
ABSTRACT
Objectives: Hereditary spherocytosis (HS) is a genetic heterogeneous disorder characterized by sphere-shaped erythrocytes on peripheral blood smear with a few clinical manifestations. As an important red cell membrane protein, ankyrin 1 can interact with transmembrane proteins and the membrane skeleton and mutations in the ankyrin 1 (ANK1) genes affect about half of all patients with HS. The purpose of this study was to investigate a Chinese Han family with HS to find out the causative gene mutation and explore the genotype–phenotype correlation which can provide the basis for the pathogenesis and prenatal diagnosis for this disease.
Methods: Whole exome sequencing (WES) followed by Sanger sequencing was performed on subjects with HS from a Chinese family in Shandong Province.
Results: A de novo nonsense ANK1 mutation (c.796G > T, p.Glu266X), a single-nucleotide change from G to T which caused a substitution from glutamic acid to an immature stop codon at codon 266, was identified.
Discussion: Our finding suggested that a de novo nonsense mutation in ANK1 may be causative to HS which plays an important role in supplementing the mutational spectrum of the ANK1 and explaining the mechanism of HS. Our study also indicated that WES can be an effective and accurate diagnostic tool in the discovery of causative mutations in genetic heterogeneous Mendelian disorders.
Introduction
Hereditary spherocytosis (HS) is an inherited disorder characterized by sphere-shaped erythrocytes on peripheral blood smear with a few clinical manifestations including anemia, jaundice, reticulocytosis, gallstones and splenomegaly [Citation1]. HS is predominantly caused by defects in erythrocyte membrane proteins, particularly including ankyrin 1, Band 3, α-spectrin, β-spectrin, and protein 4.2 [Citation2], encoded by ankyrin 1 (ANK1) gene, solute carrier family 4, member 1 (SLC4A1) gene, spectrin, alpha, erythrocytic 1 (SPTA1) gene, spectrin, beta, erythrocytic (SPTB) gene and erythrocyte membrane protein band 4.2 (EPB42) gene, respectively [Citation3]. These genes, if mutated, can lead to the deformation of red cells so that double-concave disc-shaped red blood cells become spherical, fragile red blood cells [Citation4,Citation5]. Then, these erythrocytes cannot pass through the splenic sinusoids easily where they could be selectively trapped and destroyed [Citation6]. With high genetic heterogeneity, HS is inherited in an autosomal dominant (AD) manner in most of the patients due to mutations in ANK1, SPTB or SLC4A1, while the inheritance of the remaining patients is autosomal recessive (AR) because of either SPTA1 or EPB42 mutation [Citation7].
ANK1 mutation is the most common cause of typical dominant HS [Citation8], accounting for about half of HS patients. ANK1, located on chromosome 8p11.2, encodes erythroid ankyrin 1 [Citation9] which is an important red cell membrane protein that interacts with transmembrane proteins and the membrane skeleton of cells through spectrin, band 3, and band 4.2 proteins. In most cases, HS patients carrying different ANK1 mutations such as the nonsense, splicing or frameshift mutations have a complex relationship between genotype and phenotype due to genetic heterogeneity.
In this report, clinical features of a Chinese family with HS were described in detail and the WES was used to identify a de novo ANK1 mutation responsible for HS.
Patients and methods
Patients
This study was approved by the ethics committee of the Affiliated Hospital of Qingdao University. The subjects came from a Chinese Han family in Shandong province. The proband is a 6-year-old girl who was clinically diagnosed with HS in the Affiliated Hospital of Qingdao University (Shandong, China) while her parents had normal examination findings undertaken by the physician. She went through a series of detailed blood tests. Then the blood samples were collected from the proband, her father and mother in this family.
Methods
WES
DNA samples were extracted from 200 µl peripheral venous blood with a Qiagen DNA extraction kit (Qiagen, Hilden, Germany). We carried out WES on the proband and her parents in the family with the method of human exome capture based on the protocol from Illumina’s TruSeq Exome Enrichment Guide (SureSelectXT Target Enrichment System for Illumina Paired-End Sequencing Library, Agilent). We chose the Agilent Human All Exon 50 Mb Exome Enrichment kit as exome enrichment probe sets. The Genomic DNA libraries were prepared according to the manufacturer’s instructions (Illumina, San Diego, CA, U.S.A.). 5 mg genomic DNA was mixed in 80 ml EB buffer and the segments of DNA were randomly fragmented into hundreds of bases or shorter pieces and added specific joint at both ends. An ‘A’ base was added to the 3′ end using Klenow 3′ to 5′ exo minus and DNA fragments were ligated to the Illumina multi-PE-adaptor with Illumina Paired-End sample Preparation kit. The DNA product was amplified by PCR of 12 cycles, the reaction system included 1 ml of Illumina multi-PE primer #1 (25 mM), 1 ml of Illumina multi-PE primer #2 (0.5 mM) and 1 ml of Illumina index primer (25 mM).
