Abstract
It was recently shown that the region within β-spectrin responsible for interactions with ankyrin includes a lipid-binding site which displayed sensitivity to inhibition by ankyrin. We studied its structure by constructing a series of single and double spin-labeled β-spectrin-derived peptides and analyzing their spin-spin distances via electron paramagnetic resonance spectroscopy and the Fourier deconvolution method. The results indicate that the whole ankyrin-sensitive lipid-binding site of β-spectrin exhibits a helical conformation revealing a distinct 310-helix contribution at its N-terminus. The start of the helix was located five residues upstream along the sequence compared to the theoretical predictions. A model based on the obtained data provides direct evidence that the examined lipid-binding site is a highly amphipathic helix, which is correlated with the specific conformation of its N-terminal fragment.
| Abbreviations | ||
| AE1 | = | anion-exchanger 1 |
| CD | = | circular dichroism |
| EPR | = | electron paramagnetic resonance |
| FPLC | = | fast protein liquid chromatography |
| GuHCl | = | guanidinehydrochloride |
| MTSL | = | 3-methylthiosulfonyl-1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline |
| PC | = | phosphatidylcholine |
| PE | = | phosphatidylethanolamine |
| SDSL | = | site-directed spin labeling |
| SUV | = | small unilamellar vesicle |
Introduction
Spectrin is a high molecular weight flexible rod-like protein formed by the head-to-head association of two heterodimers composed of α (280 kD) and β (247 kD) subunits. It is a major component of the membrane skeleton in human erythrocytes and indeed in most metazoan cells Citation[1–3]. Each of the subunits consists largely of triple-helical repeats of approximately 106 amino acid residues Citation[4], Citation[5]. Domains of such arrangement also characterize the many proteins that comprise the spectrin superfamily Citation[6–8]. Structural studies of various triple-helical units of spectrin indicate that these are slightly distorted, left-handed coiled coils, with two parallel (A and C) and one antiparallel (B) helices Citation[9–11], and the stability of the whole structure is determined by inter-helical interactions Citation[12], Citation[13]. The properties of the repeats, their relative arrangement and orientation, and the conformational stability of the helical linkers between the repeating units are thought to be key determinants of the structural and mechanical features of spectrin Citation[10–14]. The spectrin repeats also serve as important interaction sites for multiple signaling and structural proteins Citation[6]. The latter include ankyrin, which interacts with 14th and 15th repeats of β-spectrin and links the molecule to AE1 (anion-exchanger 1) protein Citation[15], Citation[16], yielding one of the major anchors between the membrane skeleton and the lipid bilayer.
Direct spectrin-lipid interactions were observed by a number of authors Citation[17–21], and they are thought to exert an influence on the stability of the membrane. However, the high affinity lipid-binding sites seem to be confined to particular regions of spectrin Citation[20–22]. When the effect of ankyrin on the binding of erythroid spectrin to phospholipid vesicles was tested, this interaction was inhibited Citation[23]. The mechanism of this inhibition appeared to be competitive in the case of PE/PC (phosphatidylethanolamine/phosphatidylcholine) vesicles or monolayer binding by spectrin. Similar results were obtained for brain spectrin Citation[19], and it has become apparent that β-spectrin is responsible for such interactions. Most recently, an ankyrin-sensitive binding site for PE-rich lipids was located in a 38-residue N-terminal fragment of the β-spectrin ankyrin-binding domain (residues 1768-1805), and the first eight residues appeared to play a key role in this activity Citation[24]. Still, the nature of lipid binding by this region remains to be elucidated. Detailed structural information is needed to address this question.
Here, we report on the structural features of the ankyrin-sensitive lipid-binding site in erythroid β-spectrin. To achieve this, a peptide comprising the whole 14th repeat flanked by shorter fragments of adjacent units of β-spectrin (amino acid residues 1638–1845) was used for cysteine replacement, with either one or two cysteines at specific positions within the lipid-binding site. After spin label attachment to the cysteines, the peptides were used for EPR (electron paramagnetic resonance) studies and subsequent spin-spin interaction analysis via the Fourier deconvolution method Citation[25], Citation[26]. Our results indicate that the whole region exhibits a helical structure which forms helix C of the 14th repeat of β-spectrin. Moreover, the N-terminal fragment of the helix manifests a 310-helix rather than a typical α-helix conformation. Based on the measured distances, a structural model of the studied region was prepared so as to relate the specific conformation of the helix to its lipid-binding properties. Apparently, the 310 configuration at the N-terminus of the helix provokes its highly amphipathic character. The secondary structure of this fragment does not undergo any significant changes while interacting with phospholipids or detergent.
