Abstract
Ionotropic glutamate receptors belong to the superfamily of P-loop channels as well as K+, Na+, and Ca2+ channels. However, the structural similarity between ion channels of the glutamate receptors and K+ channels is a matter of discussion. The aim of this study was to analyze differences between the structures of K+ channels and glutamate receptor channels. For this purpose, homology models of NMDA and AMPA receptor channels (M2 and M3 segments) were built using X-ray structures of K+ channels as templates. The models were optimized and used to reproduce specific data on the structure of glutamate receptor channels. Particular attention was paid to the data of the binding of channel blockers and to the results of scanning mutagenesis. The modeling demonstrates that properties of glutamate receptor channel can be reproduced assuming only local structural deformations of the K+ channel templates. The most valuable differences were found in the selectivity-filter region, whereas helical parts of M2 and M3 segments could have similar spatial organization with homologous segments in K+ channels. It is concluded that the current experimental data on glutamate receptor channels does not reveal global structural differences with K+ channels.
Introduction
During the last decade the view on the structure of ion channels of glutamate receptors has been completely changed. Initially, it was believed that ionotropic glutamate receptors belong to the superfamily of ligand-gated ionic channels that also includes nicotinic acetylcholine receptors and GABA receptors. However, numerous studies have demonstrated that glutamate receptors have subunit stoichiometry and transmembrane topology similar to K+, Na+ and Ca2 + channels (see e.g., Wo & Oswald [Citation1994], Wood et al. [Citation1995], Bennett & Dingledine [Citation1995], Rosenmund et al. [Citation1998]). All the channels are tetramers or pseudo-tetramers, possess a re-entrant membrane loop (P-loop), and have the selectivity-filter region located near the turn of the P-loop (see Zhorov & Tikhonov [Citation2004]). Taken together, these findings strongly suggest overall structural similarity between pore-forming domains of glutamate receptors and K+, Na+ and Ca2 + channels.
Crystallization of K+ channels (Doyle et al. [Citation1998], Jiang et al. [Citation2002], [Citation2003]) has opened a new era in the studies of P-loop channels. The secondary structure of the pore-forming segments was unambiguously determined; mechanisms of permeability, selectivity, and gating were analyzed basing on the X-ray data. The atomic-scale data of Na+, Ca2 + , and glutamate-gated channels are unavailable and their structure is still a matter of speculation and modeling. X-ray data of potassium channels were used for modeling Na+ and Ca2 + channels (Yamaguchi et al. [Citation2003], O'Reilly et al. [Citation2006], Lipkind & Fozzard [Citation2000], [Citation2001], [Citation2003], [Citation2005], Zhorov et al. [Citation2001], Tikhonov & Zhorov [Citation2005a],Citation[b]). The usage of X-ray structures as templates in homology modeling has provided structural explanations for many specific properties of Na+ and Ca2 + channels.
It appears that P-loop channels have closer structural similarity than expected before. For example, the selectivity filter of sodium channels is significantly wider than in K+ channels. It can accommodate organic blockers like tetrodotoxin and saxitoxin. In the early models (Lipkind & Fozzard [Citation2000]), the P-loops were placed farther from the pore axis than in K+ channels to provide large pore dimensions. However, the intensive optimization of Na+ channel models has proven that binding of the toxins can be reproduced without the displacement of the P-loops (Tikhonov & Zhorov [Citation2005a]). Even the structure of the Shaker K+ channels was seriously discussed because prokaryotic K+ channels lack PVP in the S6 segment. It has been proposed that the PVP residues cause a kink in the S6 helix and lead to a significantly different structure of the open Shaker channel (del Camino et al. [Citation2000]; Webster et al. [Citation2004]). Recent crystallization of the Kv1.2 channel (Long et al. [Citation2005]) has demonstrated a kink at the PVP motif. Nevertheless, structure of the channel is more similar to the channels without the PVP motif than it was expected from experiments of Webster et al. ([Citation2004]). Careful structural modeling (Bruhova & Zhorov, [Citation2005]) demonstrated that experimental data on the intersubunit metal bridges (Webster et al. [Citation2004]) are consistent with moderate deformation of S6 segments caused by the PVP motif.
Similarity between glutamate receptor channels and K+ channels is intensively discussed (Qian & Johnson [Citation2002], Kuner et al. [Citation2003], Wollmuth & Sobolevsky [Citation2004]). Although X-ray structures of K+ channels are taken into account, but in the most cases only at the level of speculations or schematic pictures. No serious attempt was made to check the consistency of specific experimental data on glutamate receptor channels with the structures of K+ channels. Without such structural analysis the discussion seems incomplete. This has prompted me to undertake the present study. I built a series of homology models of NMDAR and AMPAR channels using X-ray structures of K+ channels as templates. All models were optimized by the Monte Carlo-minimization method. Next, the models were used to reproduce available experimental data on important elements of topography of glutamate receptor channels. The modeling demonstrates that modest and local changes in the template structures allow specific properties of the glutamate receptor channels to reproduce. It is concluded that there are no solid reasons to suggest global structural differences between glutamate receptor channels and other members of the P-loop channel superfamily.
Methods
Modeling was performed using the ZMM program package (www.zmmsoft.com). Homology models of NMDARs and AMPARs were assembled with using X-ray structures of K+ channels as templates. The starting geometry of the models was generated based on the template structure. Bond lengths, torsion and valent angles of the models were assigned template values. Side-chains of the residues that are different between the model and the template were assigned all-trans conformations. Bond lengths and valent angles were kept rigid during the calculations. Optimization of the models was performed by the Monte Carlo-minimization (MCM) method (Li & Scheraga, [Citation1987]). The AMBER force field (Weiner et al. [Citation1984]) and implicit solvent method (Lazaridis & Karplus [Citation1999]) were used for energy calculations. For a better exploration of the conformational properties of the models, the MCM trajectories were calculated at a temperature of 600 K. Trajectories were terminated when 1000 subsequent energy minimizations did not improve the energy of a model.