The Illumina HiSeq 2000 platform was used to sequence the Captured DNA libraries. The obtained sequence reads and the human genome reference sequence were consistent, and the software tool was used to find variations. The original data were analyzed by Bioinformatics. Then we performed statistical analysis of target area coverage and capture efficiency. Furthermore, the protein function was predicted to detect the potential pathogenic causative mutations for HS.
Sanger sequencing validation
Sanger sequencing was used for validation of the ANK1 variant of the proband identified by WES. Two fragments covering the coding sequence of 5′-CCCAGATAGAAACCAAGA-3′ were amplified using ANK1 primer pairs for exon 8 (Forward: TGCATTCCAGGGAAGGATGA and Reverse: GAGAAGACTGCCTATGGCATCA). The same amplification conditions were used for both primer pairs in a total volume of 25 ml: 250 nM dNTPs, 100 ng of template DNA, 0.5 mM of each primer and 1.25 U AmpliTaq Gold DNA polymerase in 1× reaction buffer (10 mM Tris–HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2). PCR amplifications consisted of an initial denaturing step at 94°C for 5 min, 35 cycles of: 94°C for 30 s, 58°C for 60 s, 72°C for 30 s and 10 min of final extension at 72°C. Amplified PCR products were purified and sequenced with the appropriate PCR primers and the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, U.S.A.) and run on an automated sequencer, ABI 3730XL (Applied Biosystems).
Results
Clinical phenotype
The affected female individual presented with a yellow complexion, jaundice and splenomegaly without other pathological symptoms or signs on the second day after birth. Then, the routine blood examination showed that she suffered from moderate hemolytic anemia with neonatal hyperbilirubinemia and sphere-shaped erythrocytes on peripheral blood smear. Blood results showed: Hb 62 g/l, RBC 2.32×1012/l, MCV 70.1fL, MCH 26.7pg, MCHC 38.0 g/dl, indirect bilirubin 53.8 μmol/l (normal range1.7–17.2 μmol/l), and reticulocytes 8.7% (normal range 0.5–1.5%). The erythrocyte osmotic fragility test was positive while both glucose-6-phosphate dehydrogenase (G-6-PD) screening test and direct Coombs’ test were negative. Then the proband was clinically diagnosed with HS and her parents had neither hyperbilirubinemia nor anemia. The propositus underwent laparoscopic splenectomy when she was 6 years old because of splenomegaly and hemolysis, after which the hemoglobin level has returned to the normal range with mild hyperbilirubinemia.
Genetic analysis
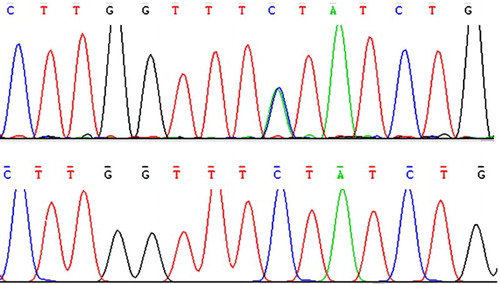
We used the WES and several filtering steps to exclude nongenetic variants by screening the database of dbSNP and 1000 genomes and a novel nonsense variant (c.796G > T, p.Glu266X) in ANK1 was found. Then, Sanger sequencing confirmation was carried out and certified the heterozygous mutation indicating a single-nucleotide changed from G to T which caused a substitution from glutamic acid to an immature stop codon at codon 266 in exon 8 of ANK1 which may cause haploid deficiency of the ANK1 protein (). According to the ESP6500 database, human genome database and dbSNP database, this mutation has never been previously demonstrated. Co-segregation analysis of this pedigree revealed that her parents did not carry this nonsense mutation, therefore, the proband has a de novo mutation in ANK1. In addition, the mutation was not observed in the 100 control individuals.
Figure 1. Sanger sequencing of PCR-amplified genomic DNA of exon 8 of the ANK1 gene confirms that the girl carried the novel mutation which caused a substitution from glutamic acid to an immature stop codon at codon 266, c.796G > T (p.Glu266X). Her parents’ control is shown on the below.