Materials and methods
Site-directed mutagenesis, protein isolation, spin labeling and characterization of mutated peptides
Substitutions were introduced into the pRSET vector containing the gene encoding erythrocyte β-spectrin-derived peptide (amino acid residues 1638-1845 according to the sequence gi:134798) with a Quick-Change site-directed mutagenesis kit (Stratagene, La Jolla, USA). Five single-cysteine and 12 double-cysteine variants were constructed and the mutations were confirmed via DNA sequencing. The peptides were expressed in Escherichia coli BL21(DE3)pLysE cells (Invitrogen), and purified as described before Citation[24].
The peptides were incubated overnight with dithiotreitol (10 mM), and the latter was removed prior to spin labeling using an FPLC (fast protein liquid chromatography) system equipped with a Bio-Silect SEC 250-5 (Bio-Rad, Hercules, USA) column. A 20-fold excess of MTSL 3-methylthiosulfonyl-1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline, (Toronto Research Chemicals, North York, Canada) Citation[27] was used to interact with the peptides. The samples were concentrated and the reaction was allowed to proceed overnight in the presence of GuHCl (guanidinehydrochloride). The addition of the latter helped to improve spin labeling efficiency; however, it did not affect the measured distances. Eventually, unattached MTSL and GuHCl was removed by another run of the Bio-Silect column.
The structural properties of thus-treated peptides were preserved, as confirmed by CD (circular dichroism) measurements performed on a Jasco J-715 CD spectrometer. A thermostat-controlled cell with a 0.1-cm path length was used at 10°C, or from 10 to 70°C in 10°C increments and the protein samples were in a 20 mM Tris-HCl/100 mM NaCl, pH 8 buffer. Helical contents were calculated from values of the amide nΠ* transition at 222 nm ([κ]222), using a value of -36,000 degrees·cm2·dmol−1 to represent 100% α-helical content.
The protein concentration was determined via absorbance measurements at 280 nm on a Cary 1E (Varian) UV-Vis spectrometer or via the Bradford method (Bio-Rad). After spin labeling, the free cysteine concentration was assayed with Ellman's reagent (DTNB) (P-L Biochemicals, Milwaukee, USA), and the spin-labeling efficiency was calculated as follows: 100% [(total concentration of cysteines) - (concentration of free cysteines)]/(total concentration of cysteines).
Monolayer experiments, performed via the Wilhelmy technique on a Nima (Nima Technology, Coventry, UK) tensiometer with a Teflon trough of 24-cm2 surface area, were carried out as described elsewhere Citation[24]. PE/PC (3/2 mol/mol) SUV (small unilamellar vesicle) liposomes were prepared by lipid-film hydratation (with a 20 mM Tris-HCl/100 mM NaCl, pH 7.5 buffer) and size extrusion through filters with 100-nm pores.
EPR measurements
EPR spectra were obtained on a Bruker ESP 300E 9 GHz spectrometer. Samples of approximately 150–320 µM in a 20 mM Tris-HCl/100 mM NaCl, pH 7.5 buffer (alternatively with 2% octyl-glucoside or 20 mg/ml PE/PC liposomes) were frozen in liquid nitrogen in a Dewar flask. EPR spectra were acquired using a 1 G field modulation amplitude at 100 kHz and a 0.2–2 mW incident microwave power. A magnetic sweep width of 200 G was used. Each time, a total number of nine scans were accumulated. All the measurements were performed at least in triplicate.
Spin-label separation calculations
The distances between two interacting spin labels were determined using the Fourier deconvolution method Citation[25], Citation[26]. The EPR spectrum of the two interacting spins in doubly labeled peptides was treated as a convolution of the spectrum of non-interacting spins in the singly labeled peptides with a dipolar broadening function. With two assumptions, namely that the broadening function was utilized to estimate average splitting values (<2B > ) for doubly labeled peptides, and that the coupling between the two spins was presumed to be axially symmetric, the mean spin-spin distance (<r > ) was determined according to the equation: <r > =[(0.75)(3/2geβµo/4π)/ < 2B > ]1/3, where ge is the isotropic g value for electrons, β is the electron Bohr magneton, and µo is the permittivity of a vacuum. The cutoff frequency for the determination of the Gaussian fit broadening function in Fourier space was at points 20–40; thus, the reported average distances were accompanied by error ranges observed for several cutoff frequencies applied to several data sets (see ). All the numerical data was analyzed using Origin (Microcal, Northampton, USA) and Excel (Microsoft Corporation) software.
Structure prediction and analysis
For all the theoretical analyses, the sequence of human erythroid β-spectrin (accession number gi:134798) was used. The structural models of β-spectrin-derived peptides were predicted using the Robetta server (http://robetta.bakerlab.org/) Citation[28]. The secondary structures and physicochemical properties were analyzed using GOR4 and ProtParam (http://www.expasy.org) and the SMART tool Citation[29] (http://smart.embl-heidelberg.de).
