
ABSTRACT
Objectives: To demonstrate the incidence, characteristics, treatment and outcomes of patients with therapy-related myelodysplastic syndromes and therapy-related acute myeloid leukaemia (t-MDS/AML) in a tertiary referral centre.
Methods: Patients meeting the diagnostic criteria for t-MDS/AML from 2003 to 2014 were reviewed to analyse their diagnostic features, details of antecedent disorder and treatment, approach to management and survival.
Results: 39 patients who developed t-MDS/AML were identified with incidence of 8.7%. Median age and gender distribution were similar to de novo MDS but t-MDS/AML patients had greater degree of cytopenia and adverse karyotypes. Time to development of t-MDS/AML was shortest for patients with antecedent haematological malignancy compared to solid tumours and autoimmune disorders (46, 85 and 109 months). Patients with prior acute leukaemia had the shortest latency and poor overall survival. Treatment options included best supportive care (56%), Azacitidine (31%) or intensive chemotherapy/allogeneic transplant (13%). Median OS of all patients was 14 months. Survival declined markedly after two years and 5-year OS was 13.8%. Longer survival was associated with blast count <5% at diagnosis, previous haematological disorder, lower risk IPSS-R and a normal karyotype. Four out of five patients who received intensive therapy/transplant remain alive with median OS of 14 months. Median OS of Azacitidine-treated group was 11 months.
Discussion: t-MDS/AML patients showed unique characteristics which influenced their treatment and outcomes. IPSS-R may be useful in risk-adapted treatment approaches and can predict outcomes. Survival remains poor but improved outcomes were seen with allogeneic transplantation. Azacitidine may be effective in patients unfit for intensive therapies.
Introduction
Therapy-related myeloid neoplasms (t-MN) are defined by the WHO 2008 Classification as therapy-related acute myeloid leukaemia (t-AML), therapy-related myelodysplastic syndromes (t-MDS) and therapy-related myelodysplastic syndrome/myeloproliferative neoplasms (t-MDS/MPN) occurring as a late complication of cytotoxic chemotherapy and/or radiation therapy administered for a prior neoplastic or non-neoplastic disorder. The incidence is 10–20% of all MDS, AML and MDS/MPN and 5–20% of patients have had cytotoxic therapy for a non-neoplastic disorder [Citation1]. The incidence of t-MN is rising as a result of an increasing number of cancer survivors at risk and changes in treatment regimens including the increasing use of cytotoxic agents for non-neoplastic disorders [Citation2].
This distinct clinical entity arises as a direct consequence of mutational events induced by cytotoxic therapy or the selection of a myeloid clone with increased risk of mutational events [Citation3]. A causal relationship is implied but the exact mechanisms remain to be proven. The University of Chicago series of 306 patients with t-MN published in 2008 highlighted the importance of this unique but heterogeneous disorder, describing the sub-types, risk factors, genetic mechanisms and cooperating mutations in its development, prognostic indicators and suggested therapeutic pathways [Citation4]. Overall, the prognosis of t-MDS is poor due to rapid progression to AML and relative resistance to conventional therapies [Citation3]. Treatment of t-MN with conventional therapy is associated with a uniformly poor prognosis with a median survival of 8.6 months for t-MDS and 6.9 months for t-AML. Current research is focused on elucidating the mechanisms of disease and developing risk prediction and risk reduction strategies [Citation5].
The aim of this study was to establish the incidence of t-MN in a tertiary referral Haematology-Oncology centre, to identify clinical characteristics and prognostic factors and to assess survival with currently available treatment options.
