Abstract
Membrane translocation is a crucial issue when addressing the activity of both cell-penetrating and antimicrobial peptides. Translocation is responsible for the therapeutic potential of cell-penetrating peptides as drug carriers and can dictate the killing mechanisms, selectivity and efficiency of antimicrobial peptides. It is essential to evaluate if the internalization of cell-penetrating peptides is mediated by endocytosis and if it is able to internalize attached cargoes. The mode of action of an antimicrobial peptide cannot be fully understood if it is not known whether the peptide acts exclusively at the membrane level or also at the cytoplasm. Therefore, experimental methods to evaluate and quantify translocation processes are of first importance. In this work, over 20 methods described in the literature for the assessment of peptide translocation in vivo and in vitro, with and without attached macromolecular cargoes, are discussed and their applicability, advantages and disadvantages reviewed. In addition, a classification of these methods is proposed, based on common approaches to detect translocation.
Introduction
The introduction of hydrophilic molecules into mammalian cells has become a key strategy for the investigation of intracellular processes and drug therapy. Several methods have been devised to achieve this goal, but they all have limitations Citation[1]. Some peptides are able to translocate across cell membranes with low toxicity Citation[2–5] and to mediate cellular uptake of proteins Citation[3] and other macromolecules Citation[6], Citation[7]. These peptides are known as cell-penetrating peptides (CPPs) but the CPP designation is now generalized for all the peptides with the ability to translocate across membranes even when cargo uptake properties are unknown. They include natural, newly synthesized and chimeric peptides Citation[8]. Beside the ability to translocate through cell membrane, these peptides are normally characterized as short, cationic, water-soluble peptides with high efficiency and low cytotoxicity Citation[9]. The mechanism used by CPPs to pass the membrane is not well understood and is a controversial topic in the literature. At the beginning the internalization mechanism of CPPs was generalized and considered to be endocytosis-independent Citation[2], Citation[5]. Nowadays there is experimental evidence for both endocytic and non-endocytic routes (for further information see Citation[9], Citation[10]). Considering the information available in the literature a unique mechanism for all the peptides does not seem reasonable and more than one mechanism can operate for a single peptide.
Ribosomal antimicrobial peptides, or AMPs, are efficient antibiotics against a variety of microbial pathogens Citation[11–14]. These peptides are usually short and cationic Citation[15] such as CPPs, but they are also characterized by a strong interaction with cellular membranes, and by the ability to selectively attack pathogens without disturbing host cell integrity Citation[16], Citation[17]. AMPs are potential candidates of a new antibiotic generation against multiresistant bacterial strains Citation[16].
Despite the fact that most AMPs exert their principal activity at the membrane level Citation[15], Citation[17], translocation processes have been shown to occur for many of them Citation[18], Citation[19]. Considering this, AMPs are natural templates for designing cell-penetrating molecules, which can be developed into carriers Citation[9], Citation[20]. AMPs and their derivatives are thus very promising molecules to use both as antibiotics and as drug transporters.
To guarantee that a specific peptide works as a CPP, its ability to cross the membrane and the capacity to transport attached cargoes is of first importance. Regarding AMPs, internalization is not a prerequisite and the biological efficiency of AMPs can be evaluated without translocation assessment. However the characterization of such membrane processes is of great value both in the development of new antibiotics and in the design of templates for CPPs (Citation[10]).
In this paper we will review methods that are available to evaluate peptide uptake into cells or vesicular model membranes.
Methods used to evaluate peptide uptake
Assessment of peptide translocation without attached cargo or labelled with a fluorescent probe
In vivo methods
The CPPs internalization mechanism is a matter of vibrant discussion in the literature. To evaluate if CPPs are internalized by endocytosis the dependence of translocation on temperature, or in the presence of specific endocytic inhibitors must be assessed in vivo. The intrinsic fluorescence of Trp or Tyr residues cannot be used to investigate peptide internalization in vivo, due to the relatively large cellular background fluorescence in the near-UV. Therefore, CPPs are normally derivatized with fluorophores such as Rhodamine B (Rh) Citation[21–24], Fluorescein Citation[5], Citation[25–32] and Nitrobenzoxadiazole (NBD) Citation[24], Citation[33]. Alternatively, the properties of macromolecular cargoes can be used. In most of the methods described in this section, peptide fluorescence is the experimentally measured signal. However the need to derivatize a peptide can be a limitation not only due to experimental and costly requirements but also because the presence of a dye may alter its properties Citation[34].
Available methods for the in vivo detection of peptide internalization can be divided into three categories: quantification of cytosolic concentration, direct visualization of internalization and peptide activity analysis.
Quantification of cytoplasmic concentration
Quantification of cytoplasmic concentration is usually carried out by means of direct measurement of fluorescence emission of peptides. In these methods, elimination of the non-internalized and cell-adsorbed peptide fractions is required to avoid an overestimation of peptide internalization; consequently, extensive washing procedures, including trypsinization and centrifugation, are required Citation[21], Citation[22], Citation[29], Citation[30], Citation[35], Citation[36]. Quenching of peptide fluorescence is an alternative to annihilate non-internalized peptide signal Citation[24], Citation[33], Citation[37].
A cell lysate should be prepared and the fluorescence intensity of labelled-CPP in supernatant measured Citation[29], Citation[32]. The washing procedures can be responsible for a signal reduction, due to sample loss and fluorescence bleaching. An additional limitation is the necessity to distinguish the peptide fluorescence from the cell background fluorescence; to assure a good signal/noise ratio the fluorophore should be chosen carefully to avoid overlap with intrinsic cellular fluorescence emissions and to circumvent a fast fading of fluorescence. For instance, Trp fluorescence of peptide can hardly be used due to the intrinsic fluorescence of cellular proteins. The fluorescence of cellular NAD(P)H (∼460 nm) and FAD (∼530 nm) is an additional concern. A high amount of cellular sample should be used to provide a good emission signal and controls without fluorophore in the same conditions (e.g., cell number and washing procedures) must also be used; a calibration curve should be prepared to allow quantification of internalization. In this case, absolute quantification of the internal peptide concentration may also require information on the total cellular volume.
