
ABSTRACT
Background: Infant mortality due to sickle cell disease in sub-Saharan Africa is high, necessitating a better understanding of the modulating factors of the disease in this region.
Methods: We assessed the hereditary persistence of foetal haemoglobin and α-thalassemia. We diagnosed 787 subjects, with or without sickle cell trait, by capillary electrophoresis in the Medical Diagnostic Laboratory of the CIRMF (Franceville, Gabon).
Results: Heterocellular and pancellular forms of hereditary persistence of foetal haemoglobin occurred at low rates of 10.9 and 2.3%, respectively. The distribution of HbS levels in individuals with sickle cell trait was trimodal, showing a high percentage (52.4%) of heterozygous subjects with α-thalassemia. The distribution of HbA2 levels was bimodal in individuals without sickle cell trait, estimated to be comprised of 12 and 15% of α and β-thalassemic heterozygous subjects, respectively.
Conclusions: In sub-Saharan Africa, α-thalassemia is a far more prevalent modulating factor than hereditary persistence of foetal haemoglobin. Our study highlights the need for further investigation of thalassemia, haemoglobinopathies that are neglected in sub-Saharan Africa.
Introduction
Sickle cell disease (SCD) is caused by homozygosity for the glu6val mutation in the β-globin gene, resulting in the exclusive presence of abnormal sickle haemoglobin (HbS, an α2βS2 tetramer) at high concentrations in red blood cells. When normal adult haemoglobin (HbA, an α2β2 tetramer) is also present, HbS is found at levels of 20–45% [Citation1], defining individuals with sickle cell trait (SCT).
SCD is recognized as a public health problem by the United Nation Organisation as early-life mortality among children born in sub-Saharan Africa with SCD is 50–90% [Citation2]. The attenuating factors of the disease, and mortality, are mostly the hereditary persistence of foetal haemoglobin (HPFH) and α-thalassemia.
Foetal haemoglobin (HbF, an α2γ2 tetramer) can inhibit the deoxygenation-induced polymerization of HbS that causes the clinical effects of SCD. HbF levels >1% are defined as high and this condition is called hereditary persistence of foetal haemoglobin (HPFH). It is associated with a lower rate of acute painful episodes, less osteonecrosis, less frequent acute chest syndromes, and reduced severity of SCD. In the neighbouring country of Cameroon, a study has confirmed the association of SNPs in BCL11A and HBS1L-MYB and high foetal haemoglobin in Cameroonian SCD patients with hospitalization rates [Citation3].
Concerning α-thalassemia, one of the first reports to use gene mapping showed that SCD subjects who were α-thalassemic, due to the deletion of one or two α-globin genes, were less anaemic than those who were non-thalassemic [Citation4]. The co-inheritance of α-thalassemia and SCD is also associated with better haematological indices and lower consultation rates in Cameroonian patients [Citation5].
The definitive diagnosis of α-thalassemia requires DNA analysis, but it can be indirectly detected by examining blood indicators, such as HbA2, HbS, and iron levels. The measurement of HbA2 (an α2δ2 tetramer) levels is a useful diagnostic aid as it normally represents less than 3% of total haemoglobin, but its concentration varies in the thalassemia syndromes and several acquired diseases [Citation6].
Few studies of the factors that modulate clinical forms of SCD (α-thalassemia and HPFH) have been performed in sub-Saharan Africa. Our study focused on the level of foetal haemoglobin in subjects with or without SCT and an indirect evaluation of α-thalassemia in the same population. Our specific objectives were to determine (1) the HbF and HbA2 levels in all subjects, (2) the prevalence of HPHF forms and HbA2 ranges in all subjects, and (3) the distribution of HbS levels in subjects with SCT and of HbA2 levels in individuals without SCT.
Materials and methods
We analysed 787 routine EDTA-anticoagulant blood samples from individuals over 2 years old, received by the Medical Diagnostic Laboratory, URAM unit, CIRMF (International Centre for Medical Research) from 2011 to 2013.
Among all individuals received in the Medical Laboratory from 2011 to 2013, patients with SCD, haemoglobin C disorders, and those aged less than 24 months were excluded from the study. Children older than 24 months normally present the adult distribution of haemoglobin molecules, as the second haemoglobin switch has been completed by this age.
Capillary zone electrophoresis was performed using the Sebia Capillarys system. Electrophoresis was performed in alkaline buffer, pH 9.4, and the haemoglobins evaluated at 415 nm. The presence of HbA was required for the appearance of the zone limitations that permitted detection of structural variants. If HbA was not present in the sample, the test was repeated after premixing with a normal control sample. Quantitative results were recorded from the original sample alone, and the mixture was used for qualitative identification only.
