
ABSTRACT
Background: The natural history and its modulation by treatments administered for immune thrombocytopenia (ITP) in the clinical practice remains unknown. In addition, little information is available on the characteristics and management of ITP in Spain.
Methods: We conducted an observational, multicenter, registry in 70 Hematology Services from Spain between 2009 and 2011, which included children from 2 months of age and adults with primary ITP or another ITP diagnosed within the last 6 months (platelet count [PC] < 100 × 109/l). Patients were followed-up at 6 and 12 months.
Results: 484 patients were included (median [Q1, Q3] age 52 [29,74] years, 87.6% adults), 56% women, 10.5% with secondary ITP. Median (Q1, Q3) PC at diagnosis was 12 × 109/l (4, 32); 72% of patients had bleeding symptoms (62% cutaneous bleeding, 29% oral cavity bleeding, 18% epistaxis). 81% of patients with primary ITP received first-line treatment, mainly with corticosteroids (>6 weeks in 59% of cases), either alone (41%) or associated with intravenous immunoglobulin (33%). The response (≥30 × 109/L) to first-line treatment was 92%. A total of 19% of patients received second-line treatment and 6% additional treatments. At 12 months, 74% of primary ITP patients maintained a PC ≥ 100 × 109/L in absence of treatment (10% still had hemorrhagic manifestations).
Conclusions: Characteristics of Spanish ITP patients are comparable to those from other countries. Although a high response rate to first-line treatments is observed, at 1 year, the disease persists in around one quarter of patients. Overall therapeutic management in Spain conforms to current recommendations, except for an excessive duration of corticosteroids therapy.
Introduction
Primary immune thrombocytopenia (primary ITP, formerly known as idiopathic thrombocytopenic purpura) [Citation1] is an autoimmune disorder present in both sexes and in both childhood and adulthood. Although ITP has been a well-known hematologic disorder for more than two centuries, some aspects regarding etiologic factors or clinical behavior still remain unknown. The availability of epidemiological data on primary ITP is very limited and there is no specific data from Spain. Prevalence of ITP has been reported ranging from 4.5 to 10.5 cases per 100,000 persons in adults [Citation2] and 4.6 cases per 100,000 in children [Citation3]. In the adult population, annual incidences between 16 and 27 new patients per million have been reported, predominantly in women aged 30–59 years, with even higher values in patients of both sexes older than 60 years [Citation4,Citation5].
Since there is no specific diagnostic technique, the diagnosis must be made by exclusion of other causes of thrombocytopenia, giving uncertainty about whether it is a primary or a secondary ITP. Secondary ITPs are a group of immune-mediated thrombocytopenias associated to either an underlying disease (such as lupus erythematosus, or infections caused by human immunodeficiency virus (HIV), Helicobacter pylori, etc.) or to drug exposure [Citation1].
ITP is characterized by a low platelet count due to an increased destruction mediated by antiplatelet antibodies and a concomitant submaximal production [Citation6–12]. The main features are the appearance of petechiae, ecchymosis and not-clinically relevant hemorrhages. However, there is a risk of serious bleedings and fatal intracranial hemorrhages, which increases at platelet counts below 5–10 × 109/l [Citation5,Citation13].
Treatment of patients with ITP is primarily aimed at reaching a platelet count high and stable enough to prevent the occurrence of serious bleeding. The optimal treatment of ITP has not been yet established, and currently multiple drugs and other therapeutic approaches as splenectomy are being used, with variable responses. In addition, until the publication of the randomized controlled trials demonstrating the efficacy of thrombopoietin analogues (romiplostim and eltrombopag) [Citation14–16], the efficacy of the most frequently used ITP treatments was unclear, since the majority of data came from non-randomised studies or case series [Citation17–23]. In 1996, the American Society of Hematology published the first ITP practice guidelines [Citation24]. These recommendations on ITP diagnosis and management were updated in 2010 by an international consensus panel [Citation25], which were immediately adapted to Spain, with the aim to unify the criteria across centres [Citation26].