Oehlke et al. developed a method to discriminate membrane surface-bound, membrane-inserted and internalized peptide fractions Citation[38]. To obtain quantitative information on the peptide fraction associated with plasma membrane and the peptide fraction actually internalized, the exposed peptide was treated with diazotized 2-nitroaniline. This procedure modifies the peptide fraction bounded to the surface, without damaging the cell Citation[39]. Bound and internalized fractions were detected by HPLC analysis of cell lysates and quantified by spectrofluorimetry (5(6)-carboxyfluorescein-N-hydroxysuccinilmide ester–labelled peptide detected with excitation at 445 nm and emission at 520 nm). The fraction of surface bound peptide is strongly modified by the diazo reagent which renders the peptide completely undetectable in HPLC chromatogram (there is a strong retention of highly hydrophobic modified products on the stationary phase of the HPLC column). The fraction of peptide inserted into plasma membrane is slightly protected from the attack of the diazo reagent, so this fraction is only partially modified. The fraction of peptide inaccessible to the diazo reagent is the internalized fraction Citation[38].
Flow cytometry analysis by fluorescence activated Cell Sorter (FACS) is a tool to quantify cellular association of a fluorophore-labelled peptide. A large number of cells are analysed and dead cells are identified by propidium iodide staining Citation[21], Citation[22], Citation[30], Citation[32], Citation[33], Citation[35], Citation[40]. The cell subpopulation displaying significant fluorescence emission is identified and quantified, and the CPP uptake is measured as the cell-associated fluorescence intensity. An absolute quantification of the internalized amount of peptide requires either a comparison with a standard Citation[41] or the use of additional methods, such as the ones described above. The need to wash/quench the membrane-bound peptide fraction, also applies for flow cytometry analysis.
A kinetic evaluation of the translocation process has been obtained by testing the inaccessibility of externally added trypan blue to fluorescein-labelled peptides Citation[42]; the rationale behind this approach is shared by many methods described for in vitro translocation studies (see below).
Matrix-assisted laser desorption ionization-time-of-flight mass spectroscopy (MALDI-TOF MS) has been used to evaluate the exit of internalized peptides from cells into the culture medium Citation[32], Citation[43], to detect peptide internalization and to investigate intracellular degradation Citation[32], Citation[36], Citation[43–45]. This method allows the direct detection of the peptide and uptake quantification Citation[44]. The sample (extracellular or intracellular supernatant) has to be concentrated to improve the signal. Elmquist and Langel proposed a procedure where samples are zip-tipped, analysed by MALDI-TOF MS and compared with synthetic peptides for a quantitative analysis Citation[32], Citation[43]. Instead of synthetic peptides the quantification of the internalized CPP can be achieved with an internal standard with the same sequence but labelled with deuterium. Peptides are biotinylated and, after cell lysis, they are captured by streptavidin-coated magnetic beads. Finally, beads are washed and analyzed by MALDI-TOF MS and the absolute amount of internalized peptide is determined. This procedure is also adequate to know if the peptide has been degraded during sample preparation or inside the cell Citation[44]. Inherent disadvantages in this method are the necessity for specialized and expensive procedures, and equipment to obtain peptide quantification. Moreover, it is rather time-consuming.
Direct visualization of internalization
In early studies, CPP internalization into cells was studied by fluorescence microscopy or in fixed cells where the fluorescence emission of a peptide-derivatized probe was directly visualized Citation[5], Citation[26], Citation[33], Citation[46]. Internalization of some AMPs has also been identified by a fluorescent label and visualized under the confocal microscope. For instance, biotin-labelled polyphemusin I Citation[47] and FITC-labelled buforin II Citation[48] were detected in the cytoplasm of E. coli without damaging the membrane; FITC-labelled magainin 2 was detected on the E. coli cell wall Citation[48]. Internalization in mammalian cells was also observed with biotin-labelled LL-37 and localization in the perinuclear region was detected Citation[49]. Alternatively, the presence of peptides in cells can be detected by immunofluorescence; for example, the localization of LL-37 was visualized with a specific antibody Citation[49].
Fixation procedures can bias the location of peptides in cells as the illusionary presence of peptides inside cells can arise from the high affinity of cationic peptides to cell surface Citation[50]. Therefore, peptide internalization in cells has to be compared in fixed and non-fixed conditions Citation[27], Citation[30]. Cell treatment with trypsin to digest the peptide at cell surface Citation[27], Citation[30], Citation[32], or quenching of peptide fluorescence has to be performed to remove the interference of the membrane-bound fraction. In these conditions it is possible to distinguish a deep localization inside the cell from membrane adsorption Citation[24]. With confocal microscopy different cellular plans can be used to confirm, or refute, peptide internalization.
Peptide activity analysis
In this class of methodologies, the effect of the peptides in the cytosol or in the membrane is evaluated. AMP translocation in vivo has been inferred from observations of intracellular damage (e.g., Citation[51]) or morphological alterations of bacteria Citation[52–54].
In other reports, the peptide interaction with membranes was evaluated by electrophysiological transmembrane current measurements in oocytes Citation[55], Citation[56]. With this setup it is possible to have access to both sides of lipidic membranes and it is not necessary to derivatize the peptide with a fluorophore. However, this detection can be ambiguous because transbilayer current can arise from the activation of endogenous channels in oocyte membranes. In order to rule out this possibility these experiments should be repeated on artificial lipid membranes Citation[57]. In addition, only qualitative information about translocation can be obtained unless a quantitative relationship between transmembrane current and internalization is assumed. Lastly, the occurrence of membrane destabilizations induced by peptides is not a proof of their ability to translocate across the membrane and reach the cytoplasm.
In vitro methods
Various biophysical approaches have been used to study peptide-lipid interaction in model bilayers. When CPPs are internalized by a physically-driven translocation mechanism, peptide-lipid interactions are truly important. Application of in vitro methods to evaluate the internalization of CPP is, however, scarce; on the other hand, AMP translocation has been essentially studied using model membranes. Nevertheless, in vitro studies face some difficulties and limitations. Large unilamellar vesicles (LUV), or small unilamellar vesicles (SUV), are the most used membrane models for the in vitro study of peptide-bilayer interactions. But, detection of peptide entry into the vesicular lumen is not easy. Theoretically, in a simple setup, it is possible to encapsulate an aqueous phase entity that could report peptide proximity or interaction (a quencher, or a fluorescence resonance energy transfer (FRET) acceptor for Trp fluorescence for example) and use it to probe the translocation event. However, the feasibility of this task can be hampered by several factors (see worked examples in part 1 of the Supplementary material in the online version). Several methods have been proposed to overcome these limitations, for instance peptide-lipid FRET (e.g., Trp-labelled peptide and Dansyl-PE-labelled lipidic vesicles) and/or permanent alteration of the peptide either in or out of the vesicles (e.g., between peptide digestion by trypsin) Citation[18], Citation[19]. A classification of methods can be devised regarding the peptide location with respect to the bilayer: (i) detection of entrapped peptide lack of accessibility from the outer phase, (ii) detection of inward peptide positioning within the membrane, (iii) direct detection of peptide in the inner phase, and (iv) detection of encapsulated peptide escape. Because of the many possible combinations of techniques and experimental setups some reported methods do not fall within the given categories, therefore, a fifth group of ‘unclassified methods’ is also presented. This classification differs from the one used for the in vivo methods mostly because for in vitro a widen range of approaches to the assessment of translocation have been described.