Large deletions within the β-globin genes and some single nucleotide polymorphisms in the promoter region of the γ-globin genes result with HbF level above 5% in HPFH [Citation7]. The latter characterizes the pancellular form of HPFH, HbF being uniformly distributed among all red blood cells. In the more common form of HPFH, called heterocellular HPFH, HbF levels only increase slightly 1–4% and its genetic determinants are complex, heterogeneous and globally linked to the β-globin gene cluster [Citation7].
Indirect analysis of α-thalassemia frequencies is performed using the percentages of HbS distribution levels in subjects with sickle cell trait. Trimodality in the percentages of β-chain variants (like HbS) in heterozygous individuals with sickle cell anaemia is the effect of 2, 3 or 4 active α-globin genes (2 or 3 active genes meaning an α-thalassemia trait) [Citation8].
Statistical analysis was performed using the analysis of variance (ANOVA) and Kruskal–Wallis rank sum test with a p-value ≤0.05 considered to be significant.
Results
Hbf levels of subjects from a semi-rural area
The subjects of our study had an average HbF frequency of 0.63%, representing normal foetal haemoglobin levels. Comparisons of the HbF levels between sexes, adults and children, pregnant and non-pregnant women, and AA and AS individuals are shown in the . Individuals who were 15 years or older had a significantly higher HbF level than those of less than 15 years (p = 0.002). The difference between the HbF level in AS (0.72%) and AA individuals (0.59%) was not significant.
Table 1. Comparison of mean levels of HbF and HbA2 in different groups of Gabonese subjects living in a semi-rural area (Franceville)
We found heterocellular and pancellular forms of HPFH at a frequency of 10.9% (86/787) and 2.3% (18/787), respectively, in our study population. Being an adult was significantly associated with both forms of HPFH (p < 0.001), as were SCT (p = 0.02) and being female (p = 0.047) ().
Table 2. Distribution of HPHF forms and HbA2 ranges in different groups of Gabonese subjects living in a semi-rural area (Franceville)
Hba2 levels in subjects from a semi-rural area
The average HbA2 level in our study population was 3.04%. High levels of HbA2 were significantly associated with being male or a non-pregnant women (). SCT was strongly associated with high HbA2 mean levels (p < 0.001; ).
In our study population, 12.83% (101/787) and 13.47% (106/787) of subjects had HbA2 levels ≤2.5% and >3.5%, respectively (). The HbA2 levels were significantly different between men and women, pregnant and non-pregnant women, and AA and AS individuals ().
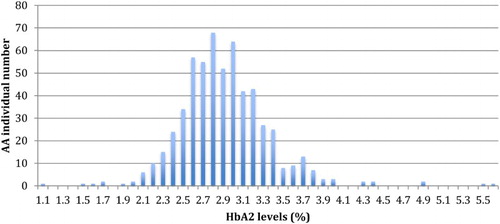
The analyses of HbA2 levels in our study population showed a bimodal distribution in individuals without SCT (). The first group represents 92.6% of the AA subjects with HbA2 levels between 1.1 and 3.5%. The second group includes subjects with HbA2 levels above 3.5%, comprising 7.4% of the AA individuals.
Figure 1. Bimodal distribution of HbA2 levels in subjects without sickle cell trait.
Hbs levels in subjects with SCT from a semi-rural area
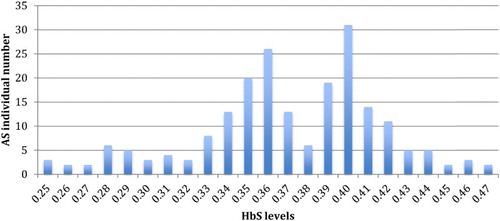
The distribution of HbS levels in individuals with SCT was trimodal (). The first group, with HbS levels between 25 and 31% (mean of 28.3%), represents individuals with two active α-globin genes (25/206 = 12.1%). The second group, with HbS levels between 32 and 37% (mean of 35.2%), consists of subjects with three active α-globin genes (83/206 = 40.3%). The last group, with HbS levels between 38 and 47% (mean of 41.3%), contained no α-thalassemic subjects (98/206 = 47.6%).
Figure 2. Trimodal distribution of HbS levels in subjects with sickle cell trait.