Discussion
In this study, we described a Chinese family with a proband affected by HS. A de novo mutation (c.796G > T, p.Glu266X) causing a premature stop codon in exon 8 of ANK1 was found by investigating this family through WES followed by Sanger sequencing to certify the relationship between the ANK1 mutation and HS. The HS patients may have asymptomatic manifestations or severe clinical symptoms which include jaundice, splenomegaly, gallstones, and hemolytic anemia [Citation10,Citation11]. In our study, the proband had jaundice and splenomegaly without other pathological symptoms such as gallstones. The diagnosis for HS was based on a series of laboratory tests and clinical findings. Then, genetic analysis was performed after splenectomy due to the persistent mild hyperbilirubinemia.
As the most common congenital hemolytic anemia due to membrane protein defects [Citation11,Citation12], HS is a group of inherited disorders with the presence of spherical-shaped erythrocytes in the peripheral blood smear [Citation13]. The clinical findings can be compensated or severe and patients with HS may need exchange transfusion when they are born and/or repeated blood transfusions subsequently [Citation8,Citation9]. ANK1 is the most common causative gene defect transmitted by a dominant mode and to date, over 23 different mutations have been found among HS patients [Citation14]. Recently, Han et al. identified a novel nonsense ANK1 mutation (p.Q1772X) in a Korean HS family which is the first report in a Korean family carrying the ANK1 mutation. The 19-year-old male patient was diagnosed with HS when he was 3 months based on spherocytosis in a peripheral blood smear. Splenectomy was performed because of splenomegaly and anemia at 6 years of age. Then, his hemoglobin levels became normal while bilirubin remained moderately elevated for several years [Citation10]. A Chinese boy carrying a heterozygous ANK1 mutation (p.Q109X) suffered from severe HS from preterm neonatal period [Citation15]. His RBC and hemoglobin levels were higher than the proband in our study and he received a RBC transfusion due to premature delivery. Previous studies indicated the insertion and deletion mutations in ANK1 identified in the promoter regions, 5′ and 3′ untranslated regions, and nonsense or missense substitutions in coding sequence [Citation16]. These mutations can influence the expression or stability of the related proteins which can cooperate with ankyrin 1 and play an important role in the erythrocyte membranous-cytoskeletal network [Citation13,Citation17]. ANK1 mutations affect about half of all patients with HS, while the mutations in protein 4.1 gene are more common in Japan [Citation18].
HS is usually diagnosed in childhood. Clinical manifestations, such as anemia, jaundice and/or splenomegaly, are the major presenting clinical features. Patients usually also have elevated reticulocyte count because of the shortened red cell lifetime. However, previous studies tried to seek effective findings on the routine complete blood count (CBC) for HS diagnosis instead of the reticulocyte count which is not analyzed automatically when CBC is requested. The mean corpuscular hemoglobin concentration (MCHC) and red cell distribution width (RDW) have been identified as important parameters [Citation19,Citation20] and incubated osmotic fragility(OF) has been regarded as confirmatory test for HS diagnosis. Although the red blood cell morphology examination is one of the most effective diagnostic methods of hemopathy, about 10% patients of HS may be misdiagnosed due to lack of typical sphere-shaped erythrocytes on peripheral blood [Citation21]. Furthermore, due to the phenotypic or genetic heterogeneity of HS, it is difficult to reach to a confirmed diagnosis especially for HS patients who have few symptoms. Therefore, genetic diagnosis become much more important as the traditional diagnostic tests could be time-consuming and may not provide feasible guidance for treatment at its most fundamental. With the rapid development of NGS, it is becoming easier to determine which candidate gene is responsible for the disease. WES has been widely used [Citation22] to identify inherited, rare gene mutations which are associated with a high risk for the development of diseases [Citation23] and has become an effective way to diagnose for diseases [Citation10] because of its timesaving, efficient and low-cost. Therefore, fewer family members may be needed with WES to diagnose rare monogenic disorders in a pedigree study [Citation24].
In conclusion, WES and Sanger sequencing were adopted to identify a de novo nonsense mutation in ANK1 associated with HS in a Chinese family. Our study indicated this novel mutation might be causative for this HS proband. Furthermore, the WES can be an effective method to diagnose complicated and heritable diseases with high genetic heterogeneity. However, the pathogenic mechanisms of this ANK1 mutation should be investigated further in depth to improve diagnosis and treatment for HS.