Methods
We reviewed our single centre database of 446 patients with MDS (n = 294) or AML (n = 152) diagnosed between January 2003 and July 2014 and identified 39 patients with a diagnosis of t-MN. This retrospective analysis involved collecting clinical, pathological and cytogenetic data at diagnosis and information regarding treatment and outcomes from the clinical and laboratory records. Anaemia is defined as haemoglobin concentration of less than 11.5 g/dL for females and 13.0 g/dL for males, neutropenia as absolute neutrophil count less than 1.5 × 109/L and thrombocytopenia as platelet count less than 150 × 109/L. Latency period was calculated as time in months from the date of primary diagnosis of cancer to the date of diagnosis of t-MDS/AML. Overall survival was calculated from the date of diagnosis of t-MN to the date of death or date of last follow-up in months. Survival was censored for one patient who was lost to follow-up and for patients who remain alive. Response to treatment was assessed according to the 2006 International Working Group (IWG) response criteria for myelodysplasia [Citation6]. Demographic characteristics of patients were described using basic statistical analysis. Overall survivals were calculated using the Kaplan–Meier method and the difference between the survival curves by log-rank analysis. Stepwise (forwards and backwards) Cox proportional hazards regression was used to determine a parsimonious model for predicting survival.
Results
Of 446 patients with MDS and AML diagnosed in our institution during the study period, we identified 39 patients (8.7%) with t-MN. The incidence of t-MDS was 11.6% (34 of 294 MDS patients) and for t-AML was 2.6% (4 of 152 AML patients). One patient had t-MDS/MPN. The median age at diagnosis was 72 years (range 37–89); 10 patients (26%) were under the age of 65. Male to female ratio was 1.3:1. Sub-classification of t-MN was described according to the WHO 2008 classification and showed 24 patients (62%) had RCMD or RA, 4(10%) had RAEB-1, 7(18%) had RAEB-2 and 4(10%) had AML.
Classification of patients according to the antecedent disorder showed 15 patients (38.4%) had prior solid tumours, 24 patients (61.5%) had haematological malignancy and 4 patients (10.3%) had autoimmune diseases. Four patients had more than one primary disorder ().
Table 1. Details of primary disorders for 39 patients with t-MN.
Twenty seven patients (69%) previously had received chemotherapy alone and five patients (13%) had radiotherapy alone (). Seven patients (18%) received combination treatment with chemotherapy and radiotherapy. 65% of the patients who had chemotherapy had received more than one cytotoxic agent. Of the chemotherapy group, 22 patients (56%) received alkylating agents and 17(44%) topoisomerase II inhibitors. Three patients had autologous stem cell transplant for myeloma following high dose Melphalan. Three patients with autoimmune disorders had received either Azathioprine or Methotrexate.
Table 2. Prior exposure to cytotoxic agents and radiotherapy for 39 patients with t-MN.
The median time to development of t-MN (latency) following the primary exposure to cytotoxic agents and/or radiation was 60 months. Latency periods for patients who received chemotherapy alone compared to those who received radiotherapy alone was 56 vs. 13 months, p = 0.45 (). Seven patients who received combined chemotherapy and radiation had a median latency period of 84 months. Latency periods for patients who had prior haematological malignancy compared to solid tumours were 46 vs. 85 months, p = 0.27. Time to development of t-MN was significantly shorter in patients who received topoisomerase II inhibitors than those who were treated with Alkylating agents (14 vs. 67 months, p = 0.007). Patients who received cytotoxic agents for autoimmune diseases had the longest latency period of 109 months.
Table 3. Summary of latency, diagnostic features and survival of 39 patients with t-MN.
The majority of the patients (68%) had more than one cytopenia at presentation and pancytopenia was observed in 15%. One-third of the patients presented with anaemia only. At presentation the median haemoglobin was 9.7 g/dL (range 7.4–15), the median absolute neutrophil count was 2.1 × 109/L (range 0.1–16.7), and the median platelet count was 106 × 109/L (range <10–631). Median marrow blast count at diagnosis was 2% and the majority (62%) of the patients had marrow blasts <5%.