Spatial similarity between models and templates was provided by flat-bottom energy penalty functions (constraints). When the deviation of a constrained atom exceeded 1 Å from its position in the template, the parabolic energy penalty was added to the conformational energy of the system. The force field for the constraints was 100 kcal*mol−1*Å−2. Constraints were imposed on alpha-carbon atoms of residues in helical parts of the models. Specific topographic characteristics of glutamate receptor channels were provided by additional ‘distance’ constraints that limit the relative disposition of specified atoms or groups. The maximal distance of clampled H-bonds was 2.2 Å. These constraints were based on available experimental data about the spatial organization of AMPAR and NMDAR channels. This system of constraints determines the consistency of a template with specific properties of glutamate receptor channels. The template is consistent with specific channel properties if the energy penalty form constraints are negligible. It indicates that the system can satisfy specific requirements without significant deviation from the template structure.
The docking of ligands was performed using a two-stage random search procedure. Initially, 5000 random positions/orientations of a ligand were generated in the vicinity of the putative binding site and shortly optimized to remove high-energy steric contacts. At the second stage, 100 lowest-energy structures were intensively optimized. Resulting structures with energies up to 10 kcal/mol from the best energy found were considered as possible conformation of the ligand-channel complex.
Results and discussion
Sequence alignment
For homology modeling the first and sometimes the key question is to align the amino-acid sequences. Although it is commonly accepted that K+, Na+ and Ca2 + channels are homologous proteins, the unambiguous alignment of their segments is impossible (see Zhorov & Tikhonov [Citation2004]). Fortunately, it is not a problem for glutamate receptor channels due to the presence of several conservative residues (). Importantly, besides the selectivity-filter region in AMPAR subunits the aligned sequences do not have insertions/deletions. Thus, the positions of amino-acid residues of glutamate receptors in K+ channel templates can be found unambiguously. All amino acid residues are numbered relatively to the N/Q/R site, which is assigned position 0.
Table I. Aligned sequences of the pore-forming domains in K+ channels and ionotropic glutamate receptors.
Selectivity filter of AMPAR channel
M2 segments of AMPAR subunits form a re-entrant membrane loop and create the narrowest part of the pore. The N-part of the segment is helical as estimated by scanning mutagenesis (Kuner et al. [Citation2001], Panchenko et al. [Citation2001]). The C-end of the segment (Q/R site and downstream) has no regular structure. These characteristics are similar to the properties of K+ channels. The main problem is to reveal the structural peculiarities that determine the differences between glutamate receptor channels and K+ channels. Unlike K+ channels, AMPAR channels do not discriminate mono-cations, and have a diameter about 7.5Å (Burnashev et al. [Citation1996]).
In the previous works (Tikhonov et al. [Citation1999], [Citation2002]) models of the AMPARs were elaborated. Initially, we have suggested that cyclic H-bonds at the N/Q/R site result in the orientation of Gln side chains that makes their oxygens inaccessible. This explained why monocationic drugs fail to block AMPAR channels. The ability of dicationic compounds of certain length to block the channels (Bolshakov et al. [Citation2000], [Citation2005]) was used to create the model of the M2 segments. We demonstrated that G + 2 could form a nucleophilic ring, thus providing interaction with terminal ammonium group of the blockers. A weakness of this model was the displacement of M2 segments from their positions in the template. This was performed to create a channel with a larger diameter than in K+ channels. In the present work I attempted to build analogous GluR1 model with M2 helices placed exactly as in the K+ channel template.
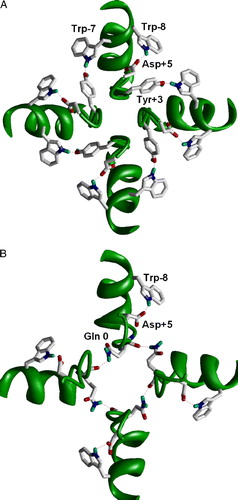
In K+ channels the selectivity filter structure is stabilized by H-bonds (A). Residues Asp + 5 and Trp-8 () are conserved among potassium channels and AMPARs. In the X-ray structures of K+ channels, there is a close disposition of these residues that allows H-bond formation. Preserving both residues in the AMPARs strongly suggests the same organization. The second stabilizing ring in potassium channels involves H-bonding of Trp-7 and Tyr + 3. Importantly, both are absent in AMPARs. Instead, in the AMPARs the intersegment H-bonds at the Q/R site (Tikhonov et al. [Citation1999], [Citation2002]) can participate in the stabilization of the structure (B). The deletion at position +4 in the AMPAR subunits and the difference in the stabilizing H-bonds can determine the difference in the pore diameters. Besides the diameter, selectivity filters of K+ channels and AMPAR channels differ by the number of lumen-facing oxygen rings. These nucleophilic rings determine interactions with permeating ions and blocking drugs. K+ channels have five such rings. Our results of probing of AMPAR channels with dicationic blockers (Magazanik et al. [Citation1997], Bolshakov et al. [Citation2000], [Citation2005]) suggest that the channel has only one lumen-facing oxygen ring (Tikhonov et al. [Citation2002]).
Figure 1. H-bonds stabilizing the selectivity-filter region in K+ channels (A) and in the model of the AMPA receptor channel (B). K+ channel is viewed from outside; the AMPAR channel is viewed from inside. The intrasubunit H-bond between Trp-8 and Asp + 5 is conserved in both types of channels. The intersubunit H-bonds between Trp-7 and Tyr + 3 is specific for K+ channels because both the donor and acceptor are absent in the AMPA receptors (see ). Instead, Gln 0 (Q/R) site form intersegment H-bonds in the AMPA receptor. This figure appears in colour in Molecular Membrane Biology online.
To generate the model I used the approach that was elaborated for modeling tetrodotoxin binding to Na+ channels (Tikhonov & Zhorov [Citation2005a]). The selectivity-filter region of the model was shaped around the dicationic adamantane derivative IEM-1460. Distance constraints were imposed to provide interaction of the terminal ammonium group of the drug with the oxygens of G + 2 residues. The stabilizing H-bonds shown in B were also clamped by distance constraints. The final model () was obtained by MCM optimization. Stability of the model was tested by subsequent optimization of the model without distance constraints. Although some deformations were observed, channel dimensions and orientation of important functional groups were not changed. The RMSD of heavy atoms between the constrained and unconstrained models (from position 0 to position +4) was 0.7 Å. Thus, model of the AMPAR channel can be built without displacement of M2 segments. The model was recently used for analysis of binding of polyamine toxins (Andersen et al. [Citation2006]).