Discussion
SCD is the principal haemoglobinopathy found in Central Africa, whereas the frequency of haemoglobinopathies C, D, and E, as well as thalassemia remains low. In Gabon, the prevalence of SCT is 21.1% among people aged over 15 years, with the prevalence by region between 12.8 and 28.2% [Citation9]. We directly analysed the prevalence of HPFH and indirectly the frequency of α-thalassemia in subjects with or without SCT.
Foetal haemoglobin levels and the prevalence of HPFH
The level of HbF in our study population was well below 1%, explained by a low frequency of the genetic determinants responsible for most of the increase in HbF levels in our population. The major consequence of such a low frequency of HbF, especially in individuals with SCT, would be a large proportion of unfavourable profiles in case of the birth of affected children. The low percentage (2.3%) of pancellular HPFH in our study population would also have the same consequences, as this form of HPFH can moderate the clinical profile of SCD.
We clearly observed an association between relatively high HbF levels and being more than 15 years old, consistent with the selection of individuals with HPFH. This raises the question of whether there is a beneficial effect of high HbF levels in other pathologies, such as malaria. Indeed, growth of the parasite was significantly delayed in vitro in cells containing foetal haemoglobin [Citation10].
No association was found in our study between HbF and sex or pregnancy, whereas other studies have shown the opposite [Citation11,Citation12]. Our study showed higher HbF levels in women, but the difference was not significant, similar to a study by Chang et al. [Citation11] Multivariate analysis showed that risk factors, such as age and sex, have a generally weak effect relative to genetic determinants. The HbF level in non-pregnant women in our study and in those in a Japanese study was similar, despite genetic differences between these two populations, confirming a standard level of 0.7% among non-pregnant women [Citation12]. The pregnant women in our study were probably mostly in the 3rd trimester, based on the Japanese study. The lack of association in our study may be due to the small sample size of pregnant women, as the mean values were similar to those reported in the Japanese study [Citation12].
There was no association between HbF levels and SCT, whereas there was a significant association between SCT and HPFH. High HbF levels have been reported in homozygous SCD subjects as a result of medullar hyperplasia [Citation13], whereas the absence of clinical signs in subjects with SCT explain the absence of such an association.
Hbs and HbA2 levels and thalassemia frequencies
The trimodal distribution of HbS () revealed that 52.4% of individuals with SCT have minor α-thalassemia with 2 or 3 active genes of α-globin. This high rate of α-thalassemia in individuals with SCT implies a higher frequency of α-thalassemia in sickle cell patients. Alpha-thalassemia, poorly studied in Gabon and Central Africa, was molecularly evaluated in one study in Gabon, in the centre of the country, and found at a frequency of 30% [Citation14]. The difference between these two frequencies may result from the presence of α-thalassemic variants other than the 3.7 deletion studied by Lell et al. [Citation14].
Our study is the first in Central Africa to show the trimodal distribution of HbS levels in subjects heterozygous for SCD (an indirect assessment of α-thalassemia frequencies). The observed distribution, especially of HbS classes, is consistent with that determined in other trimodal distributions observed in southern India and Canada [Citation15,Citation16]. Moreover the mean of HbS level observed in each three classes in our study is similar to that found by Huisman in a decisive study (28.3 vs. 28.1% in subjects with two active α-globin genes, 35.2 vs. 35.4% in individuals with three active α-globin genes and 41.3 vs. 41.2% in no α-thalassemic group) [Citation8]. This indirect method is useful as the detection of α-thalassemia in blacks is difficult because of the mild haematological changes and minor alterations in haemoglobin composition that are present [Citation17]. Indeed, the amount of available α-chain, normally in excess, is lower when α-thalassemia coexists with SCT. The positively charged α-chain combines preferentially with the negatively charged normal β-chain, rather than with the positively charged S β-chain, with a consequent reduction in the level of HbS [Citation18,Citation19].
We also evaluated the HbA2 levels in individuals without SCT to analyse α-thalassemia in these individuals. HbA2 levels ≤2.5% is one of the indicators of α-thalassemia according to several studies [Citation20,Citation21]. This rate limit is not applicable to individuals with SCT, as we showed that AS individuals had a high level of HbA2. Indeed, the α-chain is likely to combine preferentially with βA rather than with δ-chains, but when the amount of α-chain is rate limiting, combination with the δ-chain is favoured over combination with βS suggesting that the affinity of non α-chains for the α-chain is βA > δ > βS [Citation18]. The frequency of α-thalassemia would thus be estimated to be 16.7% among the individuals without SCT in our study population. However, subjects with iron deficiency must also be considered because HbA2 levels are also low in these subjects.
