
ABSTRACT
Objectives: The present review summarizes the available knowledge regarding acute and chronic kidney dysfunction in patients with paroxysmal nocturnal hemoglobinuria (PNH) focusing on its clinical features, pathophysiology and treatment.
Methods: A thorough PubMed search was performed using as main keywords: ‘paroxysmal nocturnal hemoglobinuria’, ‘acute kidney injury’, ‘chronic kidney disease’ and ‘eculizumab’.
Results: PNH’s etiopathogenesis is based on acquired mutations that lead to the reduction or absence of CD55 and CD59 complement regulators, which are responsible for some of the disease’s major clinical features, like intravascular hemolysis, cytopenias and thrombosis. PNH is often underdiagnosed, mainly due to its occasional mild manifestations and to its ability to mimic other severe clinical conditions. Various mechanisms have been proposed for the kidney damage attributed to the release of cell-free heme and free iron, including inflammatory response, oxidative stress, nitric oxide depletion, renal ischemia, membrane damage and apoptosis. Eculizumab, a terminal complement inhibitor, provides a safe and effective treatment option, especially when it is initiated early in the presence of kidney damage.
Discussion: Kidney injury is a poorly investigated clinical feature of PNH that affects a significant portion of patients. Increased awareness is needed by physicians to recognize the early signs and symptoms of acute and chronic renal insufficiency, so as to initiate the necessary therapy. It is also important to re-evaluation of PNH-specific treatments during the course of the disease.
Conclusion: Understanding the difficult but at the same time impressive mechanisms behind PNH remains a challenge for treating physicians.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, life-threatening disease caused by clonal hematopoietic stem cell evolution. It is often underdiagnosed mainly due to its occasionally mild clinical and laboratory manifestations (observed even several years after the onset of the disease) and also, due to its ability to mimic other severe clinical conditions, like the camouflage of a chameleon.
PNH’s etiopathogenesis is based on acquired mutations that lead to the reduction or absence of glycosylphosphatidylinositol (GPI)-anchored proteins. Amongst the defective GPI-anchored proteins, the loss of CD55 and CD59 complement regulators is responsible for some of the disease’s major clinical features, like intravascular hemolysis, cytopenias and thrombosis. Another clinical characteristic, frequently observed is renal dysfunction [Citation1].
In PNH, renal dysfunction often manifests as acute kidney injury (AKI) or chronic kidney disease (CKD). An accurate estimation of renal dysfunction’s prevalence and mortality in PNH is hindered by both the disease΄s rarity and by the positive effects of the complement inhibition treatment implementation. A recent study by Hillmen et al. reports that up to 65% of PNH patients present CKD (stages 1–5). Renal insufficiency (CKD stages 3–5) was observed in approximately 21% of PNH patients [Citation2]. These data are in accordance with other similar studies in the field [Citation3–5]. Renal failure has been implicated as the leading cause of death in 8–18% of the disease’s population [Citation3]. Other studies report that renal failure is responsible for an eight-fold increase in mortality in PNH patients [Citation6]. Despite its frequency, renal dysfunction’s pathophysiology is still elusive and is often neglected by physicians when treating PNH patients.
In the present review, we summarized the available knowledge regarding acute and chronic kidney dysfunction in PNH, focusing on its clinical features, its pathophysiology and the effects of the complement inhibition treatment implementation. For the purposes of the review, a thorough search of the current literature was conducted in PubMed using the following keywords: ‘paroxysmal nocturnal hemoglobinuria’, ‘acute kidney injury’, ‘chronic kidney disease’, ‘renal failure’ and ‘mechanisms’.
Clinical features of kidney injury
Since 2004, a number of classifications have been introduced for AKI, including the Risk, Injury, Failure, Loss, and End-stage renal disease (RIFLE) [Citation7], AKI Network (AKIN) [Citation8], and Kidney Disease Improving Global Outcomes (KDIGO) [Citation9] classifications. summarizes all current definitions from the above groups. Even though novel definitions have revolutionized the field, the introduction of a universal definition could be of importance in order to improve AKI identification [Citation10], especially in patients treated by physicians other than nephrologists for underlying disorders.
Table 1. Comparison of different definition criteria for AKI.
Studies reporting AKI incidence in PNH are sporadic and their results are often conflicting. In a study conducted by Hillmen et al., 14 out of 96 PNH patients had a history of acute renal failure and 5 out of 14 required hemodialysis. Among them, three patients subsequently developed CKD (chronic glomerular filtration rate (GFR) < 60 ml/minute/1.73 m2). Similar studies have reported AKI as the first clinical sign in PNH in a total of five patients [Citation11–14]. Interestingly, two of those patients presented with repeated episodes of AKI in combination with hemolytic anemia, not previously investigated. Several other cases of AKI in the PNH population have been reported in the current literature [Citation15–19].