Given the multiple controversies and uncertainties in ITP, the present epidemiological registry study was designed to know the reality of the disease in a large number of Spanish institutions with the aim of establishing, in successive phases of the project, a plan to standardize care practices. The main objectives were: to describe the clinical characteristics and diagnostic procedures in patients with ITP; to describe the therapeutic management of primary ITP in the clinical practice; to describe the response rate to each line of treatment in primary ITP patients; and to compare our results with data from other published ITP cohorts.
Patients and methods
The TIMES study was an epidemiological, observational, prospective, multicenter, registry study conducted in Hematology Services across Spain. The inclusion criteria were: children from 2 months of age and adults; presumptive diagnosis of primary ITP or another ITP diagnosed within the last 6 months; platelet count <100 × 109/l at ITP diagnosis. A presumptive diagnosis of ITP was established when the history, physical examination, complete blood count, and examination of the peripheral blood smear did not suggest other etiologies for the thrombocytopenia. To minimize sample bias, all consecutive patients attending the participating clinics who fulfilled eligibility criteria were invited to participate. The protocol was approved by an independent ethics committee, and all patients gave their written informed consent before enrollment.
The study included a baseline visit at the beginning and two follow-up visits at 6 and 12 months, independently of the clinical evolution of the disease. In case of premature discontinuation for any reason, investigators had to complete the last available information of the patient status. The following data were retrospectively collected from the medical records at each study visit through a web-based system using a secure internet server: clinical history and ITP etiology (only at baseline visit), bleeding symptoms over the previous 6 months of each visit, primary ITP treatment and response. Response criteria were set according to the standardization of Rodeghiero et al. [Citation1]: complete response, platelet count ≥100 × 109/l; partial response, platelet count ≥30 and <100 × 109/l, and at least double the baseline value; no response: platelet count <30 × 109/l or less than twice the baseline. The overall response rate (complete plus partial response) was described together with its 95% confidence interval.
Sample size (500 patients) was calculated to achieve a precision of ±2% for describing an attribute estimate with an expected proportion of 6% in the study population and a 95% confidence interval, assuming that the overall number of prevalent ITP cases in Spain is around 5000 [Citation2,Citation27,Citation28].
Descriptive analyses were provided for each variable at all the study visits. Statistical analyses were performed with the SPSS® statistical package (Chicago, IL, U.S.A.) version 21.0.
Results
Characteristics of patients with immune thrombocytopenia
A total of 484 patients from 70 Spanish hospitals were included in the study between 1 July 2009 and 30 November 2011. Of them, 361 (74.6%) were completely followed-up during 12 months ().
Figure 1. Study flow-chart.
shows the main socio-demographic and clinical characteristics of the study cohort.
Table 1. Characteristics of de novo diagnosed patients with immune thrombocytopenia (ITP) in Spain between 2009 and 2011.
Most patients (87.6%) were adults, with a median age of 52 and 58 years in primary and secondary ITP, respectively, and 55 and 62% were women, respectively. Secondary ITP was diagnosed in 10.5% (n = 51) of patients, with a wide variability in associated disorders. Systemic lupus erythematosus (9/51 cases, 17.6%) and lymphoproliferative syndromes (17.6%) were the most common underlying diseases, followed by infections ().
The most common comorbidity was hypertension, followed by recent infection, cardiovascular disease and diabetes. A total of 5.2% of patients had received drugs possibly related to ITP.
Median platelet count at baseline was below 30 × 109/l in three quarters of primary ITP patients and below 50 ×109/l in three quarters of secondary ITP patients ().