The bimodal distribution of HbA2 levels, observed for the first time in individuals without SCT, allowed us to identify a class of AA subjects whose HbA2 levels are above 3.5%. The distribution found in this group is consistent with that observed in the study of Verhovsek et al. [Citation22]. An HbA2 level >3.5% has been used in other studies for the diagnosis of subjects heterozygous for β-thalassemia [Citation23]. Few studies have diagnosed β-thalassemia subjects in Central Africa [Citation24], but this class of 7.4% of AA individuals is likely to contain mostly subjects heterozygous for β-thalassemia, although there are also probably some subjects with iron deficiency [Citation22]. Indeed, a β0-thalassemic variant has been proposed to have an African origin [Citation25].
Conclusion
Our study evaluated the frequencies of two modulating factors for SCD in Gabonese subjects with or without SCT: HPFH and α-thalassemia. We observed a low frequency of HPFH in a Gabonese semi-rural area. In contrast, we found a high prevalence of α-thalassemia, especially in individuals with SCT (birth of affected children) as a result of the trimodal distribution of the HbS percentages.
Studies on thalassemia have been rarely conducted in Central Africa, but should be performed in the context of SCD, whether the focus is on β-thalassemia, suspected in our study, or implicitly deduced α-thalassemia.
Acknowledgements
The authors thank Mr Patricien Lewobo-Mombo and Dr Jean Moutélé (CHRAB, Gabon), Dr Cyrille Bisseye and Professor Bertrand Mbatchi (LABMC, Gabon), Mr Damehan Tchelougou (LABIOGENE, Burkina Faso) and Dr Richard Onanga (URAM-CIRMF, Gabon).
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Landry-Erik Mombo, MSc, PhD, is Assistant Professor at the Department of Biology of the Faculty of Sciences (USTM, Franceville, Gabon). He is a specialist for genetic factors in tropical diseases and for hemoglobinopathies.
Gaël Mabioko-Mbembo, MSc, was a student of Master of Molecular Pathophysiology of the Faculty of Sciences (USTM, Franceville, Gabon). His subject of work at CIRMF was sickle-cell anemia.
Roland-Fabrice Kassa-Kassa, MSc, MD, is the Chief Medical Officer at URAM (CIRMF, Franceville, Gabon). He is an expert on sickle cell anemia.
Emmanuel Ontsitsagui, BSc, is the technician specializing in capillary electrophoresis at URAM (CIRMF, Franceville, Gabon).
Statiana Mboui-Ondo, Pharm D, is the Head of the Medical Analysis Department at URAM (CIRMF, Franceville, Gabon).
Leatitia Nzé-Kamsi, MSc, was a student of Master of Molecular Pathophysiology of the Faculty of Sciences (USTM, Franceville, Gabon). His subject of work at CHRAB, was hemoglobinopathies in pregnant women.
Dieudonné Nkoghé, MD, PhD, is the Director of the Unit URAM (CIRMF, Franceville, Gabon). He is seconded from the Gabonese Ministry of Health. He is an expert of the field of epidemiology.
Jacques Elion, MD, PhD, is a professor of universities (University Paris VII) and physician of the public hospitals of Paris. He is an expert of the field of Hematology and hemoglobinopathies.
Additional information
Funding
References
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med. 2013;3:a011783. doi: 10.1101/cshperspect.a011783
- Grosse SD, Odame I, Atrash HK, et al. Sickle cell disease in Africa: A neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6S4):S398–S405. doi: 10.1016/j.amepre.2011.09.013
- Wonkam A, Ngo Bitoungui VJ, Vorster AA, et al. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLoS One. 2014;9(3):e92506. doi: 10.1371/journal.pone.0092506
- Embury SH, Dozy AM, Miller J, et al. Concurrent sickle-cell anemia and α-thalassemia: effect on severity of anemia. N EngI J Med. 1982;306(5):270–274. doi: 10.1056/NEJM198202043060504
- Rumaney MB, Ngo Bitoungui VJ, Vorster AA, et al. The Co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One. 2014;9(6):e100516. doi: 10.1371/journal.pone.0100516
- Steinberg MH, Adams JG. Hemoglobin A: origin, evolution, and aftermath. Blood. 1991;l78(9):2165–2177.
