed on Hitachi 747 analyzer). Reticulocyte counts (1.1±0.4 vs. 1.2±0.5 of normal donors, expressed as % RBC) and haemoglobin determination (12.8±0.4 vs. 12.5±1.6 gr/dL) (performed by an automated analyser Sismex X6 2100, Dasit) showed no significant difference between two groups. G6PD residual activity in the red cells was 5–10% as measured spectrophotometrically at 340 nm using a SIGMA diagnostic kit (Sigma-Aldrich, Mi, Italy).
Treatment of erythrocytes
Packed cells (50 µL), prepared as described above, were resuspended (at 20% hematocrit) in an isosmotic buffer A (20 mmol L−1 Tris-HCl, pH 7.5, 125 mmol L−1 NaCl, 10 mmol L−1 KCL, 1 mmol L−1 MgCl2, 100 µg/mL streptomycin, 25 µg/mL chloramphenicol, 50 mmol L−1 glucose and 1 mmol L−1 adenosine) or a hyperosmotic buffer (buffer A containing 700 mmol L−1 sorbitol), centrifuged for 3′ at 3000 rpm discarding supernatant (to balance the cells osmotically), immediately resuspended in 200 µL of respective buffers, and incubated for 30 min at 35°C in the presence or absence of 0.3 mmol L−1 diamide. After incubation, each sample was centrifuged and the packed cells were subjected to haemolysis in 1.5 mL of hypotonic buffer containing 5 mmol L−1 sodium phosphate, pH 8; 0.02% Na N3, 30 µM phenylmethylsulfonyl fluoride (PMSF), 1 mmol L−1 sodium orthovanadate, and protease inhibitor cocktail.
Membranes were separated from the cytosol by centrifugation (20 000 g for 20 min) and washed once in a hypotonic buffer. Aliquots of membranes (10 µg) or cytosol (15 µL) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 10% gels), transferred to nitrocellulose membranes, and immunostained with the appropriate antibody.
When necessary, erythrocytes were pretreated with epiandrosterone (400 µmol L−1) or 1-chloro-2,4-dinitrobenzene (CDNB) (1 mmol L−1) or vehicle (0.5% v/v acetone or ethanol, respectively) at 35°C for 30 min, and successively subjected to diamide or hypertonic treatments as above described.
Preparation of band 3 proteolytic fragment (cdb3)
The 40/45 kDa fragment of the cytoplasmic domain of band 3 (referred to as cdb3) was obtained by α-chymotrypsin-promoted breakdown of inverted membrane vesicles derived from the ghosts and isolated by DE 52 chromatography according to (Bennett & Stenbuck [Citation1980]).
Protein kinase assay
Packed treated erythrocytes were subjected to several freeze/thaw cycles to yield a homogeneous cell lysate then subjected to microfuge at 40 000 rpm for 60 min to separate membrane from the cytoplasm. About 2 µL of each were assayed for tyrosine kinase activity at 30°C for 10 min in 30 µL incubation mixture containing 50 mmol L−1 Tris-HCl, pH 7.5, 10 mmol L−1 MnCl2 30 µmol L−1 γ[32P] ATP (specific activity 1000 cpm/pmol), 10 µmol L−1 sodium orthovanadate, and 3 µg cytoplasmic domain of band 3 (cdb3) as a substrate for tyrosine kinase activity. After incubation, the reaction was stopped by the addition of 1% SDS and 1% β-mercaptoethanol (final concentrations), and heated for 5 min at 100°C. Solubilized proteins were subjected to SDS-10%/PAGE, the gels were stained with coomassie blue and then treated with 2 mol L−1 NaOH for 60 min at 55°C, dried and autoradiographed for 2 days (Clari et al. [Citation1990]).
Protein phosphatase activity
2 µL of the cell lysate compartments obtained above were assayed for tyrosine protein phosphatase activity in 30 µL incubation mixture containing Tris-HCl 50 mmol L−1 and 3 µg [32P]cdb3 obtained as described in (Bordin et al. [Citation2002]) at 30°C for 5 min. The reaction was stopped after incubation by the addition of 1% SDS and 1% β-mercaptoethanol (final concentrations), followed by 5 min heating at 100°C. Solubilized proteins were subjected to SDS-PAGE (10% gels). The gels were stained with coomassie blue and then treated with 2 mol L−1 NaOH for 60 min at 55°C, dried and autoradiographed for 3 days.
Quantitative determination of total glutathione (GSSG + GSH) and oxidized glutathione (GSSG) content in erythrocytes
The total glutathione was determined according to the method of Tietze ([Citation1969]). Briefly, 10 µL of cytosol obtained from normal and G6PDd erythrocytes as described above, was added to the glutathione assay mixture (Tietze [Citation1969]) and analysed spectrophotometrically. GSSG content was evaluated in 10 µL cytosol incubated in a glutathione assay mixture in which vinylpyridine was added in accordance with (Teare et al. [Citation1993]). The level of GSH was determined by calculating the difference between the two determinations.
