
ABSTRACT
Insulin resistance (IR) is a state in which the tissues show a reduced sensitivity to insulin despite its normal or even increased concentration in blood. IR causes inability of peripheral tissues to respond to the hormone and to increase the glucose uptake from blood nor to raise the metabolism rate. The tissues resistance to insulin may refer to the prereceptor malfunctions, which reduce the concentration of insulin or decrease the number of insulin receptors; however, in most cases changes in signal transduction after insulin binding to the receptor are observed. The development of IR is determined by a number of genetic and environmental factors. One of the most relevant causes is obesity, which develops into a growing problem among the horse population all over the world. The accumulation of lipids in the tissues (lipotoxicity) and a mild inflammation caused by the release of proinflammatory adipokines and cytokines from the fat tissue are a common denominator that links obesity with IR. Both IR and obesity seem to cause metabolism malfunctions in horses and are the key components of equine metabolic syndrome; moreover, both may be the reason for endocrinological laminitis. The methods of prevention and treatment of IR include feeding the animals with low glycemic index food and increasing physical activity as well as pharmacological treatment, where levothyroxine sodium and metformin are of the highest importance.
Introduction
Insulin is responsible for maintaining homeostasis by controlling glucose metabolism. This hormone stimulates glucose uptake by skeletal muscle and adipose tissue cells, as well as the synthesis of glycogen in liver and muscle cells. In addition, insulin stimulates the synthesis of fatty acids and triglycerides in the liver and adipose tissue, and inhibits lipolysis. The hormone is responsible for protein anabolism, cell growth, and survival; regulates vascular endothelium; and exerts anti-inflammatory effects (Raymond Citation2008; Tymoczko et al. Citation2011). Insulin transduces signals into cells through specific insulin receptors (). Insulin receptors are present on the surface of nearly all cells, but most are found on the surface of striated muscle cells, liver cells, and adipocytes (Bhattacharya et al. Citation2007; Rojek & Niedziela Citation2009; Ye Citation2013).
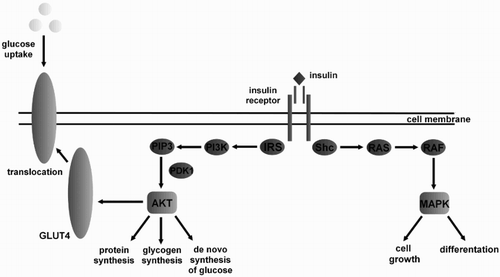
Figure 1. Mechanism of insulin action. Binding of insulin to the extracellular domain of insulin receptor initiating a signaling cascade leads to receptor autophosphorylation and tyrosine phosphorylation of intracellular protein substrates. Two main pathways are activated by insulin- the IRS (insulin receptor substrate) → PI3K (phosphatidylinositol 3-kinase) and the RAS (RAS protein) → MAPK (mitogen-activated protein kinase) pathway. Activation of the IRS pathway leads to metabolic alterations, whereas the RAS → MAPK pathway stimulating cell growth and proliferation. PIP3 – Phosphatidylinositol 3,4,5-trisphosphate; PDK1 – phosphoinositide-dependent protein kinase 1; AKT – protein kinase B; SHC – Src Homology 2 domain; GLUT4 – glucose transporter type 4 RAF – RAF protein.
Mechanisms of insulin resistance
Insulin resistance (IR) is defined as a state of reduced responsiveness of tissues to insulin, despite its normal or elevated concentration in blood; the disorder mainly occurs in adipose tissue, skeletal muscle cells, and liver (Bhattacharya et al. Citation2007; Frank & Tadros Citation2014). The consequences of IR include impaired glucose uptake by the tissues, increased efficiency of gluconeogenesis, and increased lipolysis (Treiber et al. Citation2006). Presently there is no precise clinical definition of IR in horses and ponies, and consequently, there are only few research on the pathogenesis of equine IR. The mechanisms of IR are investigated on the basis of studies on specific insulin activity and the tissues involved in human and laboratory animals.
The development of IR is caused by a number of genetic and environmental factors. One of the most important is obesity, which has a complex pathogenesis and has become a cause for concern in horses (Johnson et al. Citation2009). The development of equine obesity and IR may be caused by restricted movement and feeding of energy-dense diets rich in simple carbohydrates and starch (Kronfeld et al. Citation2005; Treiber et al. Citation2006; Johnson et al. Citation2009). Pratt et al. (Citation2006) reported a 30% decrease in insulin-dependent glucose uptake in horses fed a diet rich in non-structural carbohydrates, compared to horses fed a diet containing fibre and fat, in which sensitivity to insulin did not change. Similar results were obtained by Hoffman et al. (Citation2003), who found that sensitivity to insulin tended to decrease in horses fed a supplement rich in starch and carbohydrates.
Obesity-related IR is probably caused by the thrifty genotype; while such genes provided an advantage over other animals living in the wild and buffered seasonal food shortages, under domestication conditions the high energy efficiency becomes a burden, especially when combined with improper practices (Johnson et al. Citation2009; Tinworth et al. Citation2010; Tadros & Frank Citation2013). Evolutionarily adapted to living in a food-poor environment, horses and ponies are more predisposed to obesity and IR where food is available throughout the year (Johnson et al. Citation2009; Frank & Tadros Citation2014).