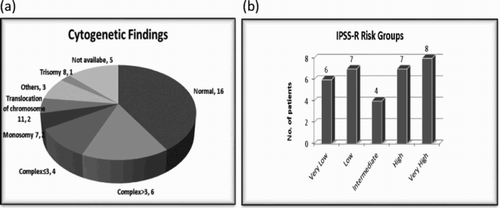
Of 34 patients for whom cytogenetic results were available, 14 patients (41%) had a normal karyotype (). Abnormal cytogenetic findings as previously described in association with t-MN were observed including monosomy 7 (n = 2), translocation involving chromosome 11 (n = 2), translocation involving chromosome 3 (n = 1) and complex ≥3 abnormalities (n = 8) [Citation2]. Twenty patients (59%) had intermediate, poor or very poor risk cytogenetics according to IPSS-R risk stratification. No patients with very good risk cytogenetics were identified ((a)). Although not formally validated in t-MN, IPSS-R prognostic scores for 32 patients were calculated from the above diagnostic information. Five patients whose cytogenetic results were not available and two patients with t-AML who had more than 30% blasts in the marrow were excluded. A range of IPSS-R categories were identified among our patients and the majority (n = 15, 47%) belonged to the high or higher risk groups ((b)).
Figure 1. Cytogenetic findings and IPSS-R risk groups of 39 patients with t-MN. (a) Patients with t-MN based on cytogenetic findings. (b) Patients with t-MN according to IPSS-R risk groups.
Five patients received intensive chemotherapy for t-MDS/AML, of whom two underwent allogeneic stem cell transplantation and remain alive at 74 and 14 months, respectively. Most of the patients (n = 22) received best supportive care with erythropoietin stimulating agents (ESA), granulocyte colony stimulating factor (G-CSF) and transfusion support. These patients were noted to be of older age (median 73 years), had poor performance status and 72% had lower risk IPSS-R.
Twelve patients received Azacitidine treatment. The patients had a median age of 71 years, had better performance status than supportive care group and had higher risk cytogenetics and IPSS/IPSS-R. They received a median of four cycles of Azacitidine (range 1–57 cycles). The overall response rate was 25% (partial response, haematological improvement and stable disease according to the IWG2006 criteria). No complete remissions were observed. Median overall survival was 11 months (range 5–57 months). Two patients remain alive on on-going therapy, 1 of whom has been on Azacitidine for 57 months. For patients who received intensive chemotherapy/stem cell transplantation, the median survival was 14 months (range 5–96 months and four out of five patients remain alive).
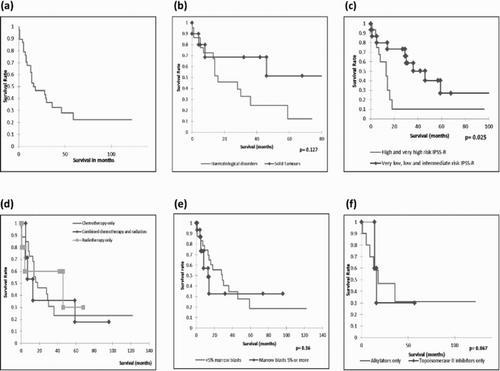
The median overall survival observed in our cohort was 14 months (range <1–122 months). Five-year overall survival was 13.8%. The survival over time showed a significant decrease in survival after 2 years ((a)). Median overall survival in t-MN group was significantly lower than that of other MDS patients in our institution (14 vs. 30 months).
Figure 2. Survival analysis of 39 patients with t-MN. (a) Overall survival of 39 patients with t-MN with time. (b) Comparison of overall survival between solid tumour and haematological malignancy groups. (c) Overall survival between lower and higher risk IPSS-R groups. (d) Overall survival and primary therapy. (e) Overall survival between higher and lower marrow blasts. (f) Overall survival between alkylating agents and topoisomerase II inhibitor-treated groups.
Three of our patients had previously undergone autologous stem cell transplant (ASCT) following high dose Melphalan for myeloma. Of these, two patients had normal karyotype and one had a complex karyotype involving both Del(5q) and Del(17p). Two patients died at 5 and 30 months, respectively, following the diagnosis of t-MN. One patient received intensive chemotherapy and allogeneic stem cell transplantation and remains alive 7 years later.
Patients with a previous history of solid tumour had a trend towards longer survival compared to those with prior haematological malignancy (37 vs. 14 months, p = 0.127) ((b)), whereas patients with prior autoimmune disease had a median survival of 24 months. Median survival based on prior treatment with radiotherapy alone, chemotherapy alone and combined chemotherapy and radiation were 42, 14 and 7 months, respectively ((d)).