Diagnostic procedures
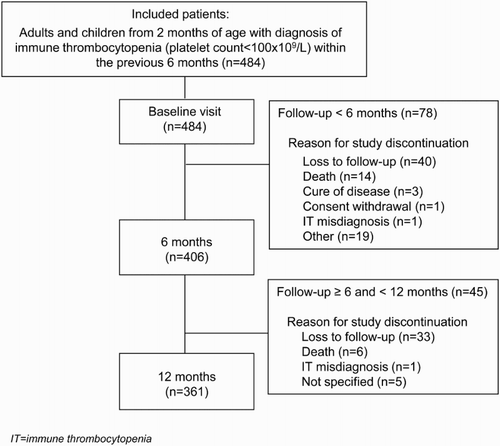
shows the main diagnostic procedures performed in addition to the initial blood count. The most commonly performed tests in both primary and secondary ITP populations were hepatitis B virus (HBV) serology (87 and 92% of patients, respectively), HIV serology (86 and 92%), hepatitis C virus (HCV) serology (83 and 92%) and antinuclear antibodies (77 and 82%).
Figure 2. Utilization and outcome of diagnostic procedures other than platelet count undertaken in the study population to confirm diagnosis of (a) primary (n = 433) and (b) secondary (n = 51) immune thrombocytopenia.
The highest percentage of positive results was observed for antinuclear antibodies in both populations (14% in primary and 37% in secondary ITP patients), followed by Helicobacter pylori in primary ITP patients (13%) and by HBV serology in secondary ITP patients (18%) (). Platelet-associated antibodies test was performed in 21% of primary ITP patients and in 41% of secondary ITP patients.
Clinical manifestations at diagnosis
Clinical manifestations at diagnosis were observed in 72% of patients with primary ITP and 67% of patients with secondary ITP, mainly cutaneous (61 and 63%, respectively) and oral cavity bleedings (29 and 24%), and epistaxis (17 and 24%). Potentially severe bleedings were rare (gastrointestinal bleedings 4 and 14%, intracranial haemorrhage 1 and 2%). Other bleeding symptoms included muscle hematoma (6 and 0%), metrorrhagia (5 and 4%) and hematuria (2 and 6%).
At diagnosis, the mean (maximum) number of episodes for each symptom in patients with primary ITP was: oral cavity bleedings, 3.1 (180); gastrointestinal bleedings, 3.1 (20); cutaneous bleeding, 1.8 (30); epistaxis, 1.5 (5); hematuria, 1.9 (7); muscle hematoma, 1.3 (5); metrorrhagia, 1.4 (2); and intracranial haemorrhage, 1.2 (2). In patients with secondary ITP these figures were: oral cavity bleedings, 1.8 (4); gastrointestinal bleedings, 1.6 (3); cutaneous bleeding, 2.5 (10); epistaxis, 2 (4); hematuria, 1 (1); metrorrhagia, 3 (5); and intracranial haemorrhage, 1 (1).
Therapeutic management of primary ITP
and summarize the main characteristics of therapeutic management in the subgroup of primary ITP patients (n = 433).
Figure 3. Therapeutic management of patients with primary ITP in Spain.
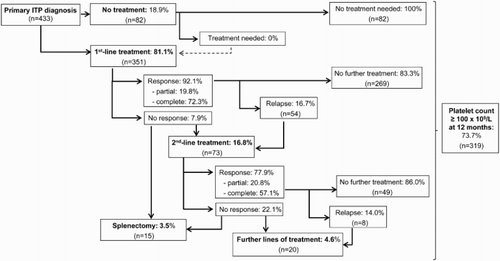
Table 2. Patterns of use of corticosteroids as first-line treatment for primary ITP.
Observation only was preferred in 18.9% of de novo diagnosed patients. None of them required treatment later on ().
The remaining 81.1% of patients received first-line treatment: 41.3% of patients received one drug, 31.6% received two drugs, 6.0% received three drugs and 2.2% received four to five drugs. No splenectomies were performed in the first-line setting.
The most common first-line drugs were corticosteroids as monotherapy (40.6%) or combined with IVIg (32.8%). 5.8% of patients received IVIg. Patients who received these treatments had lower platelet counts at baseline and more bleeding symptoms than patients who were not treated at all ().
Table 3. Characteristics of the patients with primary ITP at diagnosis according to first-line treatment.