When renal biopsies are performed in PNH patients presented with kidney injury, among the typical findings are hemosiderin deposits in tubular cells, most prominent in the proximal tubules. Interestingly, no evidence of renal arterial or vein thrombosis has been presented in relevant case reports or in larger studies conducted in PNH patients treated with eculizumab [Citation11–18,Citation20]. However, the possibility that subclinical microvascular thrombosis could contribute to renal dysfunction cannot be excluded [Citation21].
A reliable non-invasive diagnostic tool for the detection of renal cortical hemosiderosis in the PNH setting is magnetic resonance imaging (MRI). Typical findings of hemosiderin deposition in MRI scans include reversed renal cortex–medulla differentiation on T1 images and substantial loss of cortical signal intensity on T1 and T2 images [Citation22]. Such findings should raise the suspicion of AKI involvement in PNH patients that requires immediate management by supportive therapy and treatment according to the underlying cause.
CKD refers to kidney damage classified according to GFR rates in five stages () [Citation23]. CKD affects multiple organs and systems. Major clinical features include: uremia, disorders of fluid, electrolyte and acid–base balance, along with cardiopulmonary (mainly arterial hypertension), endocrine, metabolic, dermatological, gastrointestinal, immunological and hematological (anemia, hemorrhage) disorders.
Table 2. Classification of CKD.
In PNH, 21% of patients develop CKD (stages 3–5). In contrast to the general population, age did not contribute to the development of CKD in PNH patients. However, CKD at older age had a more severe clinical presentation [Citation2]. In line with these data, the most recent update from the Korean registry reported impaired renal function in 17.9% of patients not treated with eculizumab [Citation13]. In that population, the majority of patients had pre-existing acute renal failure. To our knowledge, available data in the current literature regarding renal replacement therapy in patients with end-stage renal disease due to PNH are limited. Munoz-Linares et al. [Citation5] reported that in a study conducted in a single Spanish center 3 out of 30 patients presented with severe renal insufficiency (estimated GFR < 15 ml/minute) and were in need of hemodialysis.
Fanconi syndrome is another important finding in PNH patients, caused by proximal tubular defects and CKD. Although Fanconi syndrome has been described in PNH since 1975, its incidence is probably under-estimated [Citation24–26]. In PNH patients that developed Fanconi syndrome, CKD co-existed with proximal tubular dysfunction (presented as renal glucosuria, proteinuria and aminociduria). Early recognition of Fanconi syndrome in PNH is necessary in order to implement proper treatment (calcium phosphate and calcitriol supplementation). The role of iron chelators in PNH remains to be proven [Citation26], due to its observed side effects in other chronic anemias [Citation27] (e.g. deferoxamine has been associated with reversible bilateral central serous retinopathy in the PNH setting [Citation28]).
Pathophysiology of kidney injury in PNH
Release of heme (attributed to intravascular hemolysis) is the leading cause of AKI in PNH. An acute hemolytic episode associated with massive hemoglobinuria may cause an episode of acute renal failure. The mechanisms behind this phenomenon include the following: (a) hypovolemia and renal ischemia; (b) direct toxicity of free hemoglobin in the tubular cells; (c) glomerular deposition of fibrin and (d) tubular obstruction by uric acid crystals and hemoglobin urinary casts-cylinders (the latter are products of heme reaction with Tamm–Horsfall protein (THP) – a secretory product of the cells of the distal convoluted tubules) [Citation29].
THP or uromodulin is a glycoprotein which was isolated from urine by Tamm and Horsfall in 1952. This protein is excreted by the thick ascending branch of the loop of Henle and the first part of the distal tubules. The protein has a huge molecular weight of around 7 million Daltons, with 25–40% of its weight being carbohydrates. It is a glycoprotein that in humans is encoded by the uromodulin gene. Normal daily excreted quantities range from 25 to 50 mg. When hemoglobin is present in the kidneys due to intensive hemolysis, hemoglobin casts are formed with the participation of THPs in the nephron’s section where the dilution is maximal. Even though reports of intratubular hemoglobin casts in the setting of acute hemolysis are rare in the literature, this finding may represent an under-recognized contributor to kidney disease in this clinical setting [Citation30].
Chronic hemolysis may also be a cause of chronic renal failure (CRF): repeated exposure of the renal epithelium to hemoglobin (caused by chronic hemolysis and hemoglobinuria) leads to progressively intense hemosiderin deposition (hemosiderosis) in the proximal convoluted tubules and subsequently to tubular damage. Hemosiderin cannot be excreted by filtration due to its high molecular weight and its ability to accumulate in the cells of the tubular epithelium. It can be excreted only when those cells are shed into the urine. In this case, blue-staining hemosiderin granules can be detected when Prussian blue stain is applied in the urine sediment. Renal impairment, however, may not be immediately perceived [Citation31].