Three-dimensional models of spin-labeled peptides were manually constructed using MOLDEN software Citation[30] (http://www.cmbi.ru.nl/molden/). Estimates of the distance between spin labels were obtained by manipulation of backbone torsion angles (φ and ψ) upon defined torsion angles for the R1 side chain () on a helical surface Citation[31], Citation[32], namely: χ1=-60°, χ2=-60° and χ3=-90°. As the rotational freedom of the χ4 and χ5 torsion angles produce little displacement of the nitroxide, the paramagnetic centre of the latter was considered to protrude by a length of 7 ? along the line perpendicular to the helix axis intersecting the α-carbon and β-sulphur. This is considered to be a good approximation of the average conformation of the R1 side chain of a helical protein in a frozen-solution state. The constructed models were subjected to geometry optimalization (Polak-Ribiere algorithm) and molecular modeling (run time = 1 ps, step size = 0.1 fs, simulation temperature = 300 K) using the HyperChem molecular modeling system (Hypercube, Inc.) Citation[33], within a periodic box (26.1×26.1×56.1 ?, maximum number of water molecules = 1263) in the case of the 1760-1790 fragment (see c). All the structures were visualized via YASARA (http://www.yasara.org/).
Figure 1. The structure of the R1 side chain produced by the reaction of MTSL with the sulfhydryl group of a cysteine residue. The dihedral angles associated with each bond are defined as χ1 – χ 5.
Results
Preparation and characterization of spin-labeled peptides comprising the ankyrin-binding domain of β-spectrin
In order to study the structure of the lipid-binding site of β-spectrin, we prepared a series of 12 double cysteine mutants with the corresponding five single cysteine mutants within a peptide comprising amino acid residues 1638-1845 of the β chain of human erythrocyte spectrin. The mutants were subsequently spin labeled with MTSL; thus, cysteine residues were substituted by R1 side chains (see ) Citation[27], Citation[34], yielding the following peptides: 1756R1, 1755R1/1758R1, 1756R1/1759R1, 1757R1/1760R1, 1761R1, 1761R1/1764R1, 1761R1/1765R1, 1768R1, 1768R1/1770R1, 1768R1/1771R1, 1768R1/1772R1, 1784R1, 1784R1/1787R1, 1784R1/1788R1, 1797R1, 1797R1/1800R1 and 1797R1/1801R1. Therefore, in our experiments we focused on a stretch of the sequence: 1755 ERLIDAGHSEAATIAEWKDGLNEMWADLLELIDTRMQLLAASYDLHR 1801 (see also a). CD studies indicated that all the mutated peptides were well folded, and neither cysteine replacement nor spin labeling affected their general conformation, i.e., approximately 60% helical structure, with sigmoidal-shaped thermal denaturation profiles (data not shown). As can be seen in , the CD spectra of spin-labeled peptides turned out to be nearly identical to the spectrum of the template β-spectrin peptide, yet all of these slightly differ from the spectrum of the spectrin tetramer. The regions of 206-212 nm seem to be particularly distinctive in both cases, which may reflect characteristic geometrical details of the β-spectrin fragment 1638-1845 (see ‘Discussion’ section). Monolayer experiments revealed that none of the mutants lost its affinities to PE/PC monolayers and ankyrin. Assays for free cysteines, combined with protein concentration analysis indicated that spin-labeling efficiencies were more than 85% in all cases.
Figure 2. The CD spectra of the erythroid spectrin tetramer, a β-spectrin-derived peptide comprising residues 1638-1845, and the spin-labeled peptide 1768R1/1771R1 which is representative for all the mutants prepared. All the spectra (measured at 20°C) were shown with the y-axis scale adjusted to match peak heights at 222 nm. The molar elipticity of the mutated peptides ranges from 19430 to 20700 [degrees cm2 dmol−1], which was comparable with the non-mutated template peptide. The molar elipticity for spectrin was 25929.