Patients with a normal karyotype had a longer median survival compared to those who had cytogenetic abnormalities (31 vs. 13 months, OR = 0.28 [0.11, 0.74], p = 0.01). Patients with higher risk IPSS-R also had lower survival than those with lower risk IPSS-R (10.5 vs. 31 months, OR = 0.35 [0.14, 0.88], p = 0.025) ((c)). Patients with a high marrow blast count (>5%) at diagnosis had a median overall survival of 8 months compared to 23 months in those with blast count of <5%, OR = 0.67 [0.29,1.58], p = 0.36) ((e)). In univariate analysis, normal karyotype and lower risk IPSS-R were associated with a survival advantage but high marrow blasts (>5%) at diagnosis had no effect on survival rate.
Multivariate analysis for factors predicting survival using stepwise Cox proportional hazards regression identified karyotype and prior malignancy type as statistically significant predictors for outcomes as abnormal karyotype having OR = 0.26 [0.09, 0.75], p = 0.013 and prior haematological malignancy with OR = 0.48 [0.16, 1.39], p = 0.18. Multivariate analysis using stepwise cox proportional hazards regression with variables such as IPSS-R, karyotype, marrow blast at presentation, primary tumour type and types of prior therapy showed that karyotype is the overall best predictor of survival, with prior tumour type a somewhat helpful addition if that information is available.
Discussion
t-MN is a frequently devastating outcome following treatment of prior malignancy. This study confirms data presented in previously published literature but with some significant features. The incidence and presentation of t-MN in our institution is similar to other studies. A higher proportion of younger patients were observed in t-MN group. The development of t-MN is closely influenced by the nature of preceding treatment [Citation3]. Patients who developed t-MN following treatment for solid tumour had longer latency period compared to those who had haematological disorders. There is a significantly longer median latency period in patients with non-neoplastic disorders.
Carcinoma of the breast and prostate were the most common solid tumours among our patients whereas chronic lymphoproliferative disorders were the commonest haematological disorder followed by myeloma and acute leukaemia. Latency period was considerably longer (up to 10 years) for some patients in the radiotherapy group compared to chemotherapy. We also included patients who received newer agents such as Thalidomide, Lenalidomide and Bortezomib. Although these agents are not widely recognized to be associated with t-MN, and are not classical ‘cytotoxic’ agents, the emergence of cases of t-MN in this group is of interest. However, all but one of these patients also received alkylating agents ± ASCT.
Patients with t-MN had similar degree of cytopenias but isolated neutropenia or thrombocytopenia at presentation was not seen in our cohort unlike those with de novo MDS. Most patients did not have an elevated marrow blast count at presentation. More than half of the patients had clonal chromosomal abnormalities at diagnosis and a higher proportion of patients with higher risk MDS cytogenetics were observed compared to de novo MDS group. It is notable, however, that a significant number of patients had normal karyotypes.
This group of t-MN patients were heterogeneous with regards to the IPSS-R. Patients with higher risk IPSS-R had a shorter median survival. Although prognostic scoring systems such as IPSS and IPSS-R have not been validated for this category of MDS patients, IPSS-R was able to identify two distinct groups with survival differences. This could provide a useful tool to develop a risk-adapted approach to management as described in a recent study by Nomdedeu in 2014 [Citation7].
This study also demonstrated that the overall survival of patients with t-MN who received supportive care was similar to those with de novo MDS group in our institution (28 vs. 30 months). There is some evidence of improvement in survival with current therapeutic options. However, 5-year survival remains poor. Of 12 patients who were treated with Azacitidine, one patient remains alive on continued therapy at 57 months. One patient with t-AML had intensive chemotherapy followed by Azacitidine due to persistent dysplastic changes in the marrow. If this patient is excluded from the Azacitidine treatment group, the median overall survival was found to be similar between intensive chemotherapy/transplant group and Azacitidine group. Almost all of the patients in Azacitidine treatment group had high-risk IPSS-R and high blast counts at diagnosis and were likely to have an adverse prognosis. Two studies from GFM group and Spanish group described an overall response rate of 39 and 42%, respectively, in t-MN patients treated with Azacitidine [Citation8,Citation9]. Our data also indicates that Azacitidine is an effective treatment option in patients who are not fit for intensive therapy.