A schematic representation of in vitro methods from all the classes, with focus on the fluorescence methodologies, is depicted in .
Figure 1. In vitro methods for translocation assessment, using FRET and quenching methodologies. Peptide molecules are represented by empty rectangles and additional non-translocating compounds by circles; peptide molecules that have interacted with those compounds are dashed; phospholipids labelled with an acceptor for the peptide's fluorescence are indicated by light grey headgroups. (A) Measurement of the inaccessibility of the peptide to a non-translocating entity with the ability to digest, sequester or permanently quench it; the interaction between the two prevents the peptide from acting as a FRET donor to the labelled phospholipids; small FRET reductions will indicate that the peptide was protected from that interaction; this indicates that the peptide is buried in the membrane or is located in lumen. (B) Detection of inward peptide movement; after the initial adsorption interaction of the peptide with asymmetrically acceptor-labelled vesicles (despite the representation, both inner-leaflet and outer-leaflet labelling can be used), translocation or membrane penetration processes causes the mean distance between peptide and labelled phospholipids to increase (assuming an outer-leaflet labelling) and a concomitant reduction of FRET. If the peptide has quenching capabilities, a quenchable probe can be used to symmetrically label the vesicles; translocation can then be identified by full probe quenching, which is only possible if the peptide reaches the inner bilayer surface. (C) Detection of peptide in the inner phase; in the case of translocation, digestion by encapsulated trypsin (solid circles) reduces the overall amount of peptide available for membrane interaction and, consequently, FRET between peptide and labelled-membrane; trypsin inhibition (dashed circles) is required in the outer phase, prior to peptide addition. (D) Detection of encapsulated peptide escape; in the event of translocation, the peptide fluorescence decreases as it becomes accessible to an externally added, non-translocating quencher; the quencher can, alternatively, be co-encapsulated with the peptide, and the quenching reduction monitored instead. The figure is reproduced in colour in Molecular Membrane Biology online.
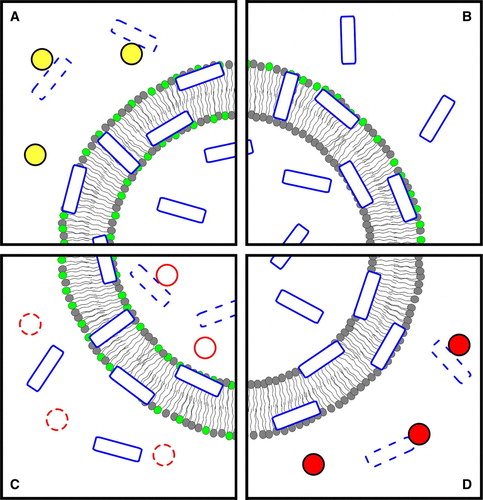
Detection of entrapped peptide lack of accessibility from the outer phase
A semi-quantitative determination of translocation can be performed by detection of peptide inaccessibility to a non-translocating entity Citation[19], Citation[24], Citation[58–60] after incubation with the model membranes (see , panel A). For example, translocation of AMP magainin 2 was identified by the observation that the percentage of inaccessible peptide increases with peptide-lipid incubation time Citation[19]. To perform this experiment trypsin was used to digest peptide after interaction with acceptor-labelled lipid vesicles. It was shown that proteolysis-dependent FRET reduction decreases with incubation time before trypsin addition. It was thus concluded that the peptide becomes inaccessible to trypsin with time. This is consistent with peptide translocation.
This concept can be used with different external components, for instance non-labelled vesicles to desorb the peptide from the labelled vesicles Citation[19], Citation[60]. An inverted setup has also been reported where the peptide (in this case, labelled with NBD or bimane fluorophores) was incubated with the non-labelled vesicles and desorption was induced by addition of acceptor-labelled anionic vesicles Citation[59] (a FRET increase, rather than a decrease, was detected in this case). The use of NBD-labelled peptides and addition of dithionite ion as the non-translocating entity is another possibility in this case, the NBD fluorescence intensity was measured without the need to label the vesicles Citation[24], Citation[59]; dithionite acts in a way similar to digestion by trypsin in that the NBD reduction/quenching is permanent. In all these approaches, an estimation of the translocation kinetics can be obtained by adding the non-translocating component at different incubation times Citation[24], Citation[59].
It should be noted that in all these methods a time-dependent measurement is required to determine the degree of peptide inaccessibility (as explained in detail in section 2 in the Supplementary material online).
These methods have a number of disadvantages: first, they are unable to discriminate membrane-bound and internalized peptide; second, in the case of lysis, which can occur for AMPs, peptide populations in inner and outer layers could be accounted as accessible, resulting in a low extent of apparent translocation; finally, not all of these methods are compatible with all peptides: for instance some AMPs are resistant to trypsin proteolysis (e.g., Polymixin B and Gramicidin S) Citation[18]. In addition, when using trypsin, extents of translocation may be overestimated if the peptide fragments also interact with the membrane.
Detection of inward peptide positioning within the membrane
In this category, the passage of the peptide to the inner leaflet of the bilayer is monitored. In all the reports where this method is used Citation[19], Citation[61], Citation[62], internalization is evaluated by the use of vesicles with asymmetric labelling. The fluorophore in membranes is a FRET acceptor for Trp-containing peptides (see , panel B).
Wimley and White developed a methylcoumarin derivatized lysophospholipid (LysoMC). This lipid is easily incorporated into bilayers (into both bilayers or only into the outer one) and is able to act as an efficient Trp-fluorescence acceptor/quencher Citation[63]. This probe was initially used to characterize the in-depth membrane distribution of Trp-containing peptides Citation[63], but more recently, Norden and co-workers have developed a method in which LysoMC is used to specifically test translocation; in addition, they described a procedure where is possible to use LysoMC to selectively label the inner layers Citation[61], Citation[62]. Experimentally, translocation information is obtained by comparison of emission spectra of inner leaflet LysoMC-labelled vesicles in the presence and absence of the peptide. The Trp residues are donors for FRET. An increase in FRET efficiency in the presence of the peptide means internalization; however, because some outer-to-inner bilayer FRET may occur, it becomes necessary to compare the obtained results with controls where translocating (tryptophan octyl esther Citation[63]) or non-translocating (Ac-18A-NH2: Ac-DWLKAFYDKVAEKLKEAF-NH2Citation[63]) Trp-containing molecules are used Citation[61], Citation[62].