![Figure 2. The CD spectra of the erythroid spectrin tetramer, a β-spectrin-derived peptide comprising residues 1638-1845, and the spin-labeled peptide 1768R1/1771R1 which is representative for all the mutants prepared. All the spectra (measured at 20°C) were shown with the y-axis scale adjusted to match peak heights at 222 nm. The molar elipticity of the mutated peptides ranges from 19430 to 20700 [degrees cm2 dmol−1], which was comparable with the non-mutated template peptide. The molar elipticity for spectrin was 25929.](/cms/asset/18c2d904-2164-4663-a302-b8dac9769b88/imbc_a_210181_f0002_b.gif)
EPR spectra
The spin-spin interaction is a function of the distance, and the latter can be deduced from the EPR spectral broadening Citation[25], Citation[35–37]. However, the line broadening caused exclusively by the spin-spin interaction is better represented in the EPR spectra taken under rigid solution conditions. Relative to the spectrum for the adequate singly labeled peptides, various extents of line broadening due to spin-spin interactions were observed in the spectra of doubly labeled peptides recorded at liquid nitrogen temperature, as revealed in . The three spectra in c, namely I1768R1/E1770R1, I1768R1/W1771R1 and I1768R1/K1772R1, respectively represent peptides labeled at the i and i + 2, i and i + 3, and i and i + 4 positions along the sequence. Of these, the spectra of I1768C/W1771C appeared to be the broadest, as the other two spectra were significantly less broadened. To test whether the broadening pattern is characteristic for the whole lipid-binding domain of β-spectrin, another three pairs of doubly labeled peptides were prepared: 1761R1/1764R1 and 1761R1/1765R1, 1784R1/1787R1 and 1784R1/1788R1, 1797R1/1800R1 and 1797R1/1801R1, respectively representing the i,i+3 and i, and i + 4 spin-labeling position scheme. As can be seen in b, the spectra of G1761R1/E1764R1 and 1761R1/1765R1 display pronounced line broadening, reflecting a similar distance pattern to that of I1768R1/W1771R1 and I1768R1/K1772R1. On the other hand, the spectra 1784R1/1787R1 and 1784R1/1788R1 (d), as well as 1797R1/1800R1 and 1797R1/1801R1 (e) display an inverted situation, where the i, i + 4 positions of the spin labels are reflected by substantial broadening, while the i, i + 3 are slightly less broadened. This may suggest that the analyzed sites along the sequence of the lipid-binding domain are not equivalent. Such differences in the extent of broadening were considerably reduced in the presence of 6 M urea, in addition to an overall decrease in line broadening in all cases (data not shown, see a). Thus, it is strongly suggested that the observed spin-spin interactions reflected structural features of the analyzed peptides. By contrast, the spectra of the mutants 1755R1/1758R1 and 1756R1/D1759R1 (both labeled in i, i + 3 positions) proved the least sensitive to denaturating conditions (a), manifesting the unstructured state. However, comparing the line broadening of native and denatured peptide 1757R1/1760R1 (a), it becomes apparent that residue 1760 is the first along the sequence, which represents the secondary arrangement of the studied domain.
Figure 3. A comparison of the low-temperature absorption EPR spectra of MTSL doubly labeled mutants with the spectra of singly labeled mutants (red lines), all originating from β-spectrin-derived polypeptide. The spectra of 6 M urea-denatured peptides (denoted as ‘ + U’) are in black. All the spectra are shown with the y-axis scale adjusted to match peak heights. The X-axis is magnetic field [Gauss]. This figure is reproduced in color in Molecular Membrane Biology online.
![Figure 3. A comparison of the low-temperature absorption EPR spectra of MTSL doubly labeled mutants with the spectra of singly labeled mutants (red lines), all originating from β-spectrin-derived polypeptide. The spectra of 6 M urea-denatured peptides (denoted as ‘ + U’) are in black. All the spectra are shown with the y-axis scale adjusted to match peak heights. The X-axis is magnetic field [Gauss]. This figure is reproduced in color in Molecular Membrane Biology online.](/cms/asset/84ac4d8d-5b8c-4c30-8a58-d6e2b1944c49/imbc_a_210181_f0003_b.jpg)
As the analyzed region of β-spectrin is considered to bind lipids Citation[24], we monitored the changes in the shape of spectra in the presence of 2% octyl-glucoside, non-ionic detergent or PE/PC liposomes. Surprisingly, no significant changes in the extent of broadening were observed (data not shown).
Estimation of spin-spin distances
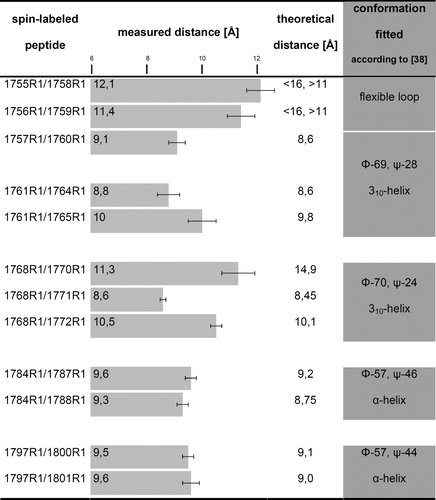
The Fourier deconvolution method Citation[25], Citation[26] allowed the quantitative examination of the structure of the lipid-binding domain of β-spectrin from the low temperature EPR spectra. Here, we determined the mean distances rather than distance distributions, as one Gaussian function was used to fit the broadening function in the Fourier space. The results are summarized in . A comparison of the spin-spin distances in I1768R1/E1770R1 with those in I1768R1/W1771R1 and 1768R1/1772R1 strongly suggests that the backbone of the N-terminal fragment of the lipid-binding domain of β-spectrin (residues 1768-1772) possesses a folded structure of helical character. This statement is based on the fact that within the helix, residues that are three apart (i, i + 3) or four apart (i, i + 4) along the sequence are spatially very close, while residues two apart (i, i + 2) are further from each other. Thus, the other peptides labeled on the i, i + 3 or i, i + 4 positions within residues 1760-1801 also form such a structure. Going into the details of the measured average distances between the labeled side chains, d(i, j), and taking into consideration the fact that an α-helix is characterized by d(i, i + 3) ≥ d(i, i + 4), whereas a 310-helix has d(i, i + 3) < d(i, i + 4) Citation[31], the only reasonable interpretation is that the analyzed domain has a significant amount of 310-helix at its N-terminus, i.e., ranging from amino acid residue 1760 to 1772. By contrast, the stretch of residues 1773-1801 appeared to be a more regular α-helix. The longer distances observed for 1755R1/1758R1 and 1756R1/1759R1 indicate the unstructured region and place the start of the helix at residue 1760.