Prednisone was the most commonly used corticosteroid (65.1%), followed by methylprednisolone (12.0%) and dexamethasone (6.2%) (). The duration of first-line corticosteroid treatment was >6 weeks in 59.5% of patients. The median number of IVIg doses was 2 (Q1, Q3: 2, 5; range: 1–13). Romiplostim was given only in 0.2% of cases, whereas rituximab or other immunosuppressants were not used as first-line therapies.
The response to first-line therapies was 92.1% (95% confidence interval 89.2–95.0%) (). The median time to response was 22 days (Q1, Q3: 7, 78; range: 0–560). Most of these ‘responder’ patients (83%; 62% of all primary ITP patients) maintained a platelet count ≥30 × 109/l at 12 months and did not require further treatment.
Only 17% of patients received second-line treatment. The median time between first-line and second-line treatment was 1 day (Q1: 0, Q3: 73, range: 0–339). The most frequent second-line treatments included rituximab (3.2%), corticosteroids (3.0%), splenectomy (2.1%), romiplostim (1.8%), corticosteroids combined with IVIg (1.7%) and IVIg as monotherapy (1.6%), although a high variability was observed. Other treatments (2.5%) included danazol, azathioprine and eltrombopag. The response rate diminished slightly to 77.9% ().
Overall, 3.5% of patients (n = 15) underwent splenectomy during the 12 months of follow-up, and only 1 of them required further treatment.
Five percent and 2% of patients received third- and fourth-line treatment, respectively (mainly romiplostim, rituximab and IVIg). The response rate in the third-line setting was 83.3%.
Change over time in platelet counts and bleeding symptoms
During follow-up, the median platelet count increased significantly in the overall cohort: 6 months: 119 × 109/l; (Q1: 66; Q3: 208; range: 0–821); 12 months: 142 × 109/l (Q1: 80; Q3: 221; range: 4–876), without relevant differences between primary and secondary ITP patients. At 12 months, approximately one quarter of patients (26.3%) still had thrombocytopenia (<100 × 109/l). No relevant changes were observed in hemoglobin levels or leukocyte count.
At 6 months, 12.7% of patients still had bleeding symptoms in the overall cohort. This percentage remained similar at 12 months (9.5%). The relative frequency of each symptom was comparable: cutaneous bleeding: 9.1% at 6 months, and 5.7% at 12 months; oral cavity bleeding: 4.0 and 3.0%; epistaxis: 3.4 and 2.7%; muscle hematoma: 0.8 and 0.2%; gastrointestinal bleeding: 0.6 and 1.0%; metrorrhagia: 1.3 and 1.2%; hematuria: 0 and 0.2%; intracranial hemorrhage: 0.4 and 0%; articular hematoma: 0 and 0%. Also the mean number of episodes (in patients with at least one episode) remained similar between the baseline–6 months and the 6–12 months periods (data not shown).
Discussion
The present study constitutes the first national Spanish registry designed to provide updated information on ITP characteristics and management in the clinical practice. According to the available epidemiological data on ITP, if the annual incidence is around 20 per million [Citation4,Citation5], during the one and half year period of recruitment in the 70 participating centres (32% of the 220 hematology services in Spain) [Citation29], with a 45 million population, the number of incident ITP cases during that period was around 1350, which implies that we managed to recruit more than one third of all new ITP cases in Spain.
The socio-demographic characteristics of our cohort were quite similar to those described in prior studies from the United Kingdom (UK) [Citation5], France [Citation30], Denmark [Citation31], Japan [Citation32], United States (US) [Citation33], and in the Pediatric and Adult Registry on Chronic ITP (PARC-ITP) Registry, which recruits patients from 31 countries worldwide (not Spain) [Citation34]. It is important to note that these previous studies only included primary ITP patients. In our study these patients accounted for 90% of the cohort, and their baseline characteristics were similar compared with those of patients with secondary ITP. For example, the median age was 52 years in our sample (58 years in patients with secondary ITP) and 56 years in the Japanese cohort [Citation32] (most of the other studies included only adult patients), with a wide age range, and the percentage of women was 56% (62%, secondary ITP cohort) compared to 55% in UK [Citation5], 60% in France [Citation30], 61% in Japan [Citation32], and 62% in Denmark [Citation31]. In the PARC-ITP Registry the percentage of women is 68% in adults and 46% in children [Citation34].