Results
Hemolysis is usually episodic in G6PD deficiency and linked to exposure to oxidizing agents underlying the key role played by G6PD/NADPH in resisting oxidant damage (Jollow & McMillan [Citation2001]).
A group of eight G6PD deficient patients characterized by a reduced G6PD activity (ranging from 5–10%) and with GSH content from 30–40% lower than in controls, was evaluated, together with 12 normal individuals.
The diamide-induced phospho-tyrosine level of membrane proteins, principally band 3 had been previously analysed and characterized in normal RBC (Brunati et al. [Citation2000]; Bordin et al. [Citation2002]). Diamide is an important laboratory oxidant, which can react with the thiol group of various proteins and GSH without inducing cell damage through free radical production (Kosower et al. [Citation1969]; Wax et al. [Citation1970]; Kosower & Kosower [Citation1995]).
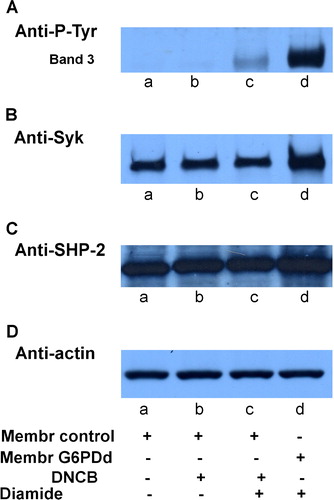
Erythrocytes from normal and G6PD-deficient patients were incubated in the presence of 0.3 mmol L−1 diamide, concentration which does not trigger band 3 Tyr-phosphorylation in normal erythrocytes ( lane c). Membranes, recovered as described in the Methods, were subjected to Western blotting and immunostained with anti-P-Tyr antibodies (panel A). It is well known that deficient cells have a higher sensitivity to oxidizing agents causing the formation of Heinz bodies and anomalies in cell morphology and deformability (Jacobasch & Rapoport [Citation1996]). Under mild oxidative stress, induced by a low concentration of diamide (0.3 mmol L−1), a high level of band 3 Tyr-phosphorylation was found in G6PDd erythrocytes (lane d), compared with their total absence in control cells (lane c).
Figure 1. Oxidative stress effect on normal and g6pdd RBC. Normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes incubated in the absence (lanes a, b) and presence (lanes c, d) of 0.3 mmol L−1 diamide. Membranes (10 µg), recovered as described in Methods, were analysed by Western blotting and immunostained with anti-P-Tyr (panel A), anti-Syk (panel B), or anti-SHP-2 (panel C) antibodies. Panel D: corresponding anti-actin immunostaining for loading control. Figure is representative of eight separate experiments.
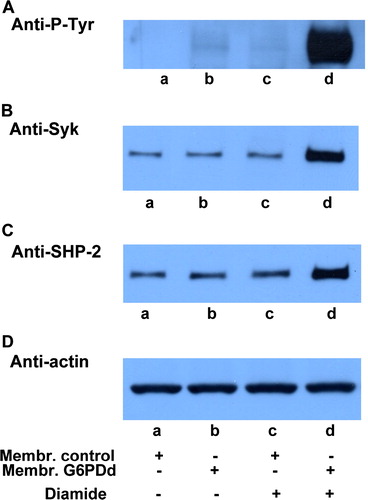
To discover the reason behind this increase, we analysed these membranes using anti-Syk (panel B) and anti-SHP-2 (panel C) antibodies and observed the status of the enzymes mainly involved in maintaining band 3 Tyr phosphorylation level balance. We had previously shown that Syk can translocate from cytosol to RBC membranes following treatment with an oxidant such as diamide, as well as with non-oxidant effector, such as NEM, being recruited to the cytoskeletal counterpart of the membrane in a P-Tyr-independent manner (Citation[Bordin et al. in press]). Panel B of clearly supports the hypothesis that, although at a lower concentration, diamide is able to induce oxidative stress in RBC from G6PD-deficient subjects in a way which is in every respect comparable with that obtained in normal cells at higher concentrations. In fact, Syk is almost three times higher (lane d) than in controls receiving the same diamide concentration (lane c) even though both membranes share the same amount of enzyme in the absence of diamide (lane a compared to lane b).
In the band 3 Tyr-phosphorylative process, protein tyrosine phosphatases energically protect the phosphotyrosine level of the protein. When phosphorylated by Lyn secondary phosphorylation in its 359 Tyr-residue (Brunati et al. [Citation2000]), band 3 becomes the docking protein for SHP-2 recruitment allowing the enzyme to take an active part in the dephosphorylation of band 3 residues 8, 21, and 904 (Bordin et al. [Citation2002]). For this reason, SHP-2 translocation may be assumed to be an indicator that secondary phosphorylation has taken place and that its extent is proportional to the number of the P-tyrosines (Bordin et al. [Citation2002]). As well as showing that, even for this second enzyme, normal and deficient RBC share the same amount in the membranes, panel C emphasizes that SHP-2 is recruited following the Tyr-phosphorylation of band 3, as expected, only in patients (lane d) who are more sensitive to diamide treatment.