Figure 4. The values of the average interspin distance for doubly labeled β-spectrin-derived polypeptide. The bar on the chart indicates experimental error. On the right, a comparison of the average interspin distances for doubly labeled β-spectrin-derived polypeptide with theoretical distances between the nitroxides of model structures which best correspond to the measured distances. As it was impossible to determine the exact R1 side chain conformation, the structures are represented by the mean values of the backbone torsion angles.
Structure modeling
Our first approach to building a model of the lipid-binding domain of β-spectrin was to define the spectrin repeats using the SMART tool Citation[7], Citation[29], and then to construct a three-dimensional model of the non-mutated template peptide using the Robetta meta-server Citation[28] (a). In accordance with the assumptions, the estimated helical structure (residues 1760-1801) should be regarded as the helix C of the 14th repeat unit of β-spectrin. Upstream along the sequence, we located the loop between helices B and C of that repeat, which was originally positioned five residues apart considering the currently available data. Thus, the theoretically predicted phasing information Citation[7] regarding helix C differs substantially from our experimental results (compare a and b).
Figure 5. A ribbon representation of the structure of the peptide comprising 1638-1845 of β-spectrin, as predicted by the Robetta server (a); green (light gray) is the analyzed region with blue (black) as the lipid-binding domain Citation[24]. The ball and stick representation of the resultant model of the whole lipid binding domain of β-spectrin with its flanking regions (b); green (gray) is the β-carbon of the first residue forming helix – A1760, which is followed by: blue (black) positively charged side chains, red (light gray) negatively charged side chains, and yellow (gray) hydrophobic side chains; the dotted line is the helix axis. The surface representation of the helix fragment comprising residues 1760-1790 (c); blue (black) are the positively charged residues, red (light gray) are the negatively charged residues. The dotted lines represent the helix axis. This figure is reproduced in color in Molecular Membrane Biology online.
![Figure 5. A ribbon representation of the structure of the peptide comprising 1638-1845 of β-spectrin, as predicted by the Robetta server (a); green (light gray) is the analyzed region with blue (black) as the lipid-binding domain Citation[24]. The ball and stick representation of the resultant model of the whole lipid binding domain of β-spectrin with its flanking regions (b); green (gray) is the β-carbon of the first residue forming helix – A1760, which is followed by: blue (black) positively charged side chains, red (light gray) negatively charged side chains, and yellow (gray) hydrophobic side chains; the dotted line is the helix axis. The surface representation of the helix fragment comprising residues 1760-1790 (c); blue (black) are the positively charged residues, red (light gray) are the negatively charged residues. The dotted lines represent the helix axis. This figure is reproduced in color in Molecular Membrane Biology online.](/cms/asset/f86995c1-0665-468f-a055-038cfc44f9b9/imbc_a_210181_f0005_b.jpg)
Secondly, we built models of doubly labeled peptides according to the measured distances between spins, complying with a predominant configuration for the R1 side chain on a helical surface Citation[31], Citation[32] (see ‘Materials and methods’ for details) using MOLDEN software Citation[30]. Manipulation of backbone torsion angles (φ and ψ), within its crystalographically realistic values for the α-helix and 310-helix Citation[38], allowed us to find a peptide chain conformation, which best represented the experimentally determined spin-spin distances. summarizes the experimentally calculated spin-spin separations and the theoretical ones which represent the best fitted secondary structure of the polypeptide chain. Based on the above, a structural model of the whole lipid-binding domain was prepared and then subjected to molecular modeling via HyperChem software Citation[33] (see b and c). The overall helical structure with an α-helix comprising residues 1774-1801, a 310-helix comprising residues 1760-1773, and a loop upstream from the latter are unique features of the resultant structure. The linker between the 14th and 15th repeat domains, assigned by SMART to residues 1795-1800, was proved to be α-helical, which is in agreement with earlier structural studies of spectrin repeats Citation[9–12]. However, the most intriguing thing is the highly amphipathic character of the helix (c), which emerges directly from its characteristic conformational pattern. Most probably, this is the origin of the high susceptibility of the helix to phospholipid mono- and bi-layers.