Matsuzaki and co-workers had already described a similar method to assess peptide translocation in which vesicles are labelled only on the outer leaflet, with the tryptophan FRET acceptor DNS-PE Citation[19]. With this method it is possible to follow the FRET change during time with a single sample and an end-point approach for each incubation time is not required. Experimentally, after peptide addition, there is an initial increase in FRET efficiency (this corresponds to a peptide partitioning to the outer leaflet); with time, a decrease in FRET efficiency is observed (which corresponds to translocation where peptide is no longer close to the FRET acceptor in the outer leaflet). With this method, a kinetic analysis of the translocation event can be carried out. Quantitative information may be obtained if additional methods are used to quantitatively relate FRET intensity to luminal peptide concentration.
With these methodologies, peptides do not need to be derivatized and small peptide and lipid concentrations can be used (0.7µM and 100µM respectively Citation[61]). With these methods, in-depth location of peptide can be erroneously measured as translocation; inaddition, they cannot be used with peptides that induce lipid flip-flop (FRET acceptor bilayer asymmetry is lost). So far this methodology has been applied only to Trp-containing peptides, although the use of others donor/acceptor pairs is also possible.
Direct detection of peptide in the inner aqueous phase
Methods based on this principle are able to detect the presence of peptides in the inner luminal aqueous phase. With this approach actual translocation, rather than just membrane internalization, is measured (see , panel C).
Three different approaches fit in this class of methods. In the first, described by Matsuzaki and co-workers Citation[19], encapsulated trypsin and a peptide-lipid FRET system (Trp-containing peptide and Dansyl chromophore incorporated in vesicles) is used: trypsin is added at the hydration stage of the preparation of symmetrically labelled vesicles; after extrusion, non-encapsulated trypsin is inhibited by a trypsin inhibitor. Upon addition of the peptide to this system, an increase in acceptor fluorescence through FRET is expected as the peptide enters the bilayer; if the peptide is not internalized afterwards, the fluorescence intensity is expected to remain constant after the initial increase. However, if the peptide translocates into the vesicle lumen, it becomes accessible to the uninhibited trypsin and is digested; assuming that the resulting fragments do not interact with the membrane, this degradation of the peptide will lead to a decrease of the amount of peptide in the bilayer and consequent reduction of FRET. A quantitative analysis of the extent of translocation can be easily carried out considering that 100% translocation implies the digestion of all the peptide molecules and absence of FRET; the fluorescence intensity of the system should be the same as in the absence of the peptide. Conversely, 0% translocation will correspond to the maximum fluorescence intensity right after the peptide addition Citation[18], Citation[19]. This method cannot be applied to proteolysis-resistent peptide and/or without Trp residues (e.g., Polymixin B and Gramicidin S) Citation[18]. At high translocation efficiency, membrane perturbation caused by peptide fragment accumulation inside vesicles can cause difficulties in internalization quantification. In addition, there is the possibility of digested fragments re-entering the membrane, which can lead to a translocation underestimation.
A second method that detects the presence of the peptide in the inner aqueous phase was recently developed by Bárány-Wallje et al. In this method the experiment is performed with planar lipid membranes (PLMs) and the distribution of fluorescently-labelled peptides, between the two compartments and partitioned in PLM, is followed by confocal fluorescence spectroscopy Citation[64]. This powerful method allows the direct scanning of fluorescence intensity and quantification of peptide in each compartment. Another advantage is the possibility to easily generate asymmetric aqueous phase conditions: a transmembrane potential can be set without the need for ionophores Citation[64]. Besides the need to label the studied peptides (and care must be taken to choose a photostable label), this method has the disadvantage of requiring an uncommon setup with a custom PLM trough.
Another possible method to evaluate peptide translocation with model membranes is to follow peptide interaction with giant vesicles (GVs). With this method direct visualization of peptide interaction with membranes is possible by confocal microscopy. The translocation of peptide across these vesicles can be confirmed by the presence of the fluorophore inside the vesicles Citation[64], Citation[65]; in this case, just as with the methodology described for fluorescence imaging of in vivo internalization, the use of confocal analysis is crucial to distinguish whether the peptide is inside the vesicle or only adsorbed at the membrane surface. As with the PLM method described above, the peptide has to be derivatized with a fluorophore, which should be carefully chosen to avoid photobleaching.
With GVs it is possible not only to directly visualize the peptide inside the vesicles but also to evaluate its effect on membrane properties: membrane integrity can be followed with a labelled lipid such as N-Rh-PE (the fluorochrome has to be different from the one used to label the peptide), and the possibility of pore formation can be evaluated in GVs loaded with two similar dyes attached to other molecules of very different sizes (for instance Alexa488-Dextran (MW 10000) and Alexa546-maleimide (MW∼1300)). This experimental setup enables direct observation of changes in the membrane permeability during the course of observations and in the case of pore formation to have information on pore size due to sequential escape of the dyes from the GVs Citation[66], Citation[67].
Detection of encapsulated peptide escape
Using a fluorescence quenching assay, Magzoub et al. had developed methodologies in which the escape of a CPP from liposomes is determined in the presence and absence of a pH gradient Citation[68] (see , Panel D). To track the peptide escape two assays based on encapsulation were carried out: (i) vesicles were prepared with the peptide inside and the Trp quencher acrylamide was added to the outer phase (the escape of peptide from liposomes was followed by fluorescence quenching of peptide Trp residues caused by acrylamide); (ii) fluorescein-labelled peptides were encapsulated with a quencher (iodide) and, in the case of translocation, a de-quenching of the fluorescein label was observed. The fluorescence intensity corresponding to 100% peptide escape was determined by vesicle lyses with Triton X-100 and the fraction of peptide escape was calculated at different incubation times Citation[68].
Both these approaches allow the quantification of translocation as well as the determination of membrane crossing kinetics. Under certain situations, however, both methods can lead to misleading conclusions: in method (i) acrylamide is excluded from membranes, so only the fraction of peptide that reaches the outer aqueous environment is accounted for (if the peptide remains essentially partitioned in the outer leaflet after crossing the membrane, a lower translocation extent is measured); in method (ii) if the peptide does not translocate but has a deep location in the bilayer, it becomes inaccessible to the encapsulated quencher and is counted as having crossed the membrane.