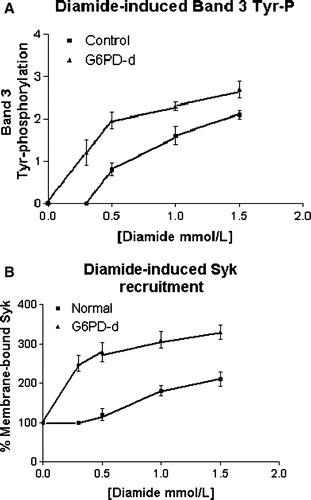
We, also, observed normal and G6PD-deficient erythrocytes in the presence of concentrations of increasing diamide. Band 3 Tyr-phosphorylation level rose in a dose-dependent manner, but it was clearly amplified in deficient compared with normal cells ( panel A), together with the concomitant recruitment of both Syk (panel B) and SHP-2 (not shown, but adequately represented by B), highlighting how the sensitivity of deficient cells to increasing oxidative stress may be represented by their enhanced band 3 Tyr-phosphorylative response.
Figure 2. Effect of increasing diamide concentration on membrane band 3 Tyr-phosphorylation (panel A) and Syk recruitment to membranes (panel B). Normal (▪) and G6PD-deficient (▴) erythrocytes incubated with 0, 0.3, 0.5, 1 and 1.5 mmol L−1 diamide, respectively. Membranes (10 µg) recovered as described in the Methods, were analyzed by Western blotting and immunorevealed with anti-P-Tyr (panel A), anti-Syk (panel B) or anti-SHP-2 (not shown but represented by panel B) antibodies, and the corresponding stains were counted in a densitometer. For the band 3-Tyr-P evaluation an arbitrary unit was chosen, whereas for enzyme recruitment, the amount of each enzyme in resting cells was used as 100% value. Results are mean of three separate experiments, with three normal and three deficient erythrocytes.
Enzyme quantification and activity and protein content
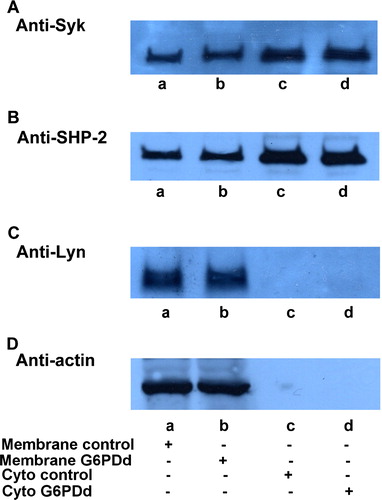
The enhanced effect of diamide in deficient cells may be due to an anomalous amount of enzyme compared to that in normal cells: the increased presence of Syk and/or Lyn as well as a lower SHP-2 content may explain the higher phosphorylation level induced in band 3 after diamide treatment since they are the key enzymes involved in this process. As shown in , membranes (lanes a, b) and cytosol (lanes c, d) from normal (lanes a, c) and favic patients (lanes b, d) have the same enzyme content, both as regards kinase such as Syk (panel A) or Lyn (panel C), and phosphatase such as SHP-2 (panel B).
Figure 3. Quantitative comparison of enzyme amount and distribution. Normal (lanes a, c) and G6PDd (lanes, b, d) erythrocytes were processed and membranes (lanes a, b) were separated from the cytosol (lanes c, d) as described in Methods. 10 µL of membranes or 30 µL cytosol were successively analysed by Western blotting and immunorevealed with anti-Syk (panel A), anti-SHP-2 (panel B) or anti-Lyn (panel C). Panel D: shows the corresponding anti-actin immunostaining for the loading control. Figure is representative of at least ten separate experiments.
With regard to enzyme activities, we subjected normal and deficient erythrocytes to numerous freeze–thaw cycles and used part of the recovered membranes (lanes a, b) and cytosol (lanes c, d) to carry out the activity assay explained in the Methods. As shown in , not even the respective kinase and phosphatase activities – which seem similar if not identical – in normal and deficient erythrocytes explain the different extent of band 3 Tyr phosphorylation.
Figure 4. Tyrosine Kinase and tyrosine Phosphatase activities in normal and G6PDd human erythrocytes.(A) Tyr-protein kinase activity. Normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes subjected to a few freeze/thaw cycles and membranes were separate from cytosol by microfuge as described in Methods. Then 2 µL of membranes (lanes a, b) and 2 µL of cytosol (lanes c, d) were incubated in basal medium of phosphorylation containing γ[32P] ATP in presence of cdb3 as described in Methods and samples were submitted to SDS PAGE. Gel was stained with Coomassie blue, subjected to 2 mol L−1 NaOH treatment, dried and autoradiographed for 2 days. Panels are representative of eight distinct experiments.(B) Tyr-protein phosphatase activity. 2 µL of membranes (lanes a, b) and 2 µL of cytosol (lanes c, d) from normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes were incubated in basal medium of dephosphorylation in presence of [32P]cdb3. Lane C: the control pattern of the loaded [32P]cdb3. Samples were submitted to SDS PAGE. Gel was stained with coomassie blue and then treated with 2 mol L−1 NaOH, dried and autoradiographed for 2 days. Panels are representative of 10 separate experiments.