Equine obesity and IR are associated with altered fat metabolism, which results in glucose intolerance, epithelial dysfunction, increased levels of procoagulation factors, and higher expression of inflammatory markers. Other diseases accompanying equine obesity and IR have been rarely described (Wylie & Collins Citation2010; Johnson et al. Citation2012; Tadros & Frank Citation2013). Type 1 diabetes mellitus (T1DM) is characterized by the lack of insulin secretion by beta cells of the pancreas as a result of their destruction (due to viral or immune-mediated disease), whereas type 2 diabetes mellitus (T2DM), characterized by relatively reduced insulin secretion, is a complex metabolic disorder that develops due to chronic hyperinsulinemia. Temporary T1DM was diagnosed in foals. T2DM occurs in older horses affected with polyuria, polydipsia and body weight loss as a consequence of pituitary pars intermedia dysfunction. Cases of bona fide DM were described in adult horses (caused by chronic pancreatitis, pituitary pars intermedia dysfunction, granulosa cell tumour, immune polyendocrinopathy, and ovarian neoplasia in pregnancy) (Durham et al. Citation2009; Johnson et al. Citation2012).
Abnormalities in insulin metabolism, such as IR and hyperinsulinemia, are manifested during the equine metabolic syndrome (EMS). Horses are at an increased risk of laminitis, and EMS is also the cause of hyperlipidaemia syndrome (hepatic lipidosis) and osteochondrosis in young horses (Johnson et al. Citation2012). Despite a strong correlation between obesity and IR, the latter and other metabolic disorders have been reported in horses with normal body weight, but such animals are often characterized by abnormal distribution of adipose tissue (Frank Citation2009).
IR causes that peripheral tissues are unable to increase glucose uptake from blood and to accelerate metabolic rate in response to the hormone. In the initial phase, to avoid long periods of elevated blood glucose concentration, beta cells of the pancreas compensate for IR by producing and secreting more insulin, leading to pathological hyperinsulinemia, which in horses is defined as a basal concentration above 30 mU/L. This means that insulin levels are continuously elevated, but appropriate amounts of glucose are still transported to the cells to maintain the glucose concentration at a normal level (60–90 mg/dl), even if it is close to the upper limit of reference values (Ralston Citation2002; Frank Citation2009; Frank et al. Citation2010). The incidence of IR in horses has not been determined, but hyperinsulinemia was found to occur in 10% of the 300 horses in Southwestern Virginia (Geor & Frank Citation2009).
Following a gradual decrease in the response of the target tissue cells to the insulin-transducted signal, when the level of glucose in blood does not drop, the organism reacts by repeatedly mobilizing the pancreas to produce and secrete an increased amount of insulin until the signal becomes strong enough to activate the transporters and to eliminate the excess amount of glucose in blood. In horses and ponies suffering from IR, the pancreas may produce such large amounts of insulin that its concentration will exceed its resting values a hundredfold (Sillence et al. Citation2007; Pawlak & Derlacz Citation2011).
Resistance of tissues to insulin may be caused by prereceptor abnormalities, which reduce the concentration of circulating insulin or decrease the number of insulin receptors. However, more frequent are changes in signal transduction following the binding of insulin to its receptor, which result from diminished receptor phosphorylation and reduced tyrosine kinase activity, and lead to lower insulin receptor substrate-1 (IRS-1) phosphorylation and decreased phosphatidylinositol 3-kinase activity. Furthermore, the abnormalities in intracellular glucose metabolism, which is regulated by enzymes such as hexokinase and glycogen synthetase, may be reduced by insulin-dependent uptake of glucose and its storage (Treiber et al. Citation2006).
Because skeletal muscles are responsible for the uptake of around 80% postprandial blood glucose, they are considered one of the most susceptible tissues to the development of IR. The molecular basis of IR in skeletal muscles is thought to be postreceptor defects in signal transduction in the insulin pathway, which result in abnormalities in glucose transporter GLUT4 translocation to the cell membrane and in non-receptor changes caused by a change in the activity of enzymatic proteins. IR in skeletal muscles impairs glucose uptake and utilization, thus decreasing muscle glycogen synthesis (Schmidt & Hickey Citation2009). By reducing the lipolytic activity of insulin in peripheral tissues, IR increases the concentration of free fatty acids (FFA) in the bloodstream. The increased pool of FFA delivered to the liver contributes to decreased hepatic insulin degradation and to uncontrolled increase in hepatic glycogenolysis and gluconeogenesis. Hepatocyte IR is also manifested in abnormal lipid metabolism. Hyperinsulinemia and hyperglycaemia promote lipogenesis and increase esterification of fatty acids to diacylglycerols (DAG), triacylglycerols (TAG), and ceramides (CER). The accumulation of lipids, mainly DAG and CER, has a direct effect on insulin signal transduction by increasing IR. DAG impairs the insulin pathway mainly as a result of increased activation of protein kinase C (PKC). CER can be generated de novo from palmitoyl-CoA and serine or by hydrolysis of sphingomyelin, and their excessive accumulation in tissues probably leads to abnormalities in insulin signal transduction, mainly through inactivation of protein kinase B (PKB) as a result of its dephosphorylation, and also by inhibiting PKB translocation to the cell membrane. In addition, CER increase the expression of proinflammatory cytokines (including tumour necrosis factor (TNF)-α), which enhance ceramide synthesis de novo and sphingolipid metabolism, thus increasing IR of the tissues (Summers Citation2006; Schmidt & Hickey Citation2009).