In these methods if the peptide translocates very efficiently a significant amount of peptide can escape from the liposome during the gel filtration step (used to remove non-encapsulated peptide); therefore, these methods are only applicable to peptides that require special conditions to translocate (such as pH or potential membrane gradients), so that membrane crossing can be triggered only after the separation procedure.
Another peptide escape assay was described by Bárány-Wallje et al. in which vesicles uniformly preloaded with peptide both in the lumen and outer medium are dialysed so that any peptide outside of the liposomes is washed away Citation[64]. If any peptide absorption/fluorescence remains after the dialysis, it can be concluded that translocation from the lumen/inner leaflet to the outer side did not occur; conversely, the occurrence of translocation can be inferred from the absence of any peptide signal. Due to the very simple setup and instrumentation requirements, this seems a good option to begin a translocation study with; however, there are a number of disadvantages that are addressed in section 3 of the Supplementary material online.
Other methods
A methodology based on a particular peptide property was developed in reference Citation[69]. For this purpose, the specific fluorescence quenching of Rh by the CPP pep-1 was taken advantage of. Vesicles labelled symmetrically with N-Rh-PE were used to evaluate fluorescence emission quenching of Rh by pep-1 in the presence and absence of a transmembrane potential. After the peptide is added to LUVs, Rh fluorescence in the outer layer is quenched. If translocation occurs, it can be identified by a further reduction in Rh fluorescence due to contact of the peptide with Rh in the inner leaflet. The effect can be improved by using labelled multilamellas vesicles (MLVs): the fraction of Rh-PE available for peptide access is smaller than if LUVs are used, therefore a much larger relative increase in quenching is expected if the peptide translocates and accesses all the phospholipids Citation[69]. Alternatively, soluble Rh can be encapsulated in vesicles and its fluorescence followed after peptide addition Citation[69].
This method allows measurements to be taken during a time course, enabling the kinetic evaluation of the translocation process. Its main disadvantage is that it is only applicable to peptides that are able to quench a given fluorophore.
Another method using MLVs has been reported Citation[60], Citation[70]: dithionite ion was externally added to NBD-PE-doped MLVs, resulting in quenching of the fluorophores in the outermost leaflet; upon the addition of a pore-forming peptide the dithionite ion will have access to the first intramembranar space of the MLVs, resulting in an increase in quenching; full NBD quenching, however, will only be attained if the peptide can translocate and form pores in the inner bilayers because only then will all the fluorophore become accessible to the dithionite ion.
This method does not require any peptide labelling or intrinsic fluorescence but, on the other hand, this can only be applied to peptides that perturb the membrane to an extent where dithionite can cross it (by a pore or otherwise). Kinetic information can be extracted but it is necessary to take into consideration the multiple membrane crossing steps.
Assessment of peptide translocation with attached cargo
The translocation process of CPPs may vary if the peptide is in free form or attached to a cargo. Differences in peptide kinetics and cell localization can occur Citation[71–73]. In the above described methodologies the only attached ‘cargoes’ were fluorophores. The nature and size of the cargoes and the nature of attachment to the peptide (whether covalent Citation[3], Citation[74] or weaker Citation[7], Citation[75]) can also be a determinant for the translocation mechanism. Given these differences, development of consistent methods to evaluate the capacity to translocate/deliver proteins across artificial/natural membranes would be fruitful to systematically confirm (or refute) vector properties of CPPs. Practically all methods described above for in vivo or in vitro testing can, in principle, be extended to assess the translocation of CPP-cargo complexes, although no reports on such usage were found; however, some properties of the macromolecule attached to the peptide are additional advantages to detect and quantify translocation, opening the possibility for more sensitive methodologies. Three methods that have been reported for the in vivo studies and one for the in vitro studies of the internalization of CPP-cargo complexes are discussed here; while these can easily fit into the above classification, they are presented separately to underscore their importance in confirming the vector properties of a CPP.
Morris et al. had observed the efficiency of pep-1 to deliver GFP Citation[75] and a similar approach demonstrated the ability of the AMP buforin 2 to translocate across bilayers with a macromolecular cargo Citation[76]. Recombinant proteins with CPP and GFP domains have also been produced Citation[77–79]. Instead of GFP, Wadia et al. labelled TAT-Cre recombinase by coupling it to Alexa 488 or Alexa 546 fluorophores Citation[80]. GFP or Cre recombinase translocation is followed by its intrinsic fluorescence; this is in all aspects identical to what was previously described for the in vivo direct visualization of internalization. Another possibility to measure Cre recombinase translocation mediated by TAT is by recombination reporter assay, where the expression of enhanced GFP (EGFP) is dependent on TAT-Cre transduction into cells, followed by nuclear import. The signal of EGFP is followed and measured as a consequence of TAT-Cre import Citation[80].
Detection of β-Gal delivery into cells was also reported. Translocation was assessed by monitoring the X-Gal staining enzymatic activity of β-Gal Citation[75]. The detection of the enzymatic activity of the cargo not only allows a sensitive detection of the internalized CPP-cargo complex, but also tests whether CPP-mediated translocation can occur without cargo damage/denaturation. A quantification of β-Gal internalization was developed by means of its enzymatic activity on the substrate 4-methylumbelliferone β-D-galactopyranoside (MUG), a non fluorescent substrate that is converted to 4-methylumbelliferone (4-MU), which is a fluorescent product. 4-MU production enables quantification of protein internalization from its activity Citation[37], Citation[81].
Another possibility to evaluate protein delivery efficiency is by immunofluorescence, using a primary antibody raised against the carried protein and a fluorescently labelled secondary antibody, as was preformed with β-Gal Citation[81]. This method is independent of particular cargo properties; therefore, it is suited to study the transport of cargoes that do not have intrinsic fluorescence or detectable enzymatic activity. With these methodologies it is possible to obtain cell transfection efficiency, but not the quantification of peptide/protein internalization.
Protein uptake was also evaluated in vitro with pep-1/β-Gal, by taking advantage of the β-Gal enzymatic activity and 4-MU fluorescent properties (see ). Briefly, after peptide/protein incubation with LUVs, trypsin was added externally to digest the non-incorporated peptide and β-Gal; afterwards, phenylmethanesulfonyl fluoride (PMSF) was added to inhibit trypsin. To induce LUV permeabilization and leakage of the incorporated β-Gal, Triton X-100 was added, and enzymatic activity of β-Gal was determined by MUG hydrolysis Citation[81]. Intrinsic cargo fluorescence and/or enzymatic activity are specific properties that can be used for application of a similar methodology to other peptides/proteins complexes.