![Figure 4. Tyrosine Kinase and tyrosine Phosphatase activities in normal and G6PDd human erythrocytes.(A) Tyr-protein kinase activity. Normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes subjected to a few freeze/thaw cycles and membranes were separate from cytosol by microfuge as described in Methods. Then 2 µL of membranes (lanes a, b) and 2 µL of cytosol (lanes c, d) were incubated in basal medium of phosphorylation containing γ[32P] ATP in presence of cdb3 as described in Methods and samples were submitted to SDS PAGE. Gel was stained with Coomassie blue, subjected to 2 mol L−1 NaOH treatment, dried and autoradiographed for 2 days. Panels are representative of eight distinct experiments.(B) Tyr-protein phosphatase activity. 2 µL of membranes (lanes a, b) and 2 µL of cytosol (lanes c, d) from normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes were incubated in basal medium of dephosphorylation in presence of [32P]cdb3. Lane C: the control pattern of the loaded [32P]cdb3. Samples were submitted to SDS PAGE. Gel was stained with coomassie blue and then treated with 2 mol L−1 NaOH, dried and autoradiographed for 2 days. Panels are representative of 10 separate experiments.](/cms/asset/d9141841-5c57-4d34-8685-fe1045fdd7e4/imbc_a_123350_f0004_b.jpg)
Similarly, an anomalous membrane protein pattern in normal and deficient cells may explain the biochemical basis of this disease. However, the Coomassie blue chromatographic pattern did not show any detectable difference in membrane protein composition (data not shown), thus excluding the possibility that enhanced oxidant-sensitive band 3 Tyr-phosphorylation involves a macro-anomaly of membrane protein content.
Hypertonic stress
We compared the ability of normal and G6PDd erythrocytes to adapt to hypertonic stress conditions by incubating both in a hyperosmotic buffer (1 osm L−1) which may trigger band 3 Tyr-phosphorylation (Minetti et al. [Citation1998]). The corresponding membranes were immunorevealed with anti-P-Tyr antibodies (, panel A), showing that RBC in patients are also more susceptible to this different stress, as demonstrated by the higher extent of membrane band 3 phosphorylation (lane d compared with normal RBC, lane c).
Figure 5. Hyperosmosis-induced membrane band 3 Tyr-phosphorylation in normal and G6PD deficient erythrocytes. Normal (lanes a, c) and G6PDd (lanes b, d) erythrocytes were incubated in isosmotic (lanes a, b) or hyperosmotic (lanes c, d) conditions. Membranes (10 µg), recovered as described in Methods, were analysed by Western blotting and immunorevealed with anti-P-Tyr (panel A), anti-Syk (panel B), anti-SHP-2 (panel C) antibodies. Panel D: corresponding anti-actin immunostaining for the loading control. Panels are representative of eight distinct experiments.
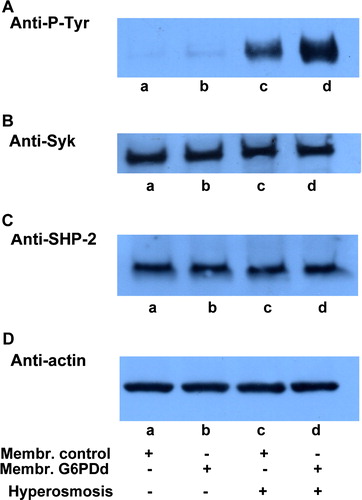
Neither Syk (panel B) nor SHP-2 (panel C) recruitment took place in normal or patient cells in these conditions.
Antioxidant cellular defence involvement in G6PD deficiency
Red cells from G6PD-deficient individuals are known to have enhanced susceptibility to haemolytic drugs and, when challenged in vitro, reveal greater GSH instability and a greater propensity to form Heinz bodies as compared with red cells from normal donors (Jacobasch & Rapoport [Citation1996]).
To test GSH involvement in the band 3 Tyr-phosphorylation process, normal erythrocytes were treated with 1 mmol L−1 CDNB, which completely depletes cellular content of glutathione (data not shown) (Awasthi et al. [Citation1981]; Chiu et al. [Citation1993]).
When erythrocytes, pretreated with CDNB, were incubated with 0.3 mmol L−1 diamide, only a slight labelling of band 3 Tyr-residues was induced (, lane c), certainly more than what is found in CDNB-untreated RBC (lane a), but practically nothing compared with that observed in G6PDd cells (lane d).
Figure 6. Effect of CDNB induced depletion of GSH on diamide treatment.Normal (lanes a–c) and G6PDd (lanes d) erythrocytes were pre-treated in presence (lanes b, c) or absence (lanes a, d) of 1 mmol L−1 CDNB, followed by incubation in presence (lanes c, d) or absence (lanes a, b) of 0.3 mmol L−1 diamide as described in Methods. Membranes (10 µg) were analysed by Western blotting and immunorevealed with anti-P-Tyr (panel A), anti-Syk (panel B), or anti-SHP-2 (panel C) antibodies. Panel D: corresponding anti-actin immunostaining for loading control. Panels are representative of eight distinct experiments.