Discussion
As it has been shown that a 38-residue N-terminal fragment of the β-spectrin ankyrin-binding domain includes the binding site for PE-rich lipids, detailed structural information is essential for understanding the mechanism of its interaction with lipids. To achieve this, we applied SDSL (site-directed spin labeling) EPR combined with the Fourier deconvolution method to study the secondary structure of the domain and its potential changes during lipid binding with high precision. The method has proven to be effective in the range of 7–25 Å Citation[25], Citation[39], Citation[40] with a standard deviation of 0.9 Å for the entire range Citation[25]. In the same way, the helical content of the α-spectrin tetramerization region was successfully evaluated Citation[41]. EPR methods with doubly spin-labeled peptides were also verified in determining the presence of the α-helix and 310-helix in protein structures Citation[42]. Double labeling at positions i,i+3 and i, i + 4 can be used to characterize the local helix geometry, as molecular dynamics simulations confirmed that the average distances, d(i, j), between the nitroxides of two R1 side chains are: d(i, i + 3) ≥ d(i, i + 4) for an α-helix, and d(i, i + 3) < d(i, i + 4) for a 310-helix Citation[37]. It should also be emphasized that substitution with an R1 side chain does not perturb the helical structure, but exhibits helix-stabilizing characteristics similarly to A, L and M residues Citation[43].
As some authors reported that freezing proteins could affect their conformations Citation[35], we also tested whether the presented EPR results follow the native structure of the domain. By measuring the spin-spin distances of several representative peptides in 40% sucrose at room temperature (data not shown), we obtained very similar results, albeit with somewhat reduced precision. Therefore, in all probability, our data does not reflect freezing-related changes.
In our study, we estimated the secondary structure of the lipid-binding domain of β-spectrin as a single α-helix with an evident contribution of a 310-helix within its 14 N-terminal residue segment. Generally, no disallowed region between the α and 310 conformation is observed in the Ramachandran plot, and the two helical forms are energetically very close; therefore, one helix may be gradually transformed to the other, maintaining a near-helical conformation of the chain throughout Citation[44], Citation[45]. Although not yet found within spectrin repeats, the occurrence of 310-helices is not rare, and they are routinely found at the ends of α-helices Citation[38] or even as longer, independent conformational units Citation[45], Citation[46]. Analyzing the sequence of the 310-helix motif of the studied lipid-binding domain, especially the stretch 1768 IAEWKD 1773, and taking into consideration that D, E and W have low and medium propensities for an α-helix but very high propensities for a 310-helix Citation[45], the latter conformation seems to be favored.
It has been suggested that both 310- and α-helices display similar 222-nm bands, but 310-helices are considered to have more intense 208-nm bands in their CD spectra Citation[47]. Thus, the proportion [κ]222/[κ]208≈0.97 for the peptide comprising amino acid residues 1638-1845 of the β chain of human erythrocyte spectrin and its R1-substituted mutants may indicate a 310-helix contribution in its overall α-helical structure, especially when compared to the whole spectrin tetramer, where [κ]222/[κ]208=1.05 and the observed small fraction of the 310-helix within the examined domain is diluted in a large, highly α-helical protein (see ).
Helical wheel projections of the studied domain clearly indicate that the 310-helix contribution to the structure significantly increases the amphipathicity of the whole helix (data not shown, see c). Taking into consideration the extremely high sequence identity across various species and the spectrin isoforms in the analyzed region Citation[7], it seems likely that the characteristic structure of the ankyrin-sensitive lipid-binding domain presented in this paper is related to ligand-binding activities (i.e., ankyrin and/or PE-rich lipids) Citation[7], Citation[23]. Most probably, the ability to bind lipids emerges from exposition of the hydrophobic part of the helix, which certainly may occur, although only while interacting with hydrophobic compounds. While the helix geometry does not change significantly in the presence of non-ionic detergent or PE/PC liposomes, some lipid-induced perturbations of the coiled-coil structure cannot be ruled out. Indeed, the preliminary results of our inter-helical SDSL experiments on the 14th repeat domain of β-spectrin may suggest such rearrangements (unpublished data). In general, inter-helical interactions are fundamental for spectrin repeat stability, and the majority of them are van der Waals contacts (with crucial, highly conserved W residues playing a dominant role) with a small amount of electrostatic and hydrogen bond interactions Citation[12], Citation[13]. Some spectrin repeats are stabilized mainly by the latter types of interaction Citation[11], which raises the possibility that some of the negatively (and/or positively) charged groups of the structure presented here might be engaged in inter-helical interactions, thus leaving free the hydrophobic residues (at least some of them) for interaction with lipids. Further study is definitely needed to answer those questions.
As far as the physiological significance of the double activity of the examined region of β-spectrin is concerned, an ankyrin/PE model was proposed to explain the role of the affinity of the ankyrin-binding site towards PE/PC-rich domains Citation[48]. It is well known that there are situations, at least in erythrocyte membranes, characterized by an ankyrin deficiency or a reduction of its affinity for spectrin. When there is not enough functional ankyrin to saturate all the ankyrin-binding sites within spectrin molecules, these sites bind to the PE-rich domains of the inner leaflet of the membrane. Thus, alternative anchors emerge, substituting for ankyrin and preserving the mechanical properties of spectrin tetramers in the skeletal lattice.