Given the nature of this registry-based study, we cannot rule out any misdiagnosis, as suggested by the presence of splenomegaly in 6 cases (1.4%) that were labelled as primary ITP. Physicians in these cases probably considered that a slight increase in spleen size, as an isolated finding, was not enough to rule out primary ITP. A similar explanation can be argued for the few cases with a family history of thrombocytopenia, which were interpreted to be not related to ITP.
The most common ITP associated disorders were systemic lupus erythematosus (1.8% of the overall population) and lymphoproliferative syndromes (1.8%), followed by HCV/HBV/HIV infections (1.9% overall). Drug-induced ITP was very rare (0.6%). In the study by Neylon et al. [Citation5] drug-related ITP was as high as 8%, and 9% of patients had a history of recent infection, compared to 14% in our sample.
We observed a considerable number of diagnostic procedures, which in general agreed with those recommended by the recent consensus documents,[Citation25,Citation26] although the study was performed before their publication. Those with the highest rate of positive findings were Helicobacter pylori testing, antinuclear antibodies and HBV serology, observing highest rates of positives among patients with secondary ITP. It must be noted the use of platelet-associated antibodies testing in 21 and 41% of primary and secondary ITP patients, respectively. This test, according to the last recommendations, has unproven or uncertain benefit in the differential diagnosis of ITP [Citation25,Citation26].
Platelet counts at diagnosis were extremely low, with three quarters of patients below 30 × 109/l in the primary ITP cohort. This observation is entirely comparable to that reported in prior studies (74% of patients below 30 × 109/l in Denmark [Citation31]; median of 7.5 × 109/l in France [Citation30]; mean of 22 × 109/l in Japan) [Citation32]. These low levels were associated with an important number of patients with bleeding symptoms, whose type and incidence was also very similar to that reported in previous newly diagnosed ITP populations [Citation32,Citation34]. The relative frequency of each symptom was comparable at the three time points, which suggests that response to treatment improves symptomatology at all levels. Despite the improvement in platelet counts, some events of potentially serious bleedings (gastrointestinal bleeding, metrorrhagia, intracranial haemorrhage) are still seen more than 6 months after diagnosis, which should make us aware of the fact that some patients are at increased risk and must be carefully followed up over time.
In our cohort, 8 in 10 patients with primary ITP received a first-line treatment, which is slightly higher than that reported in the PARC-ITP Registry (around 7 in 10 adult patients) [Citation34]. The relative distribution of each treatment (mainly corticosteroids monotherapy, followed by corticosteroids plus IVIg) is also very similar to that reported in the PARC-ITP [Citation34]. This therapeutic approach was fairly homogeneous and, in general, conformed to the recommendations of the consensus. As expected, patients with primary ITP who received corticosteroids plus IVIg had the lowest platelet counts and worst bleeding symptoms at baseline, suggesting an adequate intensification of treatment in those patients at highest risk.
Remarkably, the duration of corticosteroid treatment was too high in the majority of patients with primary ITP (almost two thirds received them for more than 6 weeks). The response rate to the first-line treatment was very high and only 2 in 10 patients initiated a second-line treatment, much more diverse and not standardized. At 12 months 74% of patients had normal platelet counts in absence of treatment, which suggests that the disease may become persistent in around one quarter of patients, despite current management practices.
Our study has some limitations. As the study was observational, we could not establish a causal relationship between the administered treatments and the observed response rates, nor can we rule out the possibility that these responses were associated with the natural evolution of disease. Strengths of our study include its relatively large sample size and the unbiased, geographically distributed recruitment, which ensures the representativeness of our sample.