These findings show that the cell content of GSH does not seem to be a determinant in the cellular response to oxidative stress, since: (i) oxidant condition did not induce total oxidation of glutathione in either normal or deficient cells (); (ii) its complete depletion induced by CDNB cannot induce the same alteration found in the deficient cells.
Table I. Total glutathione and GSSG content in normal and G6PD-deficient erythrocytes subjected to various treatments. Erythrocytes, incubated as indicated, were haemolysed, cytosol recovered and determination of total glutathione (GSSG + GSH) mmol L−1 and oxidized glutathione (GSSG) mmol L−1 determined, all as described in Methods. Mean±SD of six experiments.
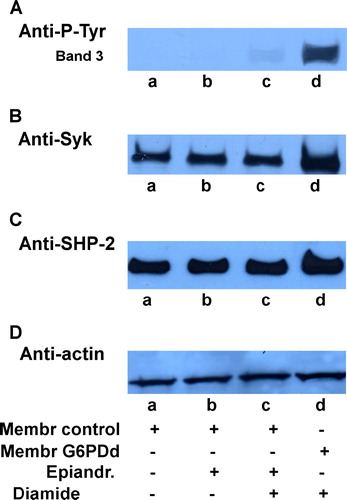
Since GSH does not account for the higher band 3 Tyr-phosphorylation present in diamide-stimulated G6PDd cells, we treated normal erythrocytes with 0.4 mmol L−1 epiandrosterone which totally inhibits G6Pdehydrogenase activity (data not shown) (Marks & Banks [Citation1960]). In this way, we attempted to reproduce in normal cells the anomalous behaviour of deficient erythrocytes (Jacobasch & Rapoport [Citation1996]). In fact, in these conditions NADPH, once oxidized, can not be reconverted in the reduced form because of the inhibition of the HMP shunt, as in the corresponding anemia.
Nevertheless, even in this case, the addition of 0.3 mmol L−1 diamide to epiandrosterone-pretreated normal erythrocytes induces barely perceptible band 3 Tyr-phosphorylation (, lane c), which cannot explain the high band 3 Tyr-P level of glucose-6-Phosphate dehydrogenase deficient patients (lane d).
Figure 7. Effect of epiandrosterone-induced G6PD activity inhibition on diamide treatment. Normal (lanes a–c) and G6PDd (lanes d) erythrocytes were pre-treated in presence (lanes b, c) or absence (lanes a, d) of 0.4 mmol L−1 epiandrosterone, followed by incubation in presence (lanes c, d) or absence (lanes a, b) of 0.3 mmol/diamide as described in Methods. Membranes (10 µg) were analysed to Western blotting and immunorevealed with anti-P-Tyr (panel A), anti-Syk (panel B) or anti-SHP-2 (panel C) antibodies. Panel D: corresponding anti-actin immunostaining for loading control. Panels are representative of eight distinct experiments.
Concomitantly, we also analysed both Tyr-kinase and phosphatase activities to define the role of the varying treatments on enzyme behaviour. Our findings revealed that only diamide, as previously indicated by Syk and SHP-2 recruitment to the membrane of G6PD deficient cells () was able partially to increase P-Tyr-kinase activity in this membrane, without affecting the membrane of normal cells. Moreover, neither CDNB, nor epiandrosterone pre-treatment was able to alter kinase and phosphatase activities in normal cells (data not shown).
In both cases, therefore, the total absence of GSH (, lane b) or G6PD inhibition (, lane b) were not sufficient to trigger band 3 Tyr-phosphorylation as in the deficiency.
Effect of epiandrosterone or CDNB on band 3 Tyr-phosphorylation under hyperosmotic stress
Since the alterations we induced in normal erythrocytes with depletion of GSH or inhibition of G6PD did not account for the deficient RBC Tyr-phosphorylative behaviour in conditions of oxidative stress, we examined whether these would induce an anomalous erythrocyte response to a morphological adjustment. We thus subjected normal erythrocytes previously pre-treated in the presence of both epiandrosterone (, lane b) and CDNB (lane c) to the same hyperosmotic stress described above (). As clearly shown, CDNB induces band 3 Tyr-phosphorylation (panel A) in normal erythrocytes subjected to osmotic stress, as observed in patient cells (lane d). Instead, G6PD inhibition, induced by epiandrosterone (lane b) does not increase the band 3 Tyr-phosphorylative pattern, which remains at the same level as that in controls.
Figure 8. Effect of CDNB and/or epiandrosterone pre-treatment on the hyperosmosis-induced membrane band 3 Tyr-phosphorylation. Normal (lanes a–c) and G6PDd (lane d) erythrocytes were pre-treated in absence (lanes a, d) or presence of 0.4 mmol L−1 epiandrosterone (lane b), or 1 mmol L−1 CDNB (lane c), and then incubated in hyperosmotic medium (lanes a–d) as described in Methods. Membranes (10 µg), were analysed by Western blotting and immunorevealed with anti-P-Tyr (panel A) anti-Syk (panel B), or anti-SHP-2 (panel C) antibodies Panel D: corresponding anti-actin immunostaining for loading control. Panels are representative of eight distinct experiments.