Figure 2. Side view on the MthK-based model of the GluR1 channel. The dicationic derivative of adamantane (IEM-1460) is docked in the narrow part of the pore. The headgroup of the drug binds at the Q/R site, whereas the terminal ammonium group interacts with Gly + 2 residues. Reproduced with permission from Andersen et al. ([Citation2006]). This figure appears in colour in Molecular Membrane Biology online.
![Figure 2. Side view on the MthK-based model of the GluR1 channel. The dicationic derivative of adamantane (IEM-1460) is docked in the narrow part of the pore. The headgroup of the drug binds at the Q/R site, whereas the terminal ammonium group interacts with Gly + 2 residues. Reproduced with permission from Andersen et al. ([Citation2006]). This figure appears in colour in Molecular Membrane Biology online.](/cms/asset/b3698a9b-a780-4264-9cc6-d39052fa0b44/imbc_a_200775_f0002_b.jpg)
Unlike K+ channels, the model has only one lumen-facing oxygen ring formed by G + 2 residues (). Obviously, one oxygen ring is lost due to a deletion in the selectivity-filter region of AMPAR (). Two rings at the N/Q/R site (backbone and side-chain oxygens) are inaccessible because they are involved in the intersegment H-bonds. In the model of the AMPAR channel the main-chain oxygen of Q + 1 residues is directed outwards. Similar orientation of the homologous residue in K+ channels was observed (Berneche & Roux [Citation2005], Lenaeus et al. [Citation2005]) (). It seems obvious that relatively wide G + 2 ring determines low selectivity of AMPAR channels. However, quantitative analysis of selectivity is a separate job that probably requires molecular dynamics simulation with explicit water and use of free energy calculations.
Figure 3. Side view on the selectivity-filter region of K+ channel and the AMPA receptor channel model. Subunit at the left represents the proposed slow-inactivated conformation (Berneche & Roux [Citation2005]). Oxygen in position +1 is turned away from the pore. In the model of the AMPA receptor channel (right) one lumen-facing oxygen is lost because of deletion (see ). The oxygen in position 0 is involved in the intersegment H-bonds and the next one is turned away. As a result, the model has only one lumen-facing oxygen ring. This figure appears in colour in Molecular Membrane Biology online.
![Figure 3. Side view on the selectivity-filter region of K+ channel and the AMPA receptor channel model. Subunit at the left represents the proposed slow-inactivated conformation (Berneche & Roux [Citation2005]). Oxygen in position +1 is turned away from the pore. In the model of the AMPA receptor channel (right) one lumen-facing oxygen is lost because of deletion (see Table I). The oxygen in position 0 is involved in the intersegment H-bonds and the next one is turned away. As a result, the model has only one lumen-facing oxygen ring. This figure appears in colour in Molecular Membrane Biology online.](/cms/asset/d43599aa-fccf-4b37-86f8-1349c9e33267/imbc_a_200775_f0003_b.jpg)
Presence of Arg in position 0 (N/Q/R site) makes the AMPAR channel impermeable for Ca2 + and insensitive to the cationic blockers (see Dingledine et al. [Citation1999]). These properties are likely caused by positive charge of the Arg side chain. Like Asn and Gln residues, side chain of Arg residue can participate in the intersegment H-bonds. Structural properties of the AMPAR channels containing R-form subunits were discussed earlier (Tikhonov et al. [Citation1999]). In the present work only Q-form AMPARs are considered.
Selectivity filter of the NMDAR channel
C-ends of M2 segments in NMDARs are helical (Kuner et al. [Citation1996]). The selectivity-filter region of the NR1 subunit exhibits high homology with K+ channels (), but this region in NR2 subunit is significantly different. It contains VPV or VPI motif and the acidic residue is shifted from position +5 to +6 (in the NR2A and NR2B subunits) or is absent (in the NR2C and NR2D subunits). It is difficult to reveal specific interactions stabilizing the spatial structure of the narrow part of NMDAR channels. This makes the conformation of the region hardly predictable. It is a paradox, but numerous channel blockers practically do not help to solve the problem. The pore of the NMDAR is only about 5.5 Å in diameter (Villarroel et al. [Citation1995]), and, as a result, the vast majority of blockers bind at the entrance of the narrow part.
Channel blockers, such as MK-801 and phencyclidine, have a ‘V-like’ shape with hydrophobic or aromatic wings and an H-bond donating amino group at the vertex (Kroemer et al. [Citation1998]). Obviously, the binding site for such drugs should be a hydrophobic ‘funnel’ with a nucleophilic narrow part. It is well demonstrated that Asn residues of the ‘N’-site create the nucleophilic narrowing. Mutations at this position affect the pore diameter, Ca2 + permeability, and the binding of channel blockers (see Dingledine et al. [Citation1999]). Thus, the ‘N’-site seems to serve as the selectivity filter of the NMDAR channel. Particular properties governed by the ‘N’-site were explained by our hypothesis that the Asn residues form a cycle by intersegment H-bonding (Tikhonov et al. [Citation1999]). Unlike Gln residues in AMPARs, the side chain oxygens of the Asn residues in the NMDAR face the pore and form a nucleophilic ring, which effectively interacts with permeating and blocking cations. The hydrophobic site of drug binding remains unidentified although several residues in the outer vestibule affect the binding of channel blockers (Kashiwagi et al. [Citation2002]).