Hepatic IR caused by the accumulation of FFA is related to the postreceptor malfunction of insulin signal. The increased translocation of PKC delta from the cytosol to plasma membrane of the hepatocyte causes the phosphorylation of serine and threonine residues in IRS-1 and its inactivation in result. The accumulation of FFA in the hepatocytes inhibits the activity of tyrosine kinase and tyrosine autophosphorylation in the insulin receptor. It was demonstrated that long-chain fatty acids block the activity and translocation of glucokinase, which leads to a rise in the concentration of glucose in hepatocytes and, consequently, to reduction in glucose uptake. The augmented flow of FFA to the liver causes an increase in the secretion of very low-density lipoproteins (VLDL), a decrease in hepatic insulin degradation, and an extended production and release of glucose from hepatocytes, which result in the development of hyperinsulinemia, hyperglycaemia, as well as dyslipidaemia. The latest research on animals indicate a considerable role of interleukin-6 (IL-6) in the development of hepatic IR (Konstantynowicz et al. Citation2011; Frank & Tadros, Citation2014; De Graaf-Roelfsema Citation2014) In horses and ponies, liver IR has been shown to induce hypertriglyceridemia. Although plasma triglycerides and VLDL are only slightly elevated in obese horses and ponies with hyperinsulinemia, they are potential indicator factors of IR in the liver. Increased plasma concentrations of gamma glutamyl transferase and hepatic lipidosis have also been reported in fat horses (Frank Citation2009, Citation2011; Frank et al. Citation2010).
At present, there are no studies in horses on sex differences in insulin sensitivity in adult animals, but report is emerging on differences in tissue sensitivity to insulin in foals of both sexes (Jellyman et al. Citation2014). Jellyman et al. (Citation2014) demonstrated some differences in the function of pancreatic beta cells between newborn mares and stallions. In particular, insulin secretion in response to exogenous glucose was much higher in fillies than in colts aged 2 and 12 weeks. The glucose clearance, which the same authors found to be unaffected by sex despite the higher levels of insulin secretion in the mares, suggests that tissue insulin sensitivity may be lower in mares than in stallions shortly after birth. The potential causes of sexual dimorphism in the function of pancreatic beta cells remain unknown. It is necessary to verify if sex differences in glucose–insulin dynamics will appear later in adult life.
Role of adipose tissue in IR
Equine obesity is becoming more and more of an issue throughout the world. It is estimated that 45% of the horse population evaluated is obese (Wyse et al. Citation2008). According to a 1998 study, 5% of the analysed horses were overweight (USDA Citation2000). A later study from 2006 demonstrated that the scale of the problem is much larger and concerns 51% of the horses (19% obese, 32% overweight) (Thatcher et al. Citation2007). The populations of horses under investigation included mostly animals used in non-competition leisure riding or kept as pets.
Development of obesity is a causative factor for IR. There are two basic theories linking obesity to IR: the down-regulation of signalling pathways induced by adipokines and cytokines produced in adipose tissue, and the accumulation of intracellular lipids in insulin-sensitive tissue such as skeletal muscle (lipotoxicity) (Tadros & Frank Citation2013). Along with muscle and liver glycogen, triglycerides stored in adipose tissue are the body's most important sources of energy. The accumulation of energy stores is regulated by the anabolic action of insulin, while their utilization during starvation, infection, injury, or stress is controlled by proinflammatory cytokines, stress hormones, and glucagon.
In the time of stress, the organism launches a cascade of neuroendocrine responses, which results in heightened secretion of glucocorticosteroids and catecholamines. The growth of the level of cortisol, catecholamines, and glucagon is directly responsible for the extended catabolism and a higher energy expenditure of the organism. The action of stress hormones is aimed at providing the energy by means of releasing such substrates as glucose, amino acids, FFA, and glycerol. In the case of an insufficient glucose supply, the organism uses glycogen from skeletal muscles and liver as the reservoir. However, those supplies are too small to cover the caloric needs of the organs which use only glucose, such as the brain, and they become exhausted very fast. The result of the action of stress hormones and glucagon is the activation of hormone-sensitive lipase in the fat cells, and consequently, the increase in lipolysis. This causes a rise in the release of FFA and in the bioaccessibility of glycerol, a substrate for glucogenesis, which provides the energy for fight or flight responses (Sillence et al. Citation2007; Peckett et al. Citation2011; Tsatsoulis et al. Citation2013).