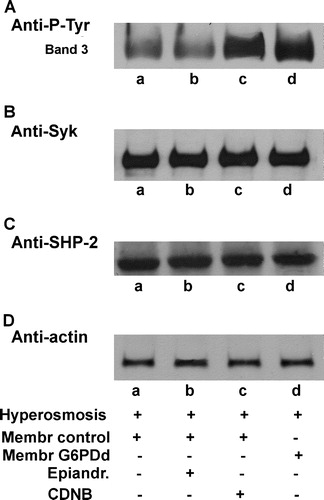
Once more, both Syk and SHP-2 location were not affected by osmotic stress either with or without any pre-treatments, as indicated by their corresponding Western blotting patterns (B and C, respectively), and by their unaltered activities (data not shown).
Discussion
G6PD in the HMP shunt regulates the production of NADPH, an obligatory substrate for several redox systems, in particular for glutathione which protects the cell from oxidative stress. It has previously been shown that conditions of oxidative stress lowering NADPH immediately raise the HMP shunt rate up to 30-fold. Red blood cells with G6PD deficiency cannot increase their shunt sufficiently during an oxidative load, and thus show a weakened cellular redox defence (Jacobasch & Rapoport [Citation1996]). Moreover, generally only 0.1% of the maximum G6PD capacity is utilized in human erythrocytes, and this is why the need for this enzyme is extremely limited unless the cells are exposed to oxidative stress. In fact, in the presence of divicine, a natural oxidizing agent found in Vicia faba beans, and in antimalarial, antipyretics or analgesic drugs, G6PD deficient patients can not provide an adequate antioxidant defence and their erythrocytes present Heinz bodies and reveal the formation of anomalies in cell morphology and deformability (Jacobasch & Rapoport [Citation1996]).
Moreover, premature cell senescence is also induced by oxidative stress that induces Hb denaturation and membrane binding of hemiochromes, Heinz body precursors, and provokes aggregation of band 3 and deposition of antibodies and complement C3c fragments. In fact, it has been described that membrane clustering of band 3 can allow immune recognition by naturally occurring antibodies, inducing antibody-dependent phagocytosis of senescent/alterate erythrocytes (Kay et al. [Citation1984]; Low et al. [Citation1985]; Schluter & Drenekhanh [Citation1986]; Lutz et al. [Citation1988]; Arese & De Flora [Citation1990]; Hebbel et al. [Citation1990]). In G6PD deficient erythrocytes, increased macrophage-mediated phagocytosis has been demonstrated in both the presence and absence of autologous serum (Horn et al. [Citation1991]), suggesting an alteration of membrane properties as a second pathway for RBC removal (Beppu et al. [Citation1987]).
Also, phospho-tyrosine level of band 3 can induce structural alterations, and this has been hypothesized to be involved in cell apoptosis, since this post-trunsductional mechanism exposes new band 3 epitopes and favour clustering and, consequently, direct membrane alteration as well as binding of multivalent ligands, both leading to hemolysis (Bottini et al. [Citation1997]).
All these facts, together with the G6PDd cell inability to response powerfully to oxidants, indicates that the physiological status of band 3 is essential for red blood cell survival/apoptosis. According to this line of evidence, we studied the alterations induced in band 3 Tyr-phosphorylation by oxidative (diamide) or hyperosmotic stress comparing normal with deficient cell responses.
The fact that G6PDd erythrocytes display an earlier band 3 Tyr-phosphorylation induced by diamide is not surprising, and represents an added parameter to the deficient cell susceptibility to oxidants.
Since the common cell defence against oxidizing conditions is based on the content of GSH and NADPH recycling, we investigated their functional role in the presence of an oxidant, hypothesizing that in the case of G6PD deficiency one or both would be determinant in the anomaly of the phosphorylation process. However, neither with pre-treatment with epiandrosterone (a condition which may resemble the disease since it inhibits G6PD activity) nor with CDNB (which depletes cellular GSH content) the addition of diamide to normal erythrocytes can induce the same phosphorylation pattern of band 3 evidenced in patients, but only a slight P-Tyr-band 3. For this reason, neither interrupted NADPH recycling, nor GSH, alone, can account for this enhanced sensitivity in the band 3 Tyr-phosphorylation of deficient cells. Our findings also exclude an important alteration in enzyme content or activity, since Syk, Lyn and SHP-2, implicated in band 3 Tyr-phosphorylation regulation, share the same distribution between membrane and cytosol and the same Tyr-protein kinase and phosphatase activity was obtained from both membrane and cytosol.