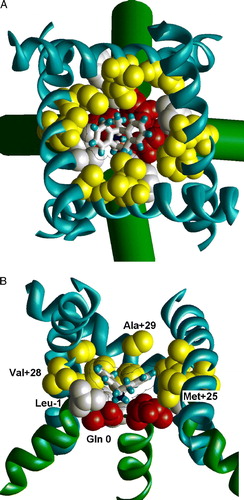
In the present work the homology model of the NMDAR channel (NR1, NR2A) was built using as template the open structure of ligand-gated K+ channel MthK. Cyclic H-bonds at the ‘N’-site (Tikhonov et al. [Citation1999]) were imposed by constraints. After the MC-minimization of the model, the binding of MK-801, phenylcyclohexyl-amine (IEM-1921), and memantine was studied by the random-docking method (see supplementary Figure at the end of the online version only). The optimal binding mode of MK801 is shown in . The drug binds in the position that overlaps with the binding site of TBA in K+ channels (Zhou et al. [Citation2001a]). In agreement with experimental data and with our previous suggestion (Tikhonov et al. [Citation1999]), the amino groups of the blockers interact with the side-chain oxygens of Asn 0 residues. Phe and Leu residues in position –1 form a funnel-like structure that binds to the hydrophobic wings of the blockers. In the M3 segment, residues in positions +25, +28 and +29 also contribute to the binding energy. The role of these residues in binding of NMDAR channel blockers was not demonstrated experimentally. Thus, this modeling study suggests a testable hypothesis about components of the binding site.
Figure 4. The optimal binding mode of MK801 in the model of the NMDA receptor channel. (A) Extracellular view. (B) Side view with one subunit removed. MK801 molecule is shown as sticks with van der Waals contour. The ligand-sensing residues are space filled. Asn residues of the ‘N’-site are shown red, residues in position –1 are shown white, and residues in positions +25, +28 and +29 are shown yellow. This figure appears in colour in Molecular Membrane Biology online.
It should be noted that the present model has significant uncertainties. In the absence of solid data about channel structure, it is not possible to analyze details of the interactions, in particular, unequal contribution of NR1 and NR2 subunits (e.g. Wollmuth et al. [Citation1996]). Significant further efforts are necessary to reproduce structure-activity relationships of the NMDAR channel blockers at the level of molecular models.
Outer vestibule of AMPAR channel
Transmembrane M3 segments of glutamate receptors are homologous to the S6 segments of K+ channels that form straight (closed channels) or kinked (open channels) helices. The main source of data about the spatial structure of M3 segments came from scanning mutagenesis (Sobolevsky et al. 2003). To check the consistency of these data with K+ channel templates the homology models of the open and closed AMPAR channel were built based on KcsA (closed) and MthK (open) structures of K+ channels. The location of the residues that are labeled is shown in .In the open channel model all residues facing the outer channel vestibule are labeled (A). An interesting fact is that several deeply located residues become accessible in the open state (B). These residues face the pocket between M2 and M3 segments. In Ca2 + and Na+ channels homologous residues control interactions with many classes of ligands and their access to the pore (see Zhorov & Tikhonov [Citation2004]).
Figure 5. Cys labeling of the M3 segment of AMPA receptor. (A and B) Open channel model. (C and D) Closed channel model. A, C and D are viewed from outside; B, side view. Labeled residues are space filled red. (A) Labeled residues line the open channel. (B) Side view on labeled residues in the M2-M3 contact. MTS reagent can reach this region through the hole above M2 helices. (C) Labeled residues L + 25, I + 28 and T + 32 face the cavity of the closed channel. (D) Labeled residues L + 35 and F + 38 are projected outwards. This figure appears in colour in Molecular Membrane Biology online.
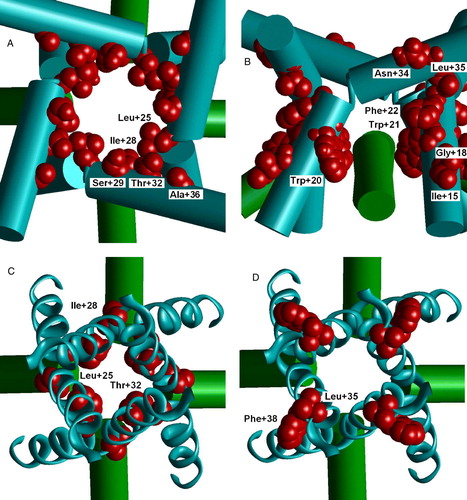
Residues L + 25, I + 28 and T + 32 face the cavity of the closed channel (C). Contrary, labeled residues L + 35 and F + 38 are projected outwards (D). Noteworthy, residues A + 33 and A + 36, which are projected inwards at the narrow region of M3 helix crossing, are not labeled. Thus, the interpretation of the Cys labeling data could be more complex than in the original paper (Sobolevsky et al. [Citation2003]). The labeled residues are not at one side of the helix in the KcsA template.
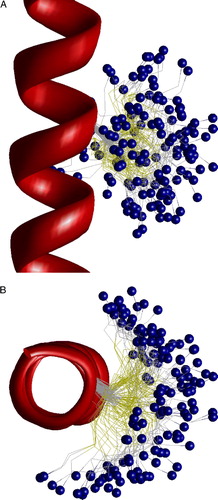
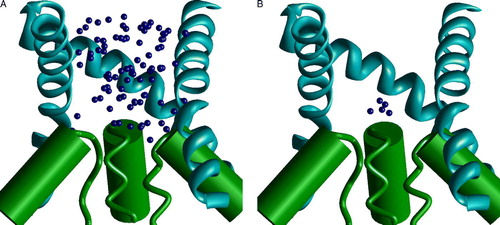
Assuming numerous limitations of the SCAM approach, straightforward interpretation of the data is not always the best one. An important problem of SCAM is the flexibility of the labels. Position of a label is often associated with the alpha carbon of the labeled residue. This assumption is particularly important in the studies of the voltage-dependence of modification rates. What is the real distance between the charged group of a label and the alpha carbon of the labeled residue? To address this question the model of 20-alanine peptide in helical conformation was built, and labeled Cys was introduced in the middle. MC-optimization of the model has produced 148 low-energy structures that are visualized in . It can be seen that the ammonium group of the label reaches neighboring turns of the helix. In the axial view the label covers a half of helix.
Figure 6. The side (A) and top (B) views of the low-energy conformations of MTSET bound to Cys residue in alpha helix. Positions of the nitrogen atom are shown as small blue spheres. Flexibility of the label allows wide distribution of its terminal group. This figure appears in colour in Molecular Membrane Biology online.