- Wood WG, Weatherall DJ, Clegg JB. Interaction of heterocellular hereditary persistence of foetal haemoglobin with β thalassaemia and sickle cell anaemia. Nature. 1976;264(5583):247–249. doi: 10.1038/264247a0
- Huisman TH. Trimodality in the percentages of beta chain variants in heterozygotes: the effect of the number of active Hbα structural loci. Hemoglobin. 1977;1(4):349–382. doi: 10.3109/03630267708996895
- Delicat-loembet LM, Elguero E, Arnathau C, et al. Prevalence of the sickle cell trait in Gabon: a nationwide study. Infect Genet Evol. 2014;25:52–56. doi: 10.1016/j.meegid.2014.04.003
- Pasvol G, Weatherall DJ, Wilson RJ, et al. Fetal haemoglobin and malaria. Lancet. 1976;1(7972):1269–1272. doi: 10.1016/S0140-6736(76)91738-4
- Chang YC, Smith KD, Moore RD, et al. An analysis of fetal hemoglobin variation in sickle cell disease: the relative contributions of the X-linked factor, beta-globin haplotypes, alpha-globin gene number, gender, and age. Blood. 1995;85(4):1111–1117.
- Yamada T, Morikawa M, Yamada T, Nishida R, Takeda M, Kawaguchi S, et al. Changes in hemoglobin F levels in pregnant women unaffected by clinical fetomaternal hemorrhage. Clin Chim Acta. 2013;415:124–127. doi: 10.1016/j.cca.2012.10.002
- Milner PF, Leibfarth JD, Ford J, et al. Increased HbF in sickle cell anemia is determined by a factor linked to the beta S gene from one parent. Blood. 1984;63(1):64–72.
- Lell B, May J, Schmidt-Ott RJ, et al. The role of red blood cell polymorphisms in resistance and susceptibility to malaria. Clin Infect Dis. 1999;28(4):794–799. doi: 10.1086/515193
- Brittenham G, Lozoff B, Harris JW, et al. Sickle cell anemia and trait in southern India: further studies. Am J Hematol. 1979;6(2):107–123. doi: 10.1002/ajh.2830060203
- Wong SC, Ali MA, Boyadjian SE. Sickle cell traits in Canada. Trimodal distribution of Hb S as a result of interaction with alpha-thalassaemia gene. Acta Haematol. 1981;65(3):157–163. doi: 10.1159/000207172
- Steinberg MH, Embury SH. Alpha-thalassemia in blacks: genetic and clinical aspects and interactions with the sickle hemoglobin gene. Blood. 1986;68(5):985–990.
- CE H, Conroy M, Jarvis M, et al. Some observations on the measurement of haemoglobin A2 and S percentages by high performance liquid chromatography in the presence and absence of alpha thalassaemia. J Clin Pathol. 2004;57(3):276–280. doi: 10.1136/jcp.2003.008037
- Felice AE, Altay CA, Milner PF, et al. The occurrence and identification of alpha-thalassemia-2 among hemoglobin S heterozygotes. Am J Clin Pathol. 1981;76(1):70–73. doi: 10.1093/ajcp/76.1.70
- Villegas A, González FA, Nieto JM, et al. Haemoglobinopathies that occur with decreased HbA2 levels: a gene mutation set involving the δ gene at a spanish centre. J Clin Pathol. 2017;70(1):75–80. doi: 10.1136/jclinpath-2016-203879
- Giordano PC, Harteveld CL. Chromatographic measurements of hemoglobin A2 in blood samples containing sickle hemoglobin. Ann Clin Lab Sci. 2000;30(4):430–431.
- Verhovsek M, So CC, O’Shea T, et al. Is HbA2 level a reliable diagnostic measurement for β-thalassemia trait in people with iron deficiency? Am J Hematol. 2012;87(1):114–116. doi: 10.1002/ajh.22188
- Ou Z, Li Q, Liu W, et al. Elevated hemoglobin A2 as a marker for β-thalassemia trait in pregnant women. Tohoku J Exp Med. 2011;223(3):223–226. doi: 10.1620/tjem.223.223
- Ranque B, Menet A, Diop IB, et al. Early renal damage in patients with sickle cell disease in sub-saharan Africa: a multinational, prospective, cross-sectional study. Lancet Haematol. 2014;1(2):e64–e73. doi: 10.1016/S2352-3026(14)00007-6
- Gonçalves J, Faustino P, Lavinha J, et al. An African origin for an “American black” beta zero-thalassemia mutation? Am J Hematol. 1994;46(4):373–374. doi: 10.1002/ajh.2830460425