Under physiological conditions, the adipose tissue stores are released as FFA, while their metabolism is regulated in peripheral tissues. A dynamic balance is maintained between triglyceride lipolysis and FFA release, as is the control of their uptake and oxidation by other tissues, especially the muscles (Guilherme et al. Citation2008; Tsatsoulis et al. Citation2013). Adipose tissue, which serves as the body's energy storage site, is also an active endocrine organ producing and releasing many hormones, metabolites, and factors that regulate appetite, inflammatory processes, and the balance of metabolic pathways, known as adipokines. These include TNF-α, IL-6, plasminogen activator inhibitor type 1, angiotensin II, adiponectin, leptin, visfatin, or resistin, which have auto-, para- and endocrine effects (Guilherme et al. Citation2008; Tsatsoulis et al. Citation2013; Ye Citation2013).
Under natural conditions horse feed is low in fat, but excessive amounts of non-structural carbohydrates may cause them to be converted to fats during lipogenesis; therefore, high energy intake, restriction of movement, and stress contribute to fat being stored in adipocytes in amounts exceeding their storage and liporegulation capacity. The accumulation of fat supplies observed during the time of chronic stress seems to be caused by a constantly elevated level of cortisol which, being an antagonist to insulin, blocks the insulin-dependent uptake of glucose by the tissues. In a short-term stress situation, the purpose of the metabolic block is to limit the glucose uptake by tissues other than the brain. On the other hand, the chronic elevation of cortisol level leads to a constant hyperglycaemia. As a result, the organism releases more insulin, which stimulates lipogenesis and deposition of fat tissue. The permanently elevated cortisol level may also induce leptin resistance, which contributes to further adipose tissue accumulation (Sillence et al. Citation2007; Peckett et al. Citation2011; Tsatsoulis et al. Citation2013).
Excess nutrients in blood may lead to overload and hypertrophy of adipose tissue, resulting in hypoxia, cell necrosis, and infiltration of adipose tissue with macrophages, which triggers the inflammatory response (Pawlak & Derlacz Citation2011; Tsatsoulis et al. Citation2013). The balance between FFA release and oxidation in peripheral tissues may be upset if the adipose tissue is dysfunctional. The muscles, liver, and pancreas convert excess fat by increasing β-oxidation, but lipids may accumulate in these tissues and contribute to changes in cell functions, including the transduction of insulin signals (Tsatsoulis et al. Citation2013; Frank & Tadros Citation2014). Equine obesity is associated with increased concentrations of FFA, which compete as an energy source, inhibit the uptake of glucose by insulin-sensitive tissues, and stimulate hepatic gluconeogenesis, thus disturbing glucose metabolism and leading to chronic hyperglycaemia (Kronfeld et al. Citation2005). FFA also stimulate pancreatic beta cells to release insulin while impairing hepatic insulin degradation. Chronic hyperglycaemia, hyperinsulinemia, and disturbed glucose transport into the cells will aggravate tissue IR (Kahn & Flier Citation2000; Vick et al. Citation2007). Research has shown that insulin sensitivity in obese horses is 80% lower than in horses with normal body weight (Hoffman et al. Citation2003).
Obesity in animals causes chronic inflammation, which plays a major role in the pathogenesis of IR (Raymond Citation2008). Dysfunctional adipose tissue reacts by secreting proinflammatory cytokines (such as IL-6, IL-1 and TNF-α) and monocyte chemoattractant protein-1 (Draznin Citation2006; Olszanecka-Glininowicz & Zahorska-Markiewicz Citation2008; Frank et al. Citation2010). Vick et al. (Citation2007) showed that obesity is positively correlated to messenger ribonucleic acid (mRNA) expression of TNF-α and interleukin-6 and TNF-α concentration in warmblood mares. Increased TNF-α concentration was also reported in ponies susceptible to the development of laminitis. The TNF-α inhibits intracellular glucose transport, enhances lipolysis, causing plasma FFA concentrations to increase, and is a reliable marker of IR. TNF-α acts in at least two sites of the insulin signalling pathway. At the insulin receptor level, it induces serine phosphorylation of IRS-1 protein and inhibits the phosphorylation of insulin receptor tyrosine kinase. Post-receptor activity involves inhibiting insulin signalling at the level of phosphatidylinositol 3-kinase and translocating GLUT-4 protein into the cell membrane, thus impairing the uptake of glucose by the cell and causing IR (Draznin Citation2006; Olszanecka-Glininowicz & Zahorska-Markiewicz Citation2008). Equine obesity is associated with quantity changes in adiponectins release – the studies reported an elevated concentration of leptin and a lowered level of adiponectin, which increases tissues’ sensitivity to insulin (Tadros & Frank Citation2013).
The weight of adipose tissue appears to determine serum leptin concentrations in the horse. Leptin release in horses has a circadian rhythm, peaking at night and declining during the daytime. However, daily variation in leptin levels may result from both feeding schedule and feed composition. Leptin level in horses increases 8–10 h after consumption of a high-carbohydrate feed and continues to increase with increasing insulin levels. The increases and decreases in leptin as well as insulin concentrations were found to be suppressed in horses eating frequent meals and in those with ad libitum access to hay or pasture grass. Adiponectin concentration in horses was found to be negatively correlated to the weight of adipose tissue, fat percentage, and body condition score. Unlike leptin, adiponectin secretion shows no diurnal rhythm and does not change depending on the composition of food (Radin et al. Citation2009).