According to Sobolevsky et al. ([Citation2005]) the voltage-dependence of Cys modification in positions +25, +28 and +32 coincide if the channel is closed. In the KcsA-based (closed) model of the AMPAR channel labels were introduced in these positions. Optimization of these models has produced structures with very similar positions of ammonium groups of the labels () in the cavity. This position is at the focus of M2 helices macrodipoles. In the MthK-based (open) model position of the label is not restricted by limited volume of the cavity and ammonium group of the label is more mobile. As a result, it should be more sensitive to the membrane voltage.
Figure 7. Side view on the superimposition of the closed channel models with L + 25, I + 28 and T + 32 substituted by MTSET bound cysteines. Due to conformational flexibility of the label, the ammonium groups of all three substitutes are located at the focus of M2 helical dipoles. This figure appears in colour in Molecular Membrane Biology online.
Outer vestibule of NMDAR channel
Although NMDAR and AMPAR channels seem to have principally similar topography of the outer vestibule (Beck et al. [Citation1999], Sobolevsky et al. [Citation2002a]), they have interesting peculiarities. SCAM analysis has suggested significant staggering of M3 segments in NR1 and NR2 subunits (Sobolevsky et al. [Citation2002b]). I do not believe that the subunits are really shifted along the pore axis: such a shift would affect many intersubunit contacts. At least, an alternative explanation should be looked for.
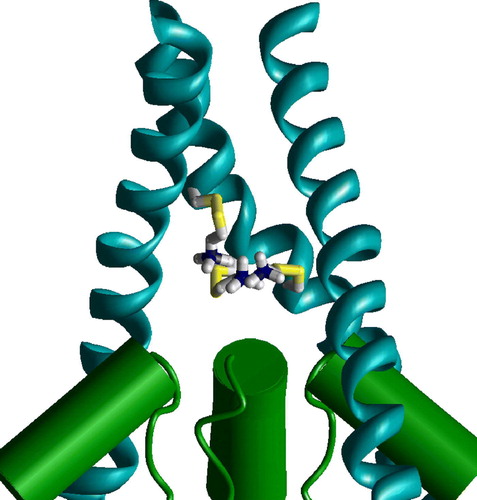
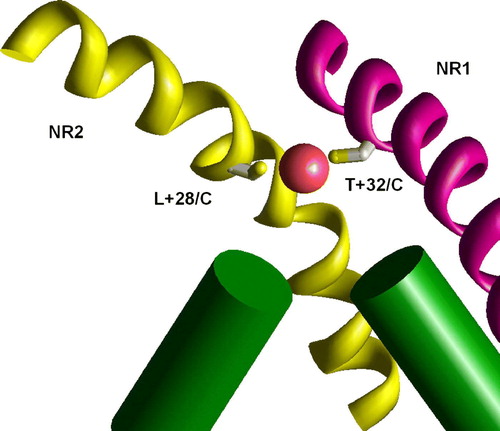
The voltage-gated (KvAP) and ligand-gated (MthK) K+ channels have significantly different vestibule structures. In the KvAP but not in MthK structure the inner helices have significant slope to the pore axis. As a result, homologous residues are in different depth in the pore. To test if this difference can account for experimental data on NMDAR channel, I have generated the NR1/NR2A model of the NMDAR channel, in which M3 segments of NR1 subunits have been modeled according to the KvAP template, whereas M3 segments of NR2 subunits have been modeled according MthK template. demonstrates that in this asymmetric model the residues A + 37 in NR1 subunit and A + 33 in NR2 subunit are exactly at one level. This agrees with experimental data (Sobolevsky et al. [Citation2002b]) about the voltage dependence of modification of the substituted cysteines. Experiments demonstrated that the double mutant T + 32/C (in NR1) L + 28/C (in NR2) is effectively blocked by Cu2 + , probably due to the ion chelation between the cysteins (Sobolevsky et al. [Citation2002b]). The asymmetrical model readily reproduces this result. The distance between the corresponding alpha carbons is about 9 Å. The model of the double mutant perfectly accommodates Cu2 + ion between Cys side chains (). Possible origin of the asymmetrical open structure is evident for heteromeric NMDAR channels. NR1 subunits bind glycine, whereas NR2 subunits bind glutamate or its agonist. Different opening forces should induce asymmetrical open structure even if the resting state is symmetrical. We do not know arrangement of subunits in NMDARs. But in any case the channel has at least one NR1 – NR2 contact. At this contact T + 32/C and L + 28/C cysteines can chelate a Cu2 + ion. Intrapore binding of the Cu2 + ion causes channel block.
Figure 8. Staggering of the M3 segments of NMDA receptor is explained by different slope. In the heteromeric model M3 segments of NR1 subunits (magenta) are KvAP-like, whereas M3 segments of NR2 subunits (yellow) are MthK-like. Residues at positions 37 (NR1) and 33 (NR2) are at the same level (space filled red). This figure appears in colour in Molecular Membrane Biology online.
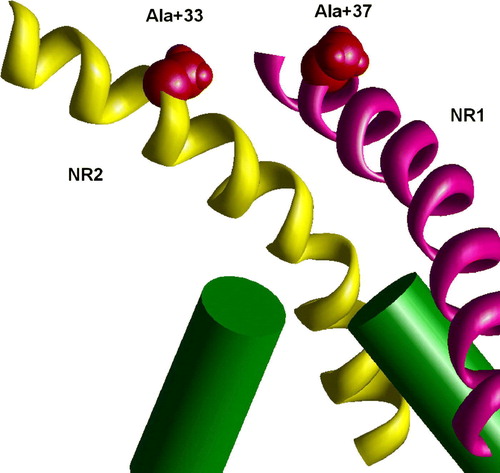
Figure 9. The binding of Cu2 + ions in the model of the double mutant NR1 T32/C NR2 L28/C. Due to different slope of M3 segments in NR1 (KvAP-like conformation, magenta) and NR2 (MthK-like conformation, yellow) these residues are at the same level and effectively bind Cu2 + ion. This figure appears in colour in Molecular Membrane Biology online.