Figure 2. To evaluate the translocation of β-Gal/pep-1 (hexagons/rectangles) complexes across model membranes the following steps were reported Citation[81]: (1) valinomycin was added to K+-loaded vesicles in Na+-buffer to create a negative transmembrane potential; the complex was added to the solution and translocated across bilayer; (2) trypsin (circles) was added to digest non-incorporated complex and free pep-1 or β-Gal (dashed hexagons/rectangles); (3) after digestion, trypsin was inhibited (dashed circles) with phenylmethylsulfonyl fluoride and TX-100 was then added to facilitate the release of internalized β-Gal and allow its quantification (digestion debris were excluded from the last two panels, and inhibited trypsin from the last, to avoid image crowding); (4) β-Gal activity was assayed by MUG (black triangles) hydrolysis to a fluorescent product, 4-MU (lighter triangles). The figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 2. To evaluate the translocation of β-Gal/pep-1 (hexagons/rectangles) complexes across model membranes the following steps were reported Citation[81]: (1) valinomycin was added to K+-loaded vesicles in Na+-buffer to create a negative transmembrane potential; the complex was added to the solution and translocated across bilayer; (2) trypsin (circles) was added to digest non-incorporated complex and free pep-1 or β-Gal (dashed hexagons/rectangles); (3) after digestion, trypsin was inhibited (dashed circles) with phenylmethylsulfonyl fluoride and TX-100 was then added to facilitate the release of internalized β-Gal and allow its quantification (digestion debris were excluded from the last two panels, and inhibited trypsin from the last, to avoid image crowding); (4) β-Gal activity was assayed by MUG (black triangles) hydrolysis to a fluorescent product, 4-MU (lighter triangles). The figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/6ae00cf0-ef92-4919-ae05-c4a6d761fbec/imbc_a_210186_f0002_b.jpg)
The classification and the general advantages and disadvantages of the application of each reviewed method are summarized in .
Table I. Classification of available methods for in vivo and in vitro translocation assessment, and respective advantages and disadvantages.
For all the methods presented here, it is possible to conclude about the ability of peptides to translocate. When further information is sought, quantitative analysis should always be carefully carried out; for this, several different complementing methodologies should be conjugated, regarding not only the actual evaluation of translocation but also to quantify the peptide distribution between the membrane and aqueous environments and to characterize the membrane topology upon peptide interaction (occurrence of membrane thinning, pore formation, lysis, etc.).
Concluding remarks
Because most methods for the assessment of membrane translocation have only been reported during the last decade, it is understandable that this field is regarded as being underdeveloped, methodology-wise Citation[18]. In addition, the description of new methods has not always been given the due emphasis by their authors, which further contributed to the lack of visibility of these procedures. Yet, over 20 methods that address the assessment of membrane translocation have been found. The authors of this work hope that the gathering of this knowledge will help others find and use the most appropriate methods for their studies. On the other hand, this compilation should by no means be regarded as an ultimate source for information on translocation assessment methodologies in a way that the development of new methods, or the improvement of already described ones, is discouraged. Rather, by exposing the strengths and weaknesses of several methodologies, it is hoped that the development of newer and better methods is encouraged and facilitated.
Supplementary Material
Problems associated with the detection of entry of peptide into vesicles
At usual lipid concentrations, used to perform in vitro studies, the volume occupied by the vesicles is very small when compared to the bulk aqueous phase: for 100 nm diameter LUVs, produced at a total lipid concentration of 1 mM, and assuming a mean membrane surface area of 0.7 nm2 per phospholipid Citation[1], the ratio of luminal volume over the total volume will be about r = 0.003 v/v. Another problem arises from the fact that these peptides usually have large molar ratio partition constants towards lipidic membranes (about 103 to 104, e.g., Citation[2], Citation[3]). This means that only a small concentration of peptide will be in the aqueous phase unless relatively low lipid concentrations are used. Given the above-mentioned obstacles, it is clear that methods based on the detection of the concentration of internalized peptide will have to deal with very low signal intensities that will likely lie below instrumental detection limit.
Comments on time-dependent measurements
It should be noted that in all the methods presented in the section Measurement of peptide inaccessibility from the outer phase a time-dependent measurement is required to determine the degree of peptide inaccessibility: because the amount of free peptide in the outer phase will decrease – as it is digested, sequestered or permanently quenched – the internalized peptide will equilibrate back to the outer phase. Consequently, two stages are expected: in the first, signal magnitude increases (here ‘signal’, in a very broad sense, can be FRET reduction, FRET increase or fluorescence reduction, depending on the particular method used), detecting the adsorbed/non-internalized peptide fraction; then a second stage ensues with a usually slower signal increase Citation[4], Citation[5], which corresponds to the detection of the internalized peptide molecules that are returning to the outer phase. At equilibrium, the signal magnitude should be approximately the same whether translocation has occurred or not. The percentage of accessible peptide is usually obtained by comparing the end of the first stage with a control where peptide, membrane and the non-translocating entity are added simultaneously Citation[4], Citation[5]. From this, it can be seen that, in a hypothetical situation where the translocation kinetics were of the same magnitude of the desorption kinetics, this method would be very difficult to apply as both stages would have the same approximate slopes and the end of the first stage would be hard to detect. On the other hand, information about translocation kinetics could, in principle, be obtained from the rate at which the peptide molecules return to the outer phase, in the second stage.
Advantages and disadvantages associated with the dialysis method
Due to the very simple setup and instrumentation requirements for this experiment (see the section Detection of encapsulated peptide escape) this seems a good option to begin a translocation study with, because membrane crossing may be ruled out immediately avoiding the need to use more complex and costly methodologies. However, in order for this method to provide a measurable absorbance/fluorescence peptide signal in the absence of translocation it is required that the amount of internalized peptide be relatively large, to compensate for the effect of the small encapsulation volume. This is only possibly to attain if the peptide partitions strongly towards the bilayer and the lipid concentration is high enough: under these conditions, upon vesicle preparation, the peptide molecules can be expected to be equally distributed by the inner and outer leaflets and very few to be left in the aqueous phase; in the absence of translocation, roughly one half of the peptide signal will remain, correspondent to the peptide molecules partitioned in the inner leaflet. Should the peptide partition weakly to the membrane or there be a small lipid concentration, the inner leaflet peptide fraction could be undetected in the absence of translocation, which would generate a false positive. This limitation has to be accounted for by quantifying the extent of membrane interaction. Other disadvantages include the lengthy dialysis process and the lack of any kind of translocation characterization, which makes this method more suited to rule out, rather than ascertain, peptide translocation.