IR as a cause of laminitis
The literature describes several mechanisms for the development of endocrine laminitis, resulting from the occurrence of IR in horses (Mcgowan Citation2008; Huntington et al. Citation2010).
Initially, impaired glucose uptake by tissues was implicated as the cause of hoof tissue damage. It results from abnormal function of the PI3K pathway and thus GLUT4 transporters that carry glucose into the cells, especially the muscle and adipose tissue cells. The potential glucose deprivation of tissues causes cell damage and death. Research on hoof explants showed that glucose is absolutely required for hoof tissues, and hoof tissue cultures in the absence of glucose caused the layers of tissue to separate rapidly, as is the case during laminitis. Research has demonstrated, however, that hoof tissue is non-insulin dependent due to the overwhelming presence of GLUT-1 transporters, which are independent of insulin. It has also been shown that insulin receptors are not present on lamellar cells but in the blood vessel walls. This provides evidence that glucose deprivation is not the cause of laminitis in insulin-resistant horses (Huntington et al. Citation2010).
The role of insulin in controlling the expansion and contraction of blood vessels may also explain the increased predisposition of insulin-resistant horses to the development of laminitis. Slow vasodilation in response to insulin occurs under the influence of nitric oxide (NO), while endothelin-1 (ET-1)- and insulin-stimulated sympathetic nervous system are responsible for vasoconstriction. Under physiological conditions, a balance is maintained between the two processes because both insulin signalling pathways (PI3K and MAPK) are equally active. NO is generated by NO synthase vascular endothelial cells and undergoes local diffusion, causing the vessels to dilate slowly. NO is secreted when the PI3K pathway is activated, whereas activation of the MAPK pathway leads to the release of ET-1 (Venugopal et al. Citation2011; Tadros & Frank Citation2013). During IR, the regulatory action of insulin on blood vessels is disturbed and the PI3K pathway is blocked, whereas the mitogen-activated pathway functions normally, which causes the blood vessels to contract. Plasma concentrations of ET-1 derived from digital blood vessels were found to increase 12 h after healthy horses were administered carbohydrates to induce laminitis. This provides evidence that digital blood vessels contract in response to carbohydrate overload, which may contribute to the development of laminitis. When fed excess dietary carbohydrates, insulin-resistant horses are predisposed to the development of laminitis due to the preponderance of vasoconstrictive processes (Frank et al. Citation2010; Huntington et al. Citation2010; Tadros & Frank Citation2013).
The pathogenesis of laminitis and other inflammatory states is accompanied by an enhanced activity of metalloproteinases. These enzymes are located in neutrophils and tissues, for example, epithelial tissue, and are responsible for the degradation and alteration of the environment of the extracellular matrix. The dynamic balance between the degradation and synthesis of collagen in the extracellular matrix in physiological conditions is sustained by the cooperation of matrix metalloproteinases (MMP) and their tissue inhibitors (tissue inhibitors of metalloproteinases). The unregulated activity of MMP, particularly MMP-2 and MMP-9, which are responsible for type IV collagen degradation, may lead to hoof tissue damage – the destruction of basement membrane – and consequently, to distal phalangeal attachment apparatus damage (Orsini et al. Citation2009; Huntington et al. Citation2010; De Laat et al. Citation2011; Katz & Bailey Citation2012). The production and later activation of MMPs are observed during local or systemic inflammatory states, ischaemia, or as a result of bacterial toxins' presence. The increased activity of MMP-2 and MMP-9 was noted during alimentary carbohydrate-induced laminitis and black walnut extract-induced laminitis.
It was thought that chronic hyperinsulinemia-related glucotoxicity, hypoperfusion, elevated release of reactive oxygen species or haemodynamic changes lead finally to MMP activation and cause insulin-induced laminitis (De Laat et al. Citation2011; Katz & Bailey Citation2012). The research on insulin-induced laminitis proved that during the developmental and acute stage of laminitis, the expression of the genes coding for MMP-2 was not triggered. Activation of the enzymes and their increased concentration in hoof lamellae were not observed either. This indicates that MMP-2 does not play a significant role in the development of insulin-induced laminitis and the presence of those enzymes in the later stages is related to the repair of the basement membrane, not its degradation. The elevated expression of MMP-9-coding gene and the increase in the concentration of inactive pro-MMP-9, which are observed during insulin-induced laminitis, are credited to the migration of polymorphonuclear granulocytes during the prodromal stage of laminitis or considered a direct cause of hyperinsulinemia; however, this matter demands further research (De Laat et al. Citation2011).
Prevention and treatment of IR in horses
Prevention and treatment of IR in horses should be based on changing the way they are managed and fed. In obese horses, efforts should be made to reduce body weight and to lower the glycaemic and insulinemic response to a meal. In this strategy, feeds with a high glycaemic index are reduced/eliminated, and the dietary content of non-structural carbohydrates is limited. Reduction of dietary digestible energy is an important factor in lowering body weight through the partial or complete elimination of access to pasture grass. The diet should be based on fibre-rich roughages (mostly hay) supplemented with minerals and vitamins (Frank et al. Citation2010; Frank Citation2011; Johnson et al. Citation2012). Improvement of tissue response to insulin is attributed to increased glucose uptake and utilization in muscles, the main site of insulin-dependent glucose uptake. This mechanism is not completely understood, but it is ascribed to the increased number of GLUT4 (the main glucose-transporting protein in muscle) and to the higher activity of insulin-signalling proteins (hexokinase) and glycogen synthase as a result of training. This increases the availability of glucose and insulin to muscle tissues, thus increasing the insulin sensitivity of skeletal muscles (Pratt et al. Citation2006; Stewart-Hunt et al. Citation2006; Schmidt & Hickey Citation2009).