Gating
The mechanism(s) of glutamate receptor channel activation is one of the most intriguing and challenging problems. In K+ channel three types of gating have been revealed. Activation gating occurs through the movement of the inner helices that opens the channel vestibule. The ‘ball-and-chain’ mechanism is believed to be responsible for fast inactivation gating. Finally, slow inactivation seemingly occurs at the level of selectivity filter. Redirection of one or more lumen-facing oxygen rings away from the lumen make the selectivity filter non-conducting.
Experimental data on the glutamate receptor channel are contradictory. State-dependence accessibility of substituted cysteines suggests that gating rearrangements are at the level of selectivity filter. Contrary, state-dependent binding of channel blocking and trapping effects gives evidence to favor of extracellular location of gate. M3 segments of glutamate receptor channels including the K+ selective one (Chen et al. [Citation1999]) lack the ‘glycine hinge’, an important determinant of K+ channel gating. On the contrary, mutations in the middle of M3 (Kohda et al. [Citation2000], Jones et al. [Citation2002]) but not in the selectivity-filter region strongly affect activation properties of glutamate receptor channels. To explain the existent paradoxes Sobolevsky et al. ([Citation2003]) suggested that the movement of the rigid M3 segments in glutamate receptors induces the movement of M2 segments. Thus, gating rearrangements in glutamate receptor channels could occur simultaneously at both levels.
Organic blockers can serve as tools to study the topography of channel binding sites (see above). They also can be used for the analysis of the gating mechanisms. Open-channel blockers of NMDARs bind in the outer vestibule near the selectivity filter. Thus, they could interact with both putative gating regions. Indeed, various interrelations between the binding of blockers and the channel gate have been observed. Compounds like MK801 and phencyclidine do not prevent channel closure and can be trapped in the closed channel (MacDonald et al. [Citation1991]). Contrary, 9-aminoacridne (Benveniste & Mayer [Citation1995]) and tetrapenthylammonium (Sobolevsky et al. [Citation1999]) prevent channel closure. This effect is often called the ‘foot-in-the-door’ blockade. Intermediate situation of ‘partial trapping’ has been observed for memantine (Blanpied et al. [Citation1997]) and some other drugs (Mealing et al. [Citation2001]; Bolshakov et al. [Citation2003]).
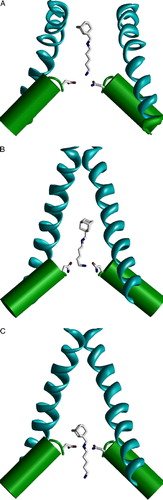
Trapping of dicationic derivatives of adamantane is voltage-dependent: at depolarized voltages they bind to some shallow site and exhibit foot-in-the-door blockade; hyperpolarization induces binding to a deep site and trapping (Antonov & Johnson [Citation1996]). The authors also demonstrated that increasing the bulkiness of the terminal ammonium group prevents the deep binding mode and trapping of the blockers in the closed NMDAR channels. For dicationic blockers like IEM-1754, two binding modes are possible: with secondary (deep mode) or terminal (shallow mode) amino group bound to the oxygens of the ‘N’-site asparagines near the focus of M2 helical dipoles. These binding modes were imposed by distance constraints between the side-chain oxygens in the Asn0 residues and corresponding amino group of the blocker. If the terminal amino group of the blocker is bound to the ‘N’-site asparagines, the molecule should prevent channel closure: in the open- and closed-channel models conformations of IEM-1754 are different (A and B). This difference is because the limited space of the cavity of the closed-channel model requires folding of the pentamethelene chain to accommodate the molecule. This conformational change of the ligand is energetically expensive because the distance between the charged amino groups is decreased. In the deep mode, only the adamantane moiety is located in the cavity of the closed channel (C). Obviously, this mode does not prevent channel closure and trapping inside the closed channel. It is clear from the model, that trapping is possible only if the ‘tail’ part enters the selectivity filter of the open channel and remains there in the closed state. Indeed, increasing the terminal group prevents the penetration through the selectivity filter, and such compounds cannot be trapped (Antonov & Johnson [Citation1996], Bolshakov et al. [Citation2003]). Thus, modeling based on the K+ channel templates reproduces the experimental data. It allows us to conclude that the selectivity filter of the NMDAR channel is not collapsed in the closed state.
Figure 10. Binding of the dicationic adamantane derivative IEM-1754 to the open and closed models of the NMDA receptor channel. Asn residues of the ‘N’-site are shown as sticks. (A and B) IEM-1754 binds to the N site by the terminal amino group. Binding modes in the open channel model (A) and in the closed channel model (B) are different due to the limited space in the closed channel cavity. The permeation of the tail through the selectivity filter allows binding of the secondary amino group to the ‘N’-site (C). Unlike the shallow binding mode (A), this deep binding mode permits channel closure and trapping. This figure appears in colour in Molecular Membrane Biology online.
It should be noted, that in K+ channels the gate at the selectivity filter level does not work as a steric plug. Rather, local rearrangements can make the selectivity-filter hydrophobic and thus, impermeable (Zhou et al. [Citation2001b], Zhou & MacKinnon [Citation2003], Berneche & Roux [Citation2005], Lenaeus et al. [Citation2005]). Another possible gating mechanism involves the misalignment of the P-loop helices as it is seen in the KirBac structure (Kuo et al. [Citation2003]). If helical dipoles are not focused in the center of the pore, the loss of combining the dipole electrostatic effect may result in the destabilization of an ion. In both cases, the spatial changes are modest. For example, the displacement of the C-ends of pore helices in the KirBac structure does not exceed 2 Å. Thus, large-scale conformational rearrangements likely occur at the level of the extracellular gate but not at the level of the selectivity filter. Nevertheless, small changes of the spatial structure in the selectivity filter can be of critical functional importance. For example, they can affect H-bonds between the N/Q/R site residues, thus changing the disposition of important groups in the selectivity filter.