References
- Wadia JS, Becker-Hapak M, Dowdy SF. Protein transport. Cell-penetrating peptides, processes and applications. 1 st ed, Ü Langel. CRC Press, New York 2002; 365–375
- Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 1994; 269: 10444–10450
- Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA 1994; 91: 664–668
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999; 285: 1569–1572
- Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 1997; 272: 16010–16017
- Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol 1995; 128: 919–927
- Morris MC, Vidal P, Chaloin L, Heitz F, Divita G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res 1997; 25: 2730–2736
- Lindgren M, Hallbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci 2000; 21: 99–103
- Magzoub M, Graslund A. Cell-penetrating peptides: from inception to application. Q Rev Biophys 2004; 37: 147–195
- Henriques ST, Melo MN, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they?. Biochem J 2006; 399: 1–7
- Koczulla AR, Bals R. Antimicrobial peptides: current status and therapeutic potential. Drugs 2003; 63: 389–406
- Tossi, A, Mitaritonna, N, Tarantino, C, Giangaspero, A, Sandri, L, Winterstein, KA. Antimicrobial Sequences Database. 2003.
- Powers JP, Hancock RE. The relationship between peptide structure and antibacterial activity. Peptides 2003; 24: 1681–1691
- Hancock RE. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect Dis 2001; 1: 156–164
- Hancock RE. Peptide antibiotics. Lancet 1997; 349: 418–422
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature 2002; 415: 389–395
- Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 2003; 55: 27–55
- Zhang L, Rozek A, Hancock RE. Interaction of cationic antimicrobial peptides with model membranes. J Biol Chem 2001; 276: 35714–35722
- Matsuzaki K, Murase O, Fujii N, Miyajima K. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 1995; 34: 6521–6526
- Fischer R, Fotin-Mleczek M, Hufnagel H, Brock R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. Chembiochem 2005; 6: 2126–2142
- Mano M, Teodosio C, Paiva A, Simoes S, Pedroso de Lima MC. On the mechanisms of the internalization of S4(13)-PV cell-penetrating peptide. Biochem J 2005; 390: 603–612
- Jones SW, Christison R, Bundell K, Voyce CJ, Brockbank SM, Newham P, Lindsay MA. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br J Pharmacol 2005; 145: 1093–1102
- Fuchs SM, Raines RT. Pathway for polyarginine entry into mammalian cells. Biochemistry 2004; 43: 2438–2444
- Drin G, Cottin S, Blanc E, Rees AR, Temsamani J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem 2003; 278: 31192–31201
- Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci USA 2000; 97: 13003–13008
- Vives E, Granier C, Prevot P, Lebleu B. Structure-activity relationship study of the plasma membrane translocating potential of a short peptide from HIV-1 Tat protein. Lett Pept Sci 1997; 4: 429–436
- Thoren PE, Persson D, Isakson P, Goksor M, Onfelt A, Norden B. Uptake of analogs of penetratin, Tat(48-60) and oligoarginine in live cells. Biochem Biophys Res Commun 2003; 307: 100–107
- Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem 2002; 277: 2437–2443
- Saalik P, Elmquist A, Hansen M, Padari K, Saar K, Viht K, Langel U, Pooga M. Protein cargo delivery properties of cell-penetrating peptides. A comparative study. Bioconjug Chem 2004; 15: 1246–1253
- Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem 2003; 278: 585–590
- Potocky TB, Menon AK, Gellman SH. Cytoplasmic and nuclear delivery of a TAT-derived peptide and a beta-peptide after endocytic uptake into HeLa cells. J Biol Chem 2003; 278: 50188–50194
- Fischer R, Kohler K, Fotin-Mleczek M, Brock R. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J Biol Chem 2004; 279: 12625–12635
- Drin G, Mazel M, Clair P, Mathieu D, Kaczorek M, Temsamani J. Physico-chemical requirements for cellular uptake of pAntp peptide. Role of lipid-binding affinity. Eur J Biochem 2001; 268: 1304–1314
- Szeto HH, Schiller PW, Zhao K, Luo G. Fluorescent dyes alter intracellular targeting and function of cell-penetrating tetrapeptides. Faseb J 2005; 19: 118–120
- Boisseau S, Mabrouk K, Ram N, Garmy N, Collin V, Tadmouri A, Mikati M, Sabatier JM, Ronjat M, Fantini J, De Waard M. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim Biophys Acta 2006; 1758: 308–319
- Balayssac S, Burlina F, Convert O, Bolbach G, Chassaing G, Lequin O. Comparison of penetratin and other homeodomain-derived cell-penetrating peptides: interaction in a membrane-mimicking environment and cellular uptake efficiency. Biochemistry 2006; 45: 1408–1420
- Henriques ST, Costa J, Castanho MA. Re-evaluating the role of strongly charged sequences in amphipathic cell-penetrating peptides: a fluorescence study using Pep-1. FEBS Lett 2005; 579: 4498–4502
- Oehlke J, Scheller A, Wiesner B, Krause E, Beyermann M, Klauschenz E, Melzig M, Bienert M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim Biophys Acta 1998; 1414: 127–139
- Oehlke J, Brudel M, Blasig IE. Benzoylation of sugars, polyols and amino acids in biological fluids for high-performance liquid chromatographic analysis. J Chromatogr B Biomed Appl 1994; 655: 105–111
- Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 2005; 280: 15300–15306
- Raucher D, Chilkoti A. Enhanced uptake of a thermally responsive polypeptide by tumor cells in response to its hyperthermia-mediated phase transition. Cancer Res 2001; 61: 7163–7170
- Massodi I, Bidwell GL, 3rd, Raucher D. Evaluation of cell penetrating peptides fused to elastin-like polypeptide for drug delivery. J Control Release 2005; 108: 396–408
- Elmquist A, Langel U. In vitro uptake and stability study of pVEC and its all-D analog. Biol Chem 2003; 384: 387–393
- Burlina F, Sagan S, Bolbach G, Chassaing G. Quantification of the cellular uptake of cell-penetrating peptides by MALDI-TOF mass spectrometry. Angew Chem Int Ed Engl 2005; 44: 4244–4247