Conversely, alteration of the substrate may partially explain the band 3 Tyr-phosphorylative behaviour of deficient cells. Data obtained under hyperosmotic conditions highlighted a higher phosphorylation level in patients subjected to hypertonic stress, when compared with controls. Hypertonicity has been demonstrated to induce Tyr-phosphorylation of membrane band 3 (Minetti et al. [Citation1998]) but without involving any enzyme re-localization. In fact, our data showed that both kinase Syk and Tyr-protein phosphatase SHP-2, responsible for the band 3 Tyr-P regulation, do not translocate from cytosol to membranes as they do in the presence of diamide (Bordin et al. [Citation2002]; Citation[Bordin et al. in press]). So, in these conditions, band 3 Tyr-phosphorylation is not due to inhibition of protein tyrosine phosphatase activity or to specific kinase (Syk) recruitment to the membrane. Band 3 Tyr-phosphorylation induced by hyperosmosis is probably mediated by the morphological alteration of the RBC which, besides causing them to lose their biconcavity and assuming a flattened and crenated shape (Minetti et al. [Citation1998]), would modify their membrane structure, making band 3 more accessible to kinase rather than to phosphatase activity. Interestingly, this membrane modification in G6PD deficient cells seems to be much more pronounced, as evidenced by the higher extent of band 3 Tyr-phosphorylation, thus indicating a former pre-existing structural anomaly, emphasized by the subsequent hyperosmotic stress. What is important is that the same higher phosphorylation level may be reached in normal cells by CDNB-preincubation which, besides depleting GSH content, can also induce alterations in RBC membrane structure probably through its interaction with membrane sulfhydryl groups (Chiu et al. [Citation1993]). However, since glutathione content in patients is only 30% less than in normal cells, its total depletion would be expected to induce an higher effect compared with that found in patients. Since this was not the case, it is reasonable that CDNB-induced alteration of the cell membrane is also involved, rather than only a direct effect of glutathione depletion.
In conclusion, it may be presumed that red cells in patients have an affect on membrane structure, perhaps on the protein–protein interaction, probably determined by the chronic lowered GSH content which, once subjected to exogenous stress such as shape modification by high osmotic strength, would induce a higher Tyr-phosphorylation response. That this interaction is under redox control is demonstrated by the effect of GSH depletion in the cells.
We hypothesize that a considerable membrane alteration in G6PD deficient erythrocytes –like that previously found involving deamidation of asparagine residues with subsequent methylation of aspartate in cytoskeletal components, such as ankyrin band 4.1 and band 4.2, and the integral protein band 3 (Ingrosso et al. [Citation2002]) – are crucial for the deficient cell response, not only towards oxidative but also osmotic stress, influencing the life-span of RBC.
This paper was first published online on prEview on 23 September 2005.
This work was supported by Italian Ministero dell'Università e della Ricerca Scientifica e Tecnologica (MURST). Mr. Claudio Bettella and Mr. Giancarlo Ruffato are gratefully acknowledged for supplying fresh blood from volunteers.
Related Research Data
References
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27: 1–40
- Awasthi YC, Gary HS, Dao DD, Partridge CA, Srivastava SK. Enzymatic conjugation of erythrocyte glutathione with 1-chloro-2,4-dinitrobenzene: the fate of glutathione conjugate in erythrocytes and the effect of glutathione depletion on hemoglobin. Blood 1981; 58: 733
- Bennett V, Stenbuck PJ. Association between ankyrin and the cytoplasmic domain of band 3 isolated from the human erythrocyte membrane. J Biol Chem 1980; 255: 6424–32
- Beppu M, Ochiai H, Kikugawa K. Macrophage recognition of the erythrocyte modified by oxidizing agents. Biochim Biophys Acta 1987; 930: 244–53
- Betke, K, Beutler, E, Brewer, GJ, Kirkman, HN, Luzzato, L, Motulsky, AG, Ramot, B, Siniscalco, M. 1967. Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. Report of a WHO scientific group-WHO Technical Report-Serial No 366.
- Bordin L, Brunati AM, Donella-Deana A, Baggio B, Toninello A, Clari G. Band 3 is a anchor protein and a target for SHP-2 tyrosine phosphatases in human erythrocytes. Blood 2002; 100: 276–82
- Bordin L, Clari G, Moro I, Dalla Vecchia F, Moret V. Functional link between phosphorylation state of membrane proteins and morphological changes of human erythrocyres. Biochem Biophys Res Commun 1995; 213: 305–11
- Bordin, L, Ion-Popa, F, Brunati, AM, Clari, G, Low, P. Effector-induced Syk-mediated phosphorylation in human erythrocytes. Biochim Biophys Acta, (in press).