Conclusion
Therapy-related MDS/AML is a distinct but heterogeneous group of patients who share some characteristics with de novo MDS patients. A higher proportion of adverse cytogenetics in this group may account for lower survival. Cytotoxic chemotherapy and radiation are implied as causative agents but the exact mechanism of development of t-MN and other predisposing causes remain unknown. Novel therapeutic agents may also be implicated but larger studies and longer follow-up are required to confirm this. There is a trend towards longer latency period for solid tumours compared to haematological malignancies or those treated with alkylating agents compared to topoisomerase II inhibitors. Improved survival was also associated with previous solid tumours, normal karyotype and lower marrow blast count at presentation. IPSS-R can identify patients with good outcomes and may be useful in a risk-adapted approach to management. Survival for patients with t-MN remains poor but may be improved for some patients suitable for treatment with Azacitidine or intensive chemotherapy. Azacitidine may be effective in patients with t-MN not fit for intensive therapies. Transplantation may result in long-term survival in selected patients.
Acknowledgement
The authors thank Eoin O’Connell for his assistance with statistical analysis for this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Su W. Maung completed higher specialist training in haematology in 2015 in the Royal College of Physicians of Ireland. She undertook clinical research on myelodysplastic syndromes from 2014 to 2016 and has recently submitted her thesis for MD (clinical medicine) to Trinity College, Dublin. She is now working as a consultant haematologist in the Mater Hospital, Dublin, Ireland. This paper is based on some of her works on MDS research project.
Cathie Burke is the clinical research nurse for MDS in Tallaght Hospital.
Jennifer Hayde is the senior clinical pharmacist in Tallaght Hospital.
Janice Walshe is a medical oncologist in Tallaght Hospital.
Ray McDermott is a medical oncologist in Tallaght Hospital.
Ronan Desmond is a consultant haematologist in Tallaght Hospital.
Johnny McHugh is a consultant haematologist in Tallaght Hospital.
Helen Enright is Dr Maung's mentor and supervisor. She is the clinical professor in Haematology of Trinity College, Dublin and also a consultant haematologist and the head of haematology department in Tallaght Hospital, Dublin. Professor Enright has a special interest in MDS and active member of MDS working group nationally and internationally.
ORCID
Su W. Maung http://orcid.org/0000-0002-2679-8896
Ray McDermott http://orcid.org/0000-0002-8952-4315
References
- Vardiman JW, Arber DA, Brunning RD, et al. Therapy-related myeloid neoplasms. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: IARC Press; 2008, p. 127–129.
- Larson RA. Cytogenetics, not just pervious therapy determines the course of therapy-related myeloid neoplasms. J Clin Oncol. 2012;30:2300–2302. doi: 10.1200/JCO.2011.41.1215
- Larson RA. Therapy-related myeloid neoplasms. Haematologica. 2009;94:454–459. doi: 10.3324/haematol.2008.005157
- Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52. doi: 10.1182/blood-2002-11-3343
- Bhatia S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. 2013;40(6):666–675. doi: 10.1053/j.seminoncol.2013.09.013
- Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419–425. doi: 10.1182/blood-2005-10-4149
- Nomdedeu M, Calvo X, Pereira A, et al. Clinical features and prognostic assessment in 233 patients with therapy-related myelodysplastic syndromes: The IPSS-r is a powerful predictor of outcome. 2014. American Society of Haematology (ASH) Meeting. Abstract 4636.
- Bally C, Fenaux P, Ades L. Azacitidine in the treatment of therapy-related MDS/AML: a report on 54 patients by GFM. Leukaemia Res. 2013;37(6):637–640. doi: 10.1016/j.leukres.2013.02.014
- Fianchi L, Criscuolo M, Lunghi M, et al. Outcome of therapy related myeloid neoplasms treated with Azacitidine. J Haematol Oncol. 2012;5:44. doi: 10.1186/1756-8722-5-44