In conclusion, our results indicate that the lipid-binding domain of β spectrin, previously mapped in our laboratory, possesses a helical structure and is entirely composed of helix C of the 14th repeat unit. The helix starts at A1760, which makes it five residues longer than expected from theoretical predictions, and most probably continues to form helix A of the next repeat unit. The highly amphipathic character of the helix apparently correlates with its 310 configuration at the N-terminus. The described structure does not undergo any significant changes when interacting with phospholipids or detergent.
This paper was first published online on iFirst on 15 January 2007.
Acknowledgements
We would like to thank Dr Daniel Krowarsch for his help with the CD experiments and Ms Jadwiga Kędzierska for her support with EPR data collection. We also thank Professor A. Kozubek for providing HyperChem software. This study was supported by the Polish Research Committee (KBN) grant no. 2 P04 021 27 and COST/13/2005. A.C. was additionally supported by European Social Fund (action no.z/z.02/II/z.6/04/04)
References
- Marchesi VT, Steers E. Selective solubilization of a protein component of the red cell membrane. Science 1968; 159: 203–204
- Goodman SR, Zagon IS, Kulikowski RR. Identification of a spectrin-like protein in nonerythroid cells. Proc Natl Acad Sci USA 1981; 78: 7570–7574
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 2001; 81: 1353–1392
- Speicher DW, Marchesi VT. Erythrocyte spectrin is comprised of many homologous triple helical segments. Nature 1984; 311: 177–180
- Yan Y, Winograd E, Viel A, Cronin T, Harrison SC, Branton D. Crystal structure of the repetitive segments of spectrin. Science 1993; 262: 2027–2030
- Dijnovic-Carugo K, Gautel M, Ylanne J, Young P. The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett 2002; 513: 119–123
- Baines AJ. Comprehensive analysis of all triple helical repeats in β-spectrins reveals patterns of selective evolutionary conservation. Cell Mol Biol Lett 2003; 8: 195–214
- Broderick MJF, Winder SJ. Towards a complete atomic structure of spectrin family proteins. J Struct Biol 2002; 137: 184–193
- Pascual J, Pfuhl M, Walter D, Saraste M, Nilges M. Solution structure of the spectrin repeat: a left-handed antiparallel triple-helical coiled-coil. J Mol Biol 1997; 273: 740–751
- Grum VL, Li D, MacDonald RI, Mondragon A. Structures of two repeats of spectrin suggest models of flexibility. Cell 1999; 98: 523–535
- Kusunoki H, MacDonald RI, Mondragon A. Structural insights into the stability and flexibility of unusual erythroid spectrin repeats. Structure 2004; 12: 645–656
- Kusunoki H, Minasov G, MacDonald RI, Mondragon A. Independent movement, dimerization and stability of tandem repeats of chicken brain α-spectrin. J Mol Biol 2004; 344: 495–511
- An X, Guo X, Zhang X, Baines AJ, Debnath G, Moyo D, Salomao M, Bhasini N, Johnson C, Discher D, Gratzer WB, Mohandas N. Conformational stabilities of the structural repeats of erythroid spectrin and their functional implications. J Biol Chem 2006; 281: 10527–10532
- Discher DE, Carl P. New insights into red cell network structure, elasticity and spectrin unfolding – a current review. Cell Mol Biol Lett 2001; 6: 593–606
- Bennett V, Stenbuck P. The membrane attachment protein for spectrin is associated with band 3 in human erythrocyte membrane. Nature 1979; 280: 468–473
- Stabach PR, Morrow JS. Identification and characterization of βV spectrin, a mammalian ortholog of drosophila βH spectrin. J Biol Chem 2000; 275: 21385–21395
- O'Toole PJ, Morrison IE, Cherry RJ. Investigations of spectrin-lipid interactions using fluoresceinphosphatidylethanolamine as a membrane probe. Biochim Biophys Acta 2000; 1466: 39–46
- Michalak K, Bobrowska M, Sikorski AF. Interaction of bovine erythrocyte spectrin with aminophospholipid liposomes. Gen Physiol Biophys 1993; 12: 163–170
- Diakowski W, Prychidny A, Swistak M, Nietuby( M, Bialkowska K, Szopa J, Sikorski AF. Brain spectrin (fodrin) interacts with phospholipids as revealed by intrinsic fluorescence quenching and monolayer experiments. Biochem J 1999; 338: 83–90
- An X, Guo X, Sum H, Morrow J, Gratzer W, Mohandas N. Phosphatidylserine binding sites in erythroid spectrin: location and implications for membrane stability. Biochemistry 2004; 43: 310–315