In horses there are few reports concerning the effect of training on IR and the cellular mechanisms of glucose utilization in skeletal muscles. A 7-day continuous light training was found to increase sensitivity to insulin by 60% and 48% in obese mares and in those with normal body weight, respectively. Other studies confirmed the increased GLUT4 content of horse muscles after 6 weeks of training. Stewart-Hunt et al. (Citation2006) reported a twofold increase in whole-body sensitivity to insulin, a 10-fold increase in GLUT4 content, increases in glycogen synthase and hexokinase activity, as well as a 9% increase in muscle aerobic endurance. What is more, these have persisted later on during the period of physical inactivity (Pratt et al. Citation2006; Stewart-Hunt et al. Citation2006; Schmidt & Hickey Citation2009).
There are, however, studies demonstrating that the training did not affect insulin sensitivity in horses. De Graaf-Roelfsema et al. (Citation2006) found no increase in insulin sensitivity after a long-term training (18 weeks). It is also the case in the research by Carter et al. (Citation2010), where 8 weeks of training did not alter insulin sensitivity neither in the trained horses nor in the control group. These examples show that it is not quite sure how intensive the training should be in order to bring the desired effects. Further research is needed to determine the optimal method of training insulin-resistant horses; the current recommendation is to begin training with 2–3 sessions per week (20–30 min per session), each comprising riding or trot and gallop exercise, and to increase training intensity and duration to 5–7 sessions per week (Frank Citation2009, Citation2011).
Levothyroxine sodium and metformin are mostly used to treat IR in horses. Levothyroxine sodium accelerates body weight loss by increasing the concentration of circulating thyroxine and stimulating basal metabolic rate, thus leading to increased insulin sensitivity. Metformin lowers hepatic glucose production through the activation of adenosine monophosphate-activated protein kinase , which inhibits gluconeogenesis and lipogenesis. Metformin also increases the presence of GLUT4 transporters on the surface of cell membranes within skeletal muscles (Frank et al. Citation2010, Citation2011; Rendle et al. Citation2013; Frank & Tadros Citation2014). The latest research demonstrate that despite the lower concentration of insulin and glucose in blood, this drug shows low bioaccessibility in horses and has no effect on increasing insulin sensitivity. The most recent reports in humans and laboratory animals indicate a significant impact of metformin in the intestine. The concentration of metformin is independent from its concentration in blood plasma. In human jejunum it may be even 30–300 times higher than in blood plasma. In rats, by regulating the two main transporters of glucose in the intestine, metformin delays its absorption in the intestine and increases its utilization by the intestine tissue (Rendle et al. Citation2013). Rendle et al.’s (Citation2013) research showed that oral administration of metformin in horses caused a decrease in glycaemic and insulinemic responses to orally administrated dextrose. Further research seems necessary in order to determine whether metformin accumulates in the intestine and whether it causes a similar effect on glucose absorption in the equine intestine.
The principal mode of action of thiazolidinedione drugs (e.g. pioglitazone) is to stimulate the nuclear peroxisome proliferator-activated receptor PPARγ (PPARγ agonist). This receptor regulates adipocyte differentiation, storage of fats and sensitivity to insulin. PPARγ activation increases glucose uptake in adipose tissue, muscles, and liver tissue;stimulates lipogenesis; and inhibits glycogenolysis and gluconeogenesis in the liver. To date in horses, the administration of pioglitazone had a positive effect in the form of increased mRNA expression of the insulin receptor in skeletal muscles. (Frank et al. Citation2010, Citation2011; Frank & Tadros Citation2014).Glyburide favours the release of exogenous insulin from pancreatic beta cells and could also prove useful in treating equine IR and EMS. There is one description of a case involving a horse with T2DM, where after applying glyburide combined with metformin the reduction of interstitial glucose concentration was obtained (Johnson et al. Citation2005).
Dietary supplementation of L-carnitine may have a positive effect on the metabolism of glucose, insulin and leptin in horses (Janicki & Buzała Citation2011). Dietary inclusion of L-carnitine also increases plasma leptin concentration, which is associated with increased glucose tolerance in healthy ponies (Mcclelland et al. Citation2004). The supplementation of ponies with 4 g L-carnitine for seven days caused a decrease in postprandial plasma concentrations of glucose and insulin, which is indicative of increased glucose tolerance. Plasma leptin concentration increased post-feeding when the diet was supplemented with L-carnitine. However, the increase in leptin concentration was not preceded by the increase in the concentration of insulin, which suggests that factors other than plasma insulin concentration may influence plasma leptin concentrations. Although the level of FFA and TAG did not differ significantly in response to supplemental L-carnitine under experimental conditions, further research should clarify if TAG synthesis could be responsible for the increased leptin levels (Weyenberg et al. Citation2009).