The closed-channel model readily accommodates trapping (MK801), partial trapping (memantine), and non-trapping (9-aminoacridine) blockers (data not shown). This is not surprising because dimensions of the compounds do not exceed tetrabuthylammonium, which has been crystallized inside the closed KcsA channel. Therefore, differences between the effects of these compounds on channel gating remain unclear. It is likely that the differences are due to some specific interactions with residues that undergo conformational transitions during channel gating. Recent data suggest that various blockers affect equilibrium between different states of the NMDARs (Dilmore & Johnson [Citation1998], Blanpied et al. [Citation2005], Yuan et al. [Citation2005]). Regrettably, structural determinants of the ‘foot-in-the-door’ blockade are not studied. 9-aminoacridine and its tetrahydro analog (Vorobjev & Sharonova [Citation1994]) are the only examples of potent NMDAR channel blockers that prevent closure of the gate. The pattern of residues affecting blockade by 9-aminoacridine is also not known. Without such data speculations about details of gating rearrangements seem to be premature. Even within the family of K+ channels open-channel structures demonstrate essential differences depending on the local sequence (Kv1.2 vs KvAP) or on the mechanism of activation (KvAP vs MthK).
This paper was first published online on iFirst on 19 February 2007.
Supplementary Figure 1. The random docking search of the optimal binding mode of MK801 in the model of NMDA receptor channel. Side views with one subunit hidden. (A and B) The two steps of the random-docking procedure. Blue spheres represent positions of the nitrogen atom of MK801. (A) 100 best structures obtained after short optimization of 5000 random positions of the drug molecule. Possible positions of the drug fit the outer vestibule of the channel. (B) Five best complexes obtained after long optimization of the structures shown in A. The charged amino group is bound at the focus of macrodipoles of the M2 helices.
This work was supported by grant from RFBR (04-04-49143) and by grant from program ‘Molecular and cell biology’. I am thankful to Lev Magazanik and Iva Bruhova for reading the manuscript and valuable comments.
References
- Andersen TF, Tikhonov DB, Bolcho U, Bolshakov K, Nelson JK, Pluteanu F, Mellor IR, Egebjerg J, Stromgaard K. Uncompetitive antagonism of AMPA receptors: mechanistic insights from studies of polyamine toxin derivatives. J Med Chem 2006; 49: 5414–5423
- Antonov SM, Johnson JW. Voltage-dependent interaction of open-channel blocking molecules with gating of NMDA receptors in rat cortical neurons. J Physiol 1996; 493: 425–445
- Beck C, Wollmuth LP, Seeburg PH, Sakmann B, Kuner T. NMDAR channel segments forming the extracellular vestibule inferred from the accessibility of substituted cysteines. Neuron 1999; 22: 559–570
- Bennett JA, Dingledine R. Topology profile for a glutamate receptor: three transmembrane domains and a channel-lining reentrant membrane loop. Neuron 1995; 24: 373–384
- Benveniste M, Mayer ML. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J Physiol 1995; 483: 367–384
- Berneche S, Roux B. A gate in the selectivity filter of potassium channels. Structure 2005; 13: 591–600
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol 1997; 77: 309–323
- Blanpied TA, Clarke RJ, Johnson JW. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J Neurosci 2005; 25: 3312–3322
- Bolshakov KV, Tikhonov DB, Gmiro VE, Magazanik LG. Different arrangement of hydrophobic and nucleophilic components of channel binding sites in N-methyl-D-aspartate and AMPA receptors of rat brain is revealed by channel blockade. Neurosci Lett 2000; 291: 101–104
- Bolshakov KV, Gmiro VE, Tikhonov DB, Magazanik LG. Determinants of trapping block of N-methyl-d-aspartate receptor channels. J Neurochem 2003; 87: 56–65
- Bolshakov KV, Kim KH, Potapjeva NN, Gmiro VE, Tikhonov DB, Usherwood PN, Mellor IR, Magazanik LG. Design of antagonists for NMDA and AMPA receptors. Neuropharmacology 2005; 49: 144–155
- Bruhova I, Zhorov BS. KvAP-based model of the pore region of Shaker potassium channel is consistent with cadmium- and ligand-binding experiments. Biophys J 2005; 89: 1020–1029
- Burnashev N, Villarroel A, Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J Physiol 1996; 496: 165–173
- Chen GQ, Cui C, Mayer ML, Gouaux E. Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature 1999; 402: 817–821
- del Camino D, Holmgren M, Liu Y, Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature 2000; 403: 321–325
- Dilmore JG, Johnson JW. Open channel block and alteration of N-methyl-D- aspartic acid receptor gating by an analog of phencyclidine. Biophys J 1998; 75: 1801–1816
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 1999; 51: 7–61
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998; 280: 69–77
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. The open pore conformation of potassium channels. Nature 2002; 417: 523–526
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature 2003; 423: 33–41
- Jones KS, VanDongen HM, VanDongen AM. The NMDA receptor M3 segment is a conserved transduction element coupling ligand binding to channel opening. J Neurosci 2002; 22: 2044–2053
- Kashiwagi K, Masuko T, Nguyen CD, Kuno T, Tanaka I, Igarashi K, Williams K. Channel blockers acting at N-methyl-D-aspartate receptors: differential effects of mutations in the vestibule and ion channel pore. Mol Pharmacol 2002; 61: 533–545
- Kohda K, Wang Y, Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci 2000; 3: 315–322
- Kroemer RT, Koutsilieri E, Hecht P, Liedl KR, Riederer P, Kornhuber J. Quantitative analysis of the structural requirements for blockade of the N-methyl-D-aspartate receptor at the phencyclidine binding site. J Med Chem 1998; 41: 393–400
- Kuner T, Wollmuth LP, Karlin A, Seeburg PH, Sakmann B. Structure of the NMDA receptor channel M2 segment inferred from the accessibility of substituted cysteines. Neuron 1996; 17: 343–352
- Kuner T, Beck C, Sakmann B, Seeburg PH. Channel-lining residues of the AMPA receptor M2 segment: structural environment of the Q/R site and identification of the selectivity filter. J Neurosci 2001; 21: 4162–4172
- Kuner T, Seeburg PH, Guy HR. A common architecture for K+ channels and ionotropic glutamate receptors?. Trends Neurosci 2003; 26: 27–32
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 2003; 300: 1922–1926
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins 1999; 35: 133–152
- Lenaeus MJ, Vamvouka M, Focia PJ, Gross A. Structural basis of TEA blockade in a model potassium channel. Nat Struct Biol 2005; 12: 454–459
- Li Z, Scheraga HA. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci USA 1987; 84: 6611–6615
- Lipkind GM, Fozzard HA. KcsA crystal structure as framework for a molecular model of the Na(+) channel pore. Biochemistry 2000; 39: 8161–8170
- Lipkind GM, Fozzard HA. Modeling of the outer vestibule and selectivity filter of the L-type Ca2 + channel. Biochemistry 2001; 40: 6786–6794
- Lipkind GM, Fozzard HA. Molecular modeling of interactions of dihydropyridines and phenylalkylamines with the inner pore of the L-type Ca2 + channel. Mol Pharmacol 2003; 63: 499–511
- Lipkind GM, Fozzard HA. Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol Pharmacol 2005; 68: 1611–1622
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005; 309: 897–903
- MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, Schneiderman JH, Pennefather PS. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J Physiol 1991; 432: 483–508
- Magazanik LG, Buldakova SL, Samoilova MV, Gmiro VE, Mellor IR, Usherwood PN. Block of open channels of recombinant AMPA receptors and native AMPA/kainate receptors by adamantane derivatives. J Physiol 1997; 505: 655–663
- Mealing GA, Paris J, Lou A, Edwards A, Hou ST, MacManus JP, Hakim AM, Morley P. Structural modifications to an N-methyl-D-aspartate receptor antagonist result in large differences in trapping block. J Neurochem 2001; 78: 316–324
- O'Reilly AO, Khambay BP, Williamson MS, Field LM, Wallace BA, Davies TG. 2006. Modelling insecticide binding sites at the voltage-gated sodium channel. Biochem J 396:255–263.