- Ray S, Chakrabarti A. Membrane interaction of erythroid spectrin: surface-density-dependent high-affinity binding to phosphatidylethanoloamine. Mol Membr Biol 2004; 21: 93–100
- Grzybek M, Chorzalska A, Bok E, Hryniewicz-Jankowska A, Czogalla A, Diakowski W, Sikorski AF. Spectrin-phospholipid interactions. Existence of multiple kinds of binding sites?. Chem Phys Lipids 2006; 141: 133–141
- Bialkowska K, Zembron A, Sikorski AF. Ankyrin inhibits binding of erythrocyte spectrin to phospholipid vesicles. Biochim Biophys Acta 1994; 1191: 21–26
- Hryniewicz-Jankowska A, Bok E, Dubielecka P, Chorzalska A, Diakowski W, Jezierski A, Lisowski M, Sikorski AF. Mapping of an ankyrin-sensitive, phosphatidylethanolamine/phosphatidylcholine mono- and bi-layer binding site in erythroid β-spectrin. Biochem J 2004; 382: 677–685
- Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci USA 1995; 92: 8239–8243
- Xiao W, Shin YK. EPR spectroscopic ruler: the method and its applications. Biological magnetic resonance. Volume 19: Distance measurements in biological systems by EPR, LJ Berliner, SS Eaton, GR Eaton. Kluwer, New York 2000; 249–272
- Berliner LJ, Grunwald J, Hankocszky O, Hideg K. A novel reversible thiol-specific spin label: papain active site labeling and inhibition. Anal Biochem 1982; 119: 450–455
- Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 2004; 32: 526–531
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA 1998; 95: 5857–5864
- Schaftenaar G, Noordik JH. Molden: a pre- and post-processing program for molecular and electronic structures. J Comput Aided Mol 2000; 14: 123–134
- Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry 2000; 39: 8396–8405
- Columbus L, Kalai T, Jeko J, Hideg K, Hubbell WL. Molecular motion of spin labeled side chains in α-helices: analysis by variation of side chain structure. Biochemistry 2001; 40: 3828–3846
- Froimowitz M. HyperChem: a software package for computational chemistry and molecular modeling. Biotechniques 1993; 14: 1010–1013
- Altenbach C, Marti T, Khorana HG, Hubbell WL. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science 1990; 248: 1088–1092
- Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry 2001; 40: 15471–15482
- Persson M, Harbridge JR, Hammarstrom P, Mitri R, Matensson LG, Carlsson U, Eaton GR, Eaton SS. Comparison of electron paramagnetic resonance methods to determine distances between spin labels on human carbonic anhydrase II. Biophys J 2001; 80: 2886–2897
- Fiori WR, Millhauser GL. Exploring the peptide 310-helix ↔ α-helix equilibrium with double label electron spin resonance. Biopolymers 1995; 37: 243–250
- Barlow DJ, Thornton JM. Helix geometry in proteins. J Mol Biol 1988; 201: 601–619
- Zhang F, Chen Y, Kweon DH, Kim CS, Shin YK. The four-helix bundle of the neuronal target membrane SNARE complex is neither disordered in the middle nor uncoiled at the C-terminal region. J Biol Chem 2002; 27: 24294–24298
- Xiao W, Brown LS, Needleman R, Lanyi JK, Shin YK. Light-induced rotation of a transmembrane α-helix in bacteriorhodopsin. J Mol Biol 2000; 304: 715–721
- Cherry L, Menhart N, Fung LWM. Spin label EPR structural studies of the N-terminus of α-spectrin. FEBS Lett 2001; 466: 341–345
- Fiori WR, Miick SM, Millhauser GL. Increasing sequence length favors α-helix over 310-helix in alanine-based peptides: evidence for a length-dependent structural transition. Biochemistry 1993; 32: 11957–11962
- Bolin KA, Hanson P, Wright SJ, Millhauser GL. An NMR investigation of the conformational effect of nitroxide spin labels on Ala-rich helical peptides. J Magn Reson 1998; 131: 248–253
- Toniolo C, Benedetti E. The polipeptide 310-helix. Trends Biochem Sci 1991; 16: 350–353
- Pal L, Basu G. Novel protein structural motifs containing two-turn and longer 310-helices. Protein Eng 1999; 12: 811–814
- Biron Z, Khare S, Samson AO, Hayek Y, Naider F, Anglister J. A monomeric 310-helix is formed in water by a 13-residue peptide representing the neutralizing determinant of HIV-1 on gp41. Biochemistry 2002; 41: 12687–12696
- Manning MC, Woody RW. Theoretical CD studies of polypeptide helices: examination of important electronic and geometric factors. Biopolymers 1991; 31: 569–586
- Sikorski AF, Bialkowska K. Interactions of spectrin with membrane intrinsic domain. Cell Mol Biol Lett 1996; 1: 97–104