Acknowledgement
We thank all patients for their participation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Gallagher PG, Steiner LA, Liem RI, et al. Mutation of a barrier insulator in the human ankyrin-1 gene is associated with hereditary spherocytosis. J Clin Invest. 2010;120(12):4453–4465. doi: 10.1172/JCI42240
- Hughes MR, Anderson N, Maltby S, et al. A novel ENU-generated truncation mutation lacking the spectrin-binding and C-terminal regulatory domains of Ank1 models severe hemolytic hereditary spherocytosis. Exp Hematol. 2011;39(3):305–320. 320 e1-2. doi: 10.1016/j.exphem.2010.12.009
- Park J, Jeong D-C, Yoo J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. 2016;90(1):69–78. doi: 10.1111/cge.12749
- Cooper RA, Jandl JH. The role of membrane lipids in the survival of red cells in hereditary spherocytosis. J Clin Invest. 1969;48(4):736–744. doi: 10.1172/JCI106031
- Da Costa L, Mohandas N, Sorette M, et al. Temporal differences in membrane loss lead to distinct reticulocyte features in hereditary spherocytosis and in immune hemolytic anemia. Blood. 2001;98(10):2894–2899. doi: 10.1182/blood.V98.10.2894
- Konca C, Söker M, Taş MA, et al. Hereditary spherocytosis: evaluation of 68 children. Indian J Hematol Blood Transfus. 2015;31(1):127–132. doi: 10.1007/s12288-014-0379-z
- Salas PC, Rosales JML, Milla CP, et al. A novel mutation in the beta-spectrin gene causes the activation of a cryptic 5′-splice site and the creation of a de novo 3′-splice site. Hum Genome Var. 2015;2:15029. doi: 10.1038/hgv.2015.29
- Gallagher PG. Red cell membrane disorders. Hematol Am Soc Hematol Educ Program. 2005: 13–18.
- Hassoun H, Palek J. Hereditary spherocytosis: a review of the clinical and molecular aspects of the disease. Blood Rev. 1996;10(3):129–147. doi: 10.1016/S0268-960X(96)90021-1
- Han JH, Kim S, Jang H, et al. Identification of a novel p.Q1772X ANK1 mutation in a Korean family with hereditary spherocytosis. PLoS One. 2015;10(6):e0131251. doi: 10.1371/journal.pone.0131251
- Morle L, Bozon M, Alloisio N, et al. Ankyrin Bugey: a de novo deletional frameshift variant in exon 6 of the ankyrin gene associated with spherocytosis. Am J Hematol. 1997;54(3):242–248. doi: 10.1002/(SICI)1096-8652(199703)54:3<242::AID-AJH11>3.0.CO;2-F
- Jarolim P, Rubin HL, Brabec V, et al. A nonsense mutation 1669Glu-->Ter within the regulatory domain of human erythroid ankyrin leads to a selective deficiency of the major ankyrin isoform (band 2.1) and a phenotype of autosomal dominant hereditary spherocytosis. J Clin Invest. 1995;95(3):941–947. doi: 10.1172/JCI117802
- Eber S, Lux SE. Hereditary spherocytosis – defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41(2):118–141. doi: 10.1053/j.seminhematol.2004.01.002
- Stenson PD, Ball EV, Mort M, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–581. doi: 10.1002/humu.10212
- Liu S, Jiang H, Huang L-Y, et al. A de novo ankyrin mutation (ANK1 Q109X) causing severe hereditary spherocytosis from preterm neonatal period. Ann Hematol. 2017;96(6):1067–1068. doi: 10.1007/s00277-017-2966-1
- Gallagher PG. Hematologically important mutations: ankyrin variants in hereditary spherocytosis. Blood Cells Mol Dis. 2005;35(3):345–347. doi: 10.1016/j.bcmd.2005.08.008
- Eber SW, Gonzalez JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996;13(2):214–218. doi: 10.1038/ng0696-214
- Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–265. doi: 10.1038/nature07935
- Michaels LA, Cohen AR, Zhao H, et al. Screening for hereditary spherocytosis by use of automated erythrocyte indexes. J Pediatr. 1997;130(6):957–960. doi: 10.1016/S0022-3476(97)70283-X
- Bianchi P, Fermo E, Vercellati C, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97(4):516–523. doi: 10.3324/haematol.2011.052845
- Christensen RD, Henry E. Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics. 2010;125(1):120–125. doi: 10.1542/peds.2009-0864
- Koboldt DC, Steinberg KM, Larson DE, et al. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155(1):27–38. doi: 10.1016/j.cell.2013.09.006
- Bolli N, Barcella M, Salvi E, et al. Next-generation sequencing of a family with a high penetrance of monoclonal gammopathies for the identification of candidate risk alleles. Cancer. 2017;123(19):3701–3708. doi: 10.1002/cncr.30777
- Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499