In conclusion, there is a wide variability in ITP patients. The onset of disease may occur at any age and gender and may or may not be associated to prior infections such as Helicobacter pylori or HBV. In the Spanish setting, there is also a great variability in the application of diagnostic methods of ITP, which sometimes do not fit the current recommendations. In patients with primary ITP the therapeutic approach of the first-line is fairly homogeneous and conforms to the recommendations of the consensus, except for an excessive duration of corticosteroids therapy. Second-line treatments are much more diverse and not standardized. At one year, the disease persists in around one quarter of patients, despite current therapies available. These results reflect the need for improving the therapeutic management of ITP patients in Spain through uniform implementation of international consensus recommendations.
Acknowledgments
Writing assistance was supported by Amgen and provided by Dr Neus Valveny, from TFS Develop. The authors wish to thank to all the Investigators of the TIMES Registry Study (in alphabetical order by hospital name): C. U. de Navarra, Dr José A. Páramo; CHU A Coruña, Dra. Mª Fernanda López Fernández; Fundación Jiménez Díaz, Dra. Sol Sánchez; H. Albacete, Dra. Ángela Ibáñez, Dra. María José Varó Castro; H. Arnau de Vilanova, Dra. Esther Herrera de Pablo, Dra. Carmen Benet, Dr Rafael Lluc; H. Arnau de Vilanova (Lleida), Dra. Rosa Isabel Upegui, Dr Macià; H. Carlos Haya, Dra. María Eva Mingot; H. Clínico de Salamanca, Dr José Ramón González Porras; H. Clínico de Santiago, Dra. Susana Pérez Crespo; H. Consorci Sanitari de Terrassa, Dra. Laura Vicente Folch; H. Consorci Sanitari de Terrassa, Dra. Marta Garcia Pintos; H. Costa del Sol, Dra. Maria Esperanza Moreno; H. de Cabueñes, Dra. Mª Cristina Fernández Canal; H. de Ciudad Real, Dra. Carmen Calle; H. de Donostia, Dra. Nagore Argoitia; H. de Elche, Dr Jose Antonio Molina, Dr Venancio Conesa; H. de Joan XXIII, Dr Andres Llorente; H. de la Fe, Dr Javier Palau; H. de la Plana, Dr Jose Sanchís; H. de Navarra, Dra. Mª Luisa Antelo; H. de San Eloy, Dr Jose Julio Hernández Hernández; H. de Vic, Dr Lluis Rodríguez, Dra. Camino Salgado; H. de Vinalopó, Dr Sergio Martínez Cañabate; H. del Mar, Dra. Blanca Sanchez, Francesc García; H. Doce de Octubre, Dr Carlos Grande; H. El Bierzo de Ponferrada, Dr Erik de Cabo; H. Francesc de Borja de Gandía, Dra. Maria José Lis Chulvi; H. Fundación Alcorcón, Dr Javier Peñalver, Dra. Pilar Martínez, Dra. Lucía Villalón; H. General de Guadalajara, Dra. Dunia de Miguel; H. General de Guadalajara, Dra. Nuria Golbano López; H. General de Manresa, Dra. Elena Cabezudo; H. General Yagüe, Dr Gerardo HermidaTomás José González López; H. Germans Trias i Pujol, Dr Francisco Almazán; H. Germans Trias i Pujol, Dra. Elisa Orna Montero; H. Gral de Móstoles, Dr Eriel Alexis Marcheco Pupo; H. Gral. Alicante, Dr