- Panchenko VA, Glasser CR, Mayer ML. Structural similarities between glutamate receptor channels and K(+) channels examined by scanning mutagenesis. J Gen Physiol 2001; 117: 345–360
- Qian A, Johnson JW. Channel gating of NMDA receptors. Physiol Behav 2002; 77: 577–582
- Rosenmund C, Stern-Bach Y, Stevens CF. The tetrameric structure of a glutamate receptor channel. Science 1998; 280: 1596–1599
- Sobolevsky AI, Koshelev SG, Khodorov BI. Probing of NMDA channels with fast blockers. J Neurosci 1999; 19: 10611–10626
- Sobolevsky AI, Beck C, Wollmuth LP. Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron 2002a; 33: 75–85
- Sobolevsky AI, Rooney L, Wollmuth LP. Staggering of subunits in NMDAR channels. Biophys J 2002b; 83: 3304–3314
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. Different gating mechanisms in glutamate receptor and K+ channels. J Neurosci 2003; 23: 7559–7568
- Sobolevsky AI, Yelshansky MV, Wollmuth LP. State-dependent changes in the electrostatic potential in the pore of a GluR channel. Biophys J 2005; 88: 235–242
- Tikhonov DB, Zhorov BS, Magazanik LG. Intersegment hydrogen bonds as possible structural determinants of the N/Q/R site in glutamate receptors. Biophys J 1999; 77: 1914–1926
- Tikhonov DB, Mellor JR, Usherwood PN, Magazanik LG. Modeling of the pore domain of the GLUR1 channel: homology with K(+) channel and binding of channel blockers. Biophys J 2002; 82: 1884–1893
- Tikhonov DB, Zhorov BS. Sodium channel activators: model of binding inside the pore and a possible mechanism of action. FEBS Lett 2005a; 579: 4207–4212
- Tikhonov DB, Zhorov BS. Modeling P-loops domain of sodium channel: homology with potassium channels and interaction with ligands. Biophys J 2005b; 88: 184–197
- Villarroel A, Burnashev N, Sakmann B. Dimensions of the narrow portion of a recombinant NMDA receptor channel. Biophys J 1995; 68: 866–875
- Vorobjev VS, Sharonova IN. Tetrahydroaminoacridine blocks and prolongs NMDA receptor-mediated responses in a voltage-dependent manner. Eur J Pharmacol 1994; 253: 1–8
- Webster SM, del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 2004; 428: 864–868
- Weiner SJ, Kollman PA, Case DA, Singh UC, Chio C, Alagona G, Profeta S, Weiner PK. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 1984; 106: 765–784
- Wo ZG, Oswald RE. Transmembrane topology of two Kainate receptor submits revealed by N-glycosylation. Proc Natl Acad Sci USA 1994; 91: 7154–7158
- Wollmuth LP, Kuner T, Seeburg PH, Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. J Physiol 1996; 491: 779–797
- Wollmuth LP, Sobolevsky AI. Structure and gating of the glutamate receptor ion channel. Trends Neurosci 2004; 27: 321–328
- Wood MW, VanDongen HM, VanDongen AM. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc Natl Acad Sci USA 1995; 92: 4882–4886
- Yamaguchi S, Zhorov BS, Yoshioka K, Nagao T, Ichijo H, Adachi-Akahane S. Key roles of Phe1112 and Ser1115 in the pore-forming IIIS5-S6 linker of L-type Ca2 + channel alpha1C subunit (CaV 1.2) in binding of dihydropyridines and action of Ca2 + channel agonists. Mol Pharmacol 2003; 64: 235–248
- Yuan H, Erreger K, Dravid SM, Traynelis SF. Conserved structural and functional control of N-methyl-D-aspartate receptor gating by transmembrane domain M3. J Biol Chem 2005; 280: 29708–29716
- Zhorov BS, Folkman EV, Ananthanarayanan VS. Homology model of dihydropyridine receptor: implications for L-type Ca(2 + ) channel modulation by agonists and antagonists. Arch Biochem Biophys 2001; 393: 22–41
- Zhorov BS, Tikhonov DB. Potassium, sodium, calcium and glutamate-gated channels: pore architecture and ligand action. J Neurochem 2004; 88: 782–799
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001a; 411: 657–661
- Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. 2001b. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 414:43–48.
- Zhou Y, MacKinnon R. 2003. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol 333:965–975.