- Aussedat B, Sagan S, Chassaing G, Bolbach G, Burlina F. Quantification of the efficiency of cargo delivery by peptidic and pseudo-peptidic Trojan carriers using MALDI-TOF mass spectrometry. Biochim Biophys Acta 2006; 1758: 375–383
- Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem 2001; 276: 5836–5840
- Powers JP, Martin MM, Goosney DL, Hancock RE. The antimicrobial peptide polyphemusin localizes to the cytoplasm of Escherichia coli following treatment. Antimicrob Agents Chemother 2006; 50: 1522–1524
- Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 1998; 244: 253–257
- Lau YE, Rozek A, Scott MG, Goosney DL, Davidson DJ, Hancock RE. Interaction and cellular localization of the human host defense peptide LL-37 with lung epithelial cells. Infect Immun 2005; 73: 583–591
- Lundberg M, Johansson M. Is VP22 nuclear homing an artifact?. Nat Biotechnol 2001; 19: 713–714
- Friedrich CL, Moyles D, Beveridge TJ, Hancock RE. Antibacterial action of structurally diverse cationic peptides on gram-positive bacteria. Antimicrob Agents Chemother 2000; 44: 2086–2092
- Boman HG, Agerberth B, Boman A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect Immun 1993; 61: 2978–2984
- Shi J, Ross CR, Chengappa MM, Sylte MJ, McVey DS, Blecha F. Antibacterial activity of a synthetic peptide (PR-26) derived from PR-39, a proline-arginine-rich neutrophil antimicrobial peptide. Antimicrob Agents Chemother 1996; 40: 115–121
- Subbalakshmi C, Sitaram N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol Lett 1998; 160: 91–96
- Deshayes S, Heitz A, Morris MC, Charnet P, Divita G, Heitz F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry 2004; 43: 1449–1457
- Deshayes S, Gerbal-Chaloin S, Morris MC, Aldrian-Herrada G, Charnet P, Divita G, Heitz F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim Biophys Acta 2004; 1667: 141–147
- Chaloin L, De E, Charnet P, Molle G, Heitz F. Ionic channels formed by a primary amphipathic peptide containing a signal peptide and a nuclear localization sequence. Biochim Biophys Acta 1998; 1375: 52–60
- Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992; 31: 12416–12423
- Terrone D, Sang SL, Roudaia L, Silvius JR. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry 2003; 42: 13787–13799
- Drin G, Demene H, Temsamani J, Brasseur R. Translocation of the pAntp peptide and its amphipathic analogue AP-2AL. Biochemistry 2001; 40: 1824–1834
- Persson D, Thoren PE, Esbjorner EK, Goksor M, Lincoln P, Norden B. Vesicle size-dependent translocation of penetratin analogs across lipid membranes. Biochim Biophys Acta 2004; 1665: 142–155
- Thoren PE, Persson D, Esbjorner EK, Goksor M, Lincoln P, Norden B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry 2004; 43: 3471–3489
- Wimley WC, White SH. Determining the membrane topology of peptides by fluorescence quenching. Biochemistry 2000; 39: 161–170
- Barany-Wallje E, Keller S, Serowy S, Geibel S, Pohl P, Bienert M, Dathe M. A critical reassessment of penetratin translocation across lipid membranes. Biophys J 2005; 89: 2513–2521
- Thoren PE, Persson D, Karlsson M, Norden B. The antennapedia peptide penetratin translocates across lipid bilayers – the first direct observation. FEBS Lett 2000; 482: 265–268
- Ambroggio EE, Kim DH, Separovic F, Barrow CJ, Barnham KJ, Bagatolli LA, Fidelio GD. Surface behavior and lipid interaction of Alzheimer beta-amyloid peptide 1-42: a membrane-disrupting peptide. Biophys J 2005; 88: 2706–2713
- Ambroggio EE, Separovic F, Bowie JH, Fidelio GD, Bagatolli LA. Direct visualization of membrane leakage induced by the antibiotic peptides: maculatin, citropin, and aurein. Biophys J 2005; 89: 1874–1881
- Magzoub M, Pramanik A, Graslund A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry 2005; 44: 14890–14897
- Henriques ST, Castanho MA. Consequences of nonlytic membrane perturbation to the translocation of the cell penetrating peptide pep-1 in lipidic vesicles. Biochemistry 2004; 43: 9716–9724
- Matsuzaki K, Yoneyama S, Murase O, Miyajima K. Transbilayer transport of ions and lipids coupled with mastoparan X translocation. Biochemistry 1996; 35: 8450–8456
- Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J 2004; 377: 159–169
- De Coupade C, Fittipaldi A, Chagnas V, Michel M, Carlier S, Tasciotti E, Darmon A, Ravel D, Kearsey J, Giacca M, Cailler F. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J 2005; 390: 407–418
- Silhol M, Tyagi M, Giacca M, Lebleu B, Vives E. Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur J Biochem 2002; 269: 494–501
- Pooga M, Soomets U, Hallbrink M, Valkna A, Saar K, Rezaei K, Kahl U, Hao JX, Xu XJ, Wiesenfeld-Hallin Z, Hokfelt T, Bartfai T, Langel U. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol 1998; 16: 857–861
- Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 2001; 19: 1173–1176
- Takeshima K, Chikushi A, Lee KK, Yonehara S, Matsuzaki K. Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J Biol Chem 2003; 278: 1310–1315
- Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 2001; 276: 3254–3261
- Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem 2003; 278: 34141–34149
- Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther 2003; 8: 284–294
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 2004; 10: 310–315
- Henriques ST, Costa J, Castanho MA. Translocation of beta-galactosidase mediated by the cell-penetrating peptide pep-1 into lipid vesicles and human HeLa cells is driven by membrane electrostatic potential. Biochemistry 2005; 44: 10189–10198
- Marsh D. CRC handbook of lipid bilayers. CRC Press, Boca Raton 1990
- Heerklotz H, Seelig J. Detergent-like action of the antibiotic peptide surfactin on lipid membranes. Biophys J 2001; 81: 1547–1554
- Strahilevitz J, Mor A, Nicolas P, Shai Y. Spectrum of antimicrobial activity and assembly of dermaseptin-b and its precursor form in phospholipid membranes. Biochemistry 1994; 33: 10951–1060
- Terrone D, Sang SL, Roudaia L, Silvius JR. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry 2003; 42: 13787–13799
- Matsuzaki K, Murase O, Fujii N, Miyajima K. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 1995; 34: 6521–6526