Francisco Javier Lucas; H. Gral. Castellón, Dra. Teresa Gozalvo, Dr Guillermo Cañigral; H. Gregorio Marañón, Dra. Ana Rodríguez Huerta; H. Gregorio Marañón, Dra. Cristina Pascual; H. Infanta Elena de Valdemoro, Dra. Sonia García; H. Infanta Leonor, Dr José Angel Hernández Rivas; H. Infanta Sofía, Dra. Asunción Mora; H. Infanta Sofía, Dra. Pilar Massó, Dr Eugenio Giménez, Dr Raúl Córdoba; H. Jaén, Dr Antonio Alcalá; H. Jerez, Dr Sebastián Garzón, Dra Madrigal; H. Juan Ramón Jiménez, Dr Juan Nicolás Rodríguez; H. Juan Ramón Jiménez, Dra. Maria Victoria Moreno Romero, Dr Antonio Fernández Jurado; H. La Candelaria Tenerife, Dra. Magdalena Herrera, Dra. Ana Sánchez Quintana; H. La Línea, Dr Manuel González Silva; H. La Mancha Centro (H. de Alcázar de San Juan), Dra. Victoria Angélica Godoy Vila; H. La Paz, Dra. Maite Álvarez, Ihosvany Fernández Bello; H. La Princesa, Dra. Beatriz Aguado Bueno; H. Lanzarote, Dr José Manuel Calvo; H. Niño Jesús, Dr Julián Sevilla; H. Ntra Sra. De Sonsoles, Dra. Maria Jesús Rodríguez Domínguez; H. Nuestra Señora del Prado, Dr Fernando Solano Ramos; H. Ourense, Dra. Marta Rodriguez Gomez, Dr José Luis Sastre; H. Parc Taulí, Dra. Sònia Piernas; H. Povisa, Dra. Adriana Simiele Narvarte; H. Provincial de Castellón, Dra. Susana Mollá; H. Ramón y Cajal, Dr Jesús César Pérez, Dr Juan Diego Rodriguez Gambarte; H. San Pedro de Alcántara, Dra. Nuria Bermejo; H. Sant Joan de Deu Martorell, Dra. Eva Sánchez, Dr German de las Heras; H. Sant Pau, Dr David Valcarcel, Dra. Salut Brunet, Dra. Adriana Sierra; H. Segovia, Dra. Carmen Olivier; H. Severo Ochoa, Dr Pedro Sánchez Godoy, Dra. Eva Yebra Fernández; H. Son Llàtzer, Dr Joan Bargay Lleonart; H. Universitario de Canarias, Dra. Mª José Rodríguez Salazar; H. Universitario Rafael Méndez de Lorca, Dr Raúl Pérez López; H. Universitario Santa Maria del Rosell de Cartagena, Dra. Jerónima Ibáñez Garcia; H. Vall d'Hebron, Dra. Esther Sancho, Dr Javier Bueno; H. Valle del Nalón (Langreo), Dr Jose Manuel Alonso; H. Universitario Virgen de Valme, Dr Eduardo Ríos; H. Virgen de la Luz de Cuenca, Dr Jesús Manuel Vall, Dra. María Josefa Busto Medina; H. Virgen de la Salud, Dr Angel Remacha, Dra. Elena Gutiérrez; H. Virgen del Rocío, Dra. Fátima de la Cruz.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
María Eva Mingot http://orcid.org/0000-0001-9083-855X
Eduardo Ríos http://orcid.org/l0000-0002-4072-3911
Additional information
Funding
References
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–2393. doi: 10.1182/blood-2008-07-162503
- Segal JB, Powe NR. Prevalence of immune thrombocytopenia: analyses of administrative data. J Thromb Haemost. 2006;4:2377–2383. doi: 10.1111/j.1538-7836.2006.02147.x
- Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol. 2010;85:174–180. doi: 10.1002/ajh.21833
- Frederiksen H, Schmidt K. The incidence of idiopathic thrombocytopenic purpura in adults increases with age. Blood. 1999;94:909–913.