Conclusions
IR in horses may have different underlying mechanisms. A major factor leading to the development of equine IR is obesity, which is becoming a common problem around the world. Obesity-related IR is caused by lipotoxicity, changes in insulin signal transduction, and systemic inflammation, resulting from proinflammatory cytokines secreted by adipocytes. IR leads to many metabolic disorders, which can be a cause of many equine diseases, in particular laminitis, which is often severe and may prevent such a horse from being used.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bhattacharya S, Dey D, Roy SS. 2007. Molecular mechanism of insulin resistance. J Biosci. 32:405–413.
- Carter RA, Mccutcheon J, Valle E, Meilahn EN, Geor RJ. 2010. Effects of exercise training on adiposity, insulin sensitivity, and plasma hormone and lipid concentrations in overweight or obese, insulin resistant horses. Am J Vet Res. 71:314–321.
- De Graaf-Roelfsema E. 2014. Glucose homeostasis and the enteroinsular axis in the horse: a possible role in equine metabolic syndrome. Vet J. 199:11–18.
- De Graaf-Roelfsema E, Van Ginneken ME, Van Breda E, Wijnberg ID, Keizer HA, Van Der Kolk JH. 2006. The effect of long-term exercise on glucose metabolism and peripheral insulin sensitivity in standardbred horses. Equine Vet J Supplement. 38:221–225.
- De Laat MA, Kyaw-Tanner MT, Nourian AR, Mcgowan CM, Sillence MN, Pollitt CC. 2011. The developmental and acute phases of insulin-induced laminitis involve minimal metalloproteinase activity. Vet Immunol Immunopathol. 140:275–281.
- Draznin B. 2006. Molecular mechanisms of insulin resistance: serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: the two sides of a coin. Diabetes. 55:2392–2397.
- Durham AE, Hughes KJ, Cottle HJ, Rendle DI, Boston RC. 2009. Type 2 diabetes mellitus with pancreatic beta cell dysfunction in 3 horses confirmed with minimal model analysis. Equine Vet J. 41:924–929.
- Frank N. 2009. Equine metabolic syndrome. J Equine Vet Sci. 29:259–267.
- Frank N. 2011. Equine metabolic syndrome. Vet Clin North Am Equine Pract. 27:73–92.
- Frank N, Geor RJ, Bailey SR, Durham AE, Johnson PJ. 2010. Equine metabolic syndrome. J Vet Intern Med. 24:467–475.
- Frank N, Tadros EM. 2014. Insulin dysregulation. Equine Vet J. 46:103–112.
- Geor R, Frank N. 2009. Metabolic syndrome – from human organ disease to laminar failure in equids. Vet Immunol Immunopathol. 129:151–154.
- Guilherme A, Virbasius JV, Puri V, Czech MP. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 9:367–377.
- Hoffman RM, Boston RC, Stefanovski D, Kronfeld DS, Harris PA. 2003. Obesity and diet affect glucose dynamics and insulin sensitivity in Thoroughbred geldings. J Anim Sci. 81:2333–2342.
- Huntington PJ, Pollitt C, Mcgowan C. 2010. Recent research into laminitis. NZ Equine Vet Practitioner. 34:7–20.
- Janicki B, Buzała M. 2011. L-karnityna w żywieniu koni sportowych. Med Weter. 67:824–828.
- Jellyman JK, Valenzuela OA, Allen VL, Holdstock NB, Fowden AL. 2014. Sex-associated differences in pancreatic β cell function in healthy preweaning pony foals. Equine Vet J. 15:1–7.
- Johnson PJ, Scotty NC, Wiedmeyer C, Kreeger JM. 2005. Diabetes mellitus in a domesticated Spanish Mustang. J Am Vet Med Ass. 226:584–588.
- Johnson PJ, Wiedmeyer CE, Lacarrubba A, Ganjam VK, Messer NT. 2012. Diabetes, insulin resistance, and metabolic syndrome in horses. J Diabetes Sci Technol. 6:534–540.
- Johnson PJ, Wiedmeyer CE, Messer NT, Ganjam VK. 2009. Medical implications of obesity in horses – lessons for human obesity. J Diabetes Sci Technol. 3:163–174.
- Kahn BB, Flier JS. 2000. Obesity and insulin resistance. J Clin Inves. 106:473–481.
- Katz LM, Bailey SR. 2012. A review of recent advances and current hypotheses on the pathogenesis of acute laminitis. Equine Vet J. 44:752–761.
- Konstantynowicz K, Mikłosz A, Stepek T, Chabowski A. 2011. Akumulacja lipidów (triacylo-, diacylogliceroli i ceramidów) wewnątrz hepatocytów, a rozwój insulinooporności wątrobowej. Postepy Hig Med Dosw. 65:236–243.
- Kronfeld DS, Treiber KH, Hess TM, Boston RC. 2005. Insulin resistance in the horse: definition, detection and dietetics. J Anim Sci. 83:22–31.