- Bottini E, Bottini FG, Borgiani P, Businco L. Association between ACP1 and Favism: A possible biochemical mechanism. Blood 1997; 89: 2613–15
- Brunati AM, Bordin L, Clari G, James P, Quadroni M, Baritono E, Pinna LA, Donella-Deana A. Sequential phosphorylation of protein band3 by Syk and Lyn tyrosine kinases in intact human erythrocytes, Identification of primary and secondary phosphorylation sites. Blood 2000; 96: 1550–57
- Chiu DTY, Lai KM, Xu CM, Liour SSS, Lee J, Liu TZ. Direct alteration of erythrocyte membrane properties by 1-chloro-2,4-dinitrobenzene without oxidant challenge. Exp Hematol 1993; 21: 114–18
- Clari G, Marzaro G, Moret V. Metabolic depletion effect on serine/threonine- and tyrosine-phosphorylations of membrane proteins in human erythrocytes. Biochim Biophys Acta 1990; 1023: 319–24
- De Franceschi L, Fumagalli L, Olivieri O, Corrocher R, Lowell CA, Berton G. Deficiency of Src family kinases Fgr and Hck results in activation of erythrocyte K/Cl cotransport. J Clin Invest 1997; 99: 220–27
- Harrison ML, Isaacson CC, Burg DL, Geahlen RL, Low PS. Phosphorylation of human erythrocyte band 3 by endogenous p72syk. J Biol Chem 1994; 269: 955–59
- Harrison ML, Rathinavelu P, Arese P, Geahlen RL, Low PS. Role of band 3 tyrosine phosphorylation in the regulation of erythrocyte glycolysis. J Biol Chem 1991; 266: 4104–11
- Hebbel RP. The sickle erythrocyte in double jeopardy: autoxidation and iron decompartmentalization. Semin Hematol 1990; 27: 51–69
- Horn S, Bashan N, Gopas J. Phagocytosis of phenylhydrazine oxidized and G-6-PD-deficient red blood cells: the role of cell-bound immunoglobulis. Blood 1991; 78: 1818–25
- Ingrosso D, Cimmino A, D'Angelo S, Alfinito F, Zappia V, Galletti P. Protein methylation as a marker of aspartate damage in glucose-6-phosphate dehydrogenase-deficient erythrocytes, Role of oxidative stress. Eur J Biochem 2002; 269: 2031–39
- Jacobasch G, Rapoport SM. Hemolytic anemias due to erythrocyte enzyme deficiency. Mol Aspects Med 1996; 17: 143–70
- Jollow DJ, McMillan DC. Oxidative stress, glucose-6-phosphate dehydrogenase and the red cell. Biological Reactive Intermediates VI, PM Dansette, R Snuder, M Delaforge, GG Gibson, H Greim, DJ Jollow, TJ Monks, IG Snipes. Plenum Publisher, New York 2001; 595–605
- Kay MM. Localization of senescent cell antigen on band 3. Proc Natl Acad Sci 1984; 81: 5753–57
- Kosower NS, Kosower EM, Wertheim B, Correa WS. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem Biophys Res Commun 1969; 37: 593–96
- Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol 1995; 251: 123–133
- Lee GR, Bithell TC, Foerster J, Athens JW, Lukens JN. Glucose-6-phosphate dehydrogenase deficiency and related deficiencies involving the pentose phosphate pathway and glutathione metabolism. Wintrobe's Clinical Hematology, GR Lee. Lea & Febiger, Philadelphia, PA 1993; 1: 1006–16
- Low PS, Rathinavelu P, Harrison ML. Regulation of glycolysis via reversible enzyme binding to the membrane protein band 3. J Biol Chem 1993; 268: 14627–631
- Low PS, Allen DP, Zioncheck TF, Chari P, Willardson BM, Geahlen RL, Harrison ML. Tyrosine phosphorylation of band 3 inhibits peripheral protein binding. J Biol Chem 1987; 262: 4592–96
- Low PS, Waugh SM, Zinke K, Drenckhahn D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science 1985; 227: 531–33
- Lutz HU, Fasler S, Stammler P, Bussolino F, Arese P. Naturally occurring anti-band 3 antibodies and complement in phagocytosis of oxidatively-stressed and in clearance of senescent red cells. Blood Cells 1988; 14: 175–95
- Marks PA, Banks J. Inhibition of mammalian glucose-6-phosphate dehydrogenase by steroids. Proc Natl Acad Sci USA 1960; 46: 447–52
- Minetti G, Seppi C, Ciana A, Balduini C, Low PS, Brovelli A. Characterization of the hypertonically induced tyrosine phosphorylation of erythrocyte band 3. Biochem J 1998; 335: 305–11
- Musch MW, Hubert EM, Goldstein L. Volume expansion stimulates p72syk and p56lyn in skate erythrocytes. J Biol Chem 1991; 274: 7923–28
- Schluter K, Drenekhanh D. Co-clustering of denaturated hemoglobin with band 3: its role in binding of autoantibodies against band 3 to abnormal and aged erythrocytes, Proc. Natl Acad Sci USA 1986; 83: 6137–41
- Teare JP, Punchard NA, Powell JJ, Lumb PJ, Mitchell WD, Thompson RP. Automated spectrophotometric method for determining oxidized and reduced glutathione in liver. Clin Chem 1993; 39: 686–89
- Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 1969; 27: 502–22
- Wax R, Rosenberg E, Kosower NS, Kosower EM. Effect of the thiol-oxidizing agent diamide on the growth of Escherichia coli. J Bacteriol 1970; 101: 1092–93
- Yannoukakos D, Vasseue C, Piau J-P, Wajcman H, Bursaux E. Phosphorylation sites in human erythrocyte band 3 protein. Biochim Biophys Acta 1991; 1061: 253–66
- Zipser Y, Piade A, Kosower NS. Erythrocytes thiol status regulates band3 phosphotyrosine level via oxidation/reduction of band3-associated phosphotyrosine phosphatase. FEBS Lett 1997; 406: 126–30