- Neylon AJ, Saunders PWG, Howard MR, et al. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122:966–974. doi: 10.1046/j.1365-2141.2003.04547.x
- Chong BH, Ho S-J. Autoimmune thrombocytopenia. J Thromb Haemost. 2005;3:1763–1772. doi: 10.1111/j.1538-7836.2005.01376.x
- McMillan R. Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. Semin Hematol. 2000;37:239–248. doi: 10.1016/S0037-1963(00)90102-1
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346:995–1008. doi: 10.1056/NEJMra010501
- Ballem PJ, Segal GM, Stratton JR, et al. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest. 1987;80:33–40. doi: 10.1172/JCI113060
- Houwerzijl EJ, Blom NR, van der Want JJL, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood. 2004;103:500–506. doi: 10.1182/blood-2003-01-0275
- Chang M, Nakagawa PA, Williams SA, et al. Immune thrombocytopenic purpura (ITP) plasma and purified ITP monoclonal autoantibodies inhibit megakaryocytopoiesis in vitro. Blood. 2003;102:887–895. doi: 10.1182/blood-2002-05-1475
- McMillan R, Wang L, Tomer A, et al. Suppression of in vitro megakaryocyte production by antiplatelet autoantibodies from adult patients with chronic ITP. Blood. 2004;103:1364–1369. doi: 10.1182/blood-2003-08-2672
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, et al. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001;97:2549–2554. doi: 10.1182/blood.V97.9.2549
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371:395–403. doi: 10.1016/S0140-6736(08)60203-2
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med. 2010;363:1889–1899. doi: 10.1056/NEJMoa1002625
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373:641–648. doi: 10.1016/S0140-6736(09)60402-5
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med. 2007;146:25–33. doi: 10.7326/0003-4819-146-1-200701020-00006
- Cines DB, Bussel JB. How I treat idiopathic thrombocytopenic purpura (ITP). Blood. 2005;106:2244–2251. doi: 10.1182/blood-2004-12-4598
- Blanchette V, Imbach P, Andrew M, et al. Randomised trial of intravenous immunoglobulin G, intravenous anti-D, and oral prednisone in childhood acute immune thrombocytopenic purpura. Lancet. 1994;344:703–707. doi: 10.1016/S0140-6736(94)92205-5
- Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood. 2007;109:1401–1407. doi: 10.1182/blood-2005-12-015222
- Kojouri K, Vesely SK, Terrell DR, et al. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood. 2004;104:2623–2634. doi: 10.1182/blood-2004-03-1168
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood. 2008;112:999–1004. doi: 10.1182/blood-2008-01-131029
- Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood. 2010;115:2755–2762. doi: 10.1182/blood-2009-07-229815
- George JN, Woolf SH, Raskob GE, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88:3–40.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168–186. doi: 10.1182/blood-2009-06-225565
- Sanz MÁ, Vicente García V, Fernández A, et al. [Guidelines for diagnosis, treatment and monitoring of primary immune thrombocytopenia]. Med Clínica. 2012;138:261.e1–261.e17.
- Ferri consultor clinico, 2006–2007 : Claves diagnosticas y tratamiento [Internet]. [cited 2014 Mar 13]. Available from: http://www.agapea.com/libros/Ferri-consultor-clinico-2006-2007-claves-diagnosticas-y-tratamiento-9788481749144-i.htm
- Hedman A, Henter JI, Hedlund I, et al. Prevalence and treatment of chronic idiopathic thrombocytopenic purpura of childhood in Sweden. Acta Paediatr. 1997;86:226–227. doi: 10.1111/j.1651-2227.1997.tb08876.x
- Hemoterapia AE de HY, Médicos E. Libro blanco de la hematología y hemoterapia en España. Sociedad Española de Hematol y Hemoterapia. 2012. p. 264.
- Grimaldi-Bensouda L, Michel M, Aubrun E, et al. A case-control study to assess the risk of immune thrombocytopenia associated with vaccines. Blood. 2012;120:4938–4944. doi: 10.1182/blood-2012-05-431098
- Jensen AO, Nørgaard M, Engebjerg MC, et al. Predictors for splenectomy among patients with primary chronic immune thrombocytopenia: a population-based cohort study from Denmark. Ann Hematol. 2011;90:207–212. doi: 10.1007/s00277-010-1047-5
- Kurata Y, Fujimura K, Kuwana M, et al. Epidemiology of primary immune thrombocytopenia in children and adults in Japan: a population-based study and literature review. Int J Hematol. 2011;93:329–335. doi: 10.1007/s12185-011-0791-1
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol. 2011;86:420–429. doi: 10.1111/j.1600-0609.2011.01587.x
- Kuhne T, Berchtold W, Michaels LA, et al. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011;96:1831–1837. doi: 10.3324/haematol.2011.050799