- Mcclelland GB, Kraft CS, Michaud D, Russell JC, Mueller CR, Moyes CD. 2004. Leptin and the control of respiratory gene expression in muscle. BBA Mol Basis Dis. 1688:86–93.
- Mcgowan C. 2008. The role of insulin in endocrinopathic laminitis. J Equine Vet Sci. 28:603–607.
- Olszanecka-Glininowicz M, Zahorska-Markiewicz B. 2008. Otyłość jako choroba zapalna. Postępy Hig Med Dośw. 62:249–257.
- Orsini J, Galantino-Homer H, Pollitt CC. 2009 Laminitis in horses: through the lens of systems theory. J Equine Vet Sci. 29:105–114.
- Pawlak J, Derlacz RA. 2011. Mechanizm powstawania oporności na insulinę w tkankach obwodowych. Postępy Biochemii. 57:200–206.
- Peckett AJ, Wright DC, Riddell MC. 2011. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism. 60:1500–1510.
- Pratt SE, Geor RJ, Mccutcheon LJ. 2006. Effects of dietary energy source and physical conditioning on insulin sensitivity and glucose tolerance in standardbred horses. Equine Vet J Suppl. 38:579–584.
- Radin MJ, Sharkey LC, Holycross BJ. 2009. Adipokines: a review of biological and analytical principles and an update in dogs, cats, and horses. Vet Clin Pathol. 38:136–156.
- Ralston SL. 2002. Insulin and glucose regulation. Vet Clin N Am-Equine. 18:295–304.
- Raymond JG. 2008. Metabolic predispositions to laminitis in horses and ponies: obesity, insulin resistance and metabolic syndromes. J Equine Vet Sci. 12:753–759.
- Rendle DI, Rutledge F, Hughes KJ, Heller J, Durham AE. 2013. Effects of metformin hydrochloride on blood glucose and insulin responses to oral dextrose in horses. Equine Vet J. 45:751–754.
- Rojek A, Niedziela M. 2009. Receptor insuliny i jego związek z różnymi formami insulinooporności. Postępy Biol Kom. 4:617–648.
- Schmidt SL, Hickey MS. 2009. Regulation of insulin action by diet and exercise. J Equine Vet Sci. 29:274–284.
- Sillence M, Asplin K, Pollitt C, Mcgowan C. 2007. What causes equine laminitis? The role of impaired glucose uptake. Kingston: Rural Industries Research and Development Corporation.
- Stewart-Hunt L, Geor RJ, Mccutcheon LJ. 2006. Effects of short-term training on insulin sensitivity and skeletal muscle glucose metabolism in standardbred horses. Equine Vet J Suppl. 38:226–232.
- Summers SA. 2006. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 45:42–72.
- Tadros EM, Frank N. 2013. Endocrine disorders and laminitis. Equine Vet Educ. 25:152–162.
- Thatcher C, Pleasant RS, Geor RJ, Elvinger K, Negrin K, Franklin J, Gay L, Were S. 2007. Prevalence of obesity in mature horses: An equine body condition study. The American Academy of Veterinary Nutrition, 7th Annual Clinical Nutrition and Research Symposium, Seattle, WA. 6.
- Tinworth KD, Harris PA, Sillence MN, Noble GK. 2010. Potential treatments for insulin resistance in the horse: a comparative multi-species review. Vet J. 186:282–291.
- Treiber KH, Kronfeld DS, Geor RJ. 2006. Insulin resistance in equids: possible role in laminitis. J Nutr. 136:2094–2098.
- Tsatsoulis A, Mantzaris MD, Bellou S, Andrikoula M. 2013. Insulin resistance: an adaptive mechanism becomes maladaptive in the current environment – an evolutionary perspective. Metabolism. 62:622–633.
- Tymoczko JL, BERG JM, Stryer L. 2011. Biochemistry. A short course. 2nd ed. New York, NY: W. H. Freeman.
- USDA. 2000. Lameness and Laminitis in US Horses. USDA: APHIS: VS, CEAH, National Animal Health. Fort Collins, CO: Monitoring System.
- Venugopal CS, Eades S, Holmes EP, Beadle RE. 2011. Insulin resistance in equine digital vessel rings: an in vitro model to study vascular dysfunction in equine laminitis. Equine Vet J. 43:744–749.
- Vick MM, Adams AA, Murphy BA, Sessions DA, Horohov DW, Cook RF, Fitzgerald BP. 2007. Relationships among inflammatory cytokines, obesity, and insulin sensitivity in the horse. J Anim Sci. 85:1144–1155.
- Weyenberg S Van, Buyse J, Janssens GPJ. 2009. Increased plasma leptin through L-carnitine supplementation is associated with an enhanced glucose tolerance in healthy ponies. J Anim Physiol Anim Nutr. 93:203–208.
- Wylie CE, Collins SN. 2010. Equine insulin resistance: The quest for sensitivity. Vet J. 186:275–276.
- Wyse CA, Mcnie KA, Tannahill VJ, Murray JK, Love S. 2008. Prevalence of obesity in riding horses in Scotland. Veterinary Record Vet Rec. 162:590–591.
- Ye J. 2013. Mechanisms of insulin resistance in obesity. Front Med. 7:14–24.