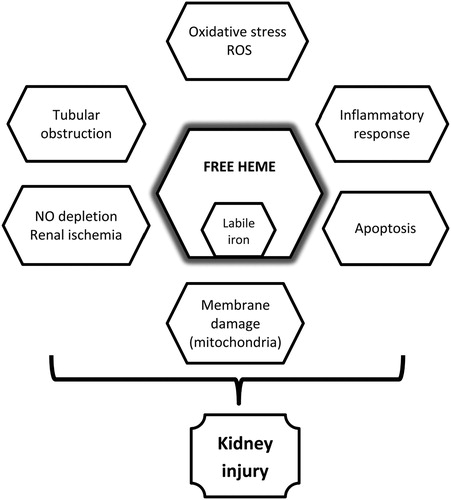
In addition, recurrent microvascular thrombosis can lead to ischemic necrosis of the renal cortex, with subsequent loss of the kidney’s ability to concentrate urine (specific gravity > 1025) and a decreased creatinine clearance. CRF is, also, associated with microinfarction on the medulla, tubular atrophy, and interstitial fibrosis and is characterized by impaired GFR [Citation32]. summarizes mechanisms of renal involvement in PNH.
Renal protection against free heme
Free heme removal
A number of mechanisms have been proposed in relation to salvaging of hemoglobin iron and to kidney’s protection against free heme’s toxic effects. summarizes the protective and damaging effects of free hemoglobin to the kidneys.
Table 3. Protective and damaging effects associated with free hemoglobin.
In classical PNH, red cell intravascular hemolysis is continuous and appears in various degrees. When hemoglobin is free in the plasma, it exists mostly as alpha/beta dimers that rapidly complex to haptoglobin (Hp), a liver-produced plasma protein. This is the first mechanism of hemoglobin iron salvage. By binding to Hp, hemoglobin avoids filtration at the glomerulus and the release of its heme moiety is prevented [Citation33]. More specifically, the haptoglobin–hemoglobin complexes are too large to be filtered by the glomerulus, so they are carried to the liver, where they are recognized by the CD163 receptors (these are membrane proteins expressed on monocyte/macrophage surfaces and hepatocytes) [Citation34–36]. The binding of the hemoglobin molecules to CD163 receptors results in the neutralization of free hemoglobin’s toxic actions. Hemoglobin, also, has the ability to up-regulate CD163’s expression. The uptake of hemoglobin by CD163 receptors not only attenuates the toxic effects of cell-free hemoglobin, but it also induces several anti-inflammatory responses, including interleukin-10 release and heme oxygenase-1 synthesis (HO-1) [Citation15]. In cases where hemolysis is accelerated, Hp molecules are depleted, due to the liver’s inability to increase Hp’s production as a response to the increased hemolysis: Hp molecules are typically adequate to salvage only a normal amount of plasma hemoglobin.
Figure 1. Schematic representation of mechanisms of renal involvement in PNH.
Once the scavenging capacity of Hp is exhausted, the excess of free hemoglobin undergoes oxidation to become methemoglobin: the ferrous iron (Fe+2) of the heme is converted to ferric iron (Fe+3). As a result metheme’s molecules are separated from globulin and bind to hemopexin, another protein produced by the liver [Citation37].
The binding of metheme to hemopexin is the second mechanism, by which kidneys are protected against the loss of iron in cases of increased hemolysis. Hemopexin is a plasma glycoprotein, consisted of a peptide chain of 439 amino acids and expressed mainly in the liver. It possesses the highest affinity for heme than any other protein with the ability to bind to heme [Citation38].
The hemopexin–metheme complexes can bound to specific hepatic receptors (such as the low-density lipoprotein receptor-related protein 1: LRP1-CD91). They are then endocytosed and degraded in the liver, by a series of enzymatic steps that result in the releasing of the iron. In contrast to Hp, hemopexin is recycled by hepatocytes and released in circulation, so a single hemopexin molecule collects and salvages more than one free metheme molecule. The production of hemopexin is constant and therefore, its levels do not fall dramatically during periods when there is increased intravascular hemolysis.
A third protection mechanism against hemoglobin’s and heme’s toxic actions is the binding of metheme to albumin. Heme binds to albumin only when hemopexin is saturated. Metheme–albumin complexes further reduce the loss of hemoglobin through urine. These complexes are probably reversible and metheme molecules soon transfer to hemopexin, due to hemopexin’s higher binding affinity to metheme than albumin’s.
There is a number of other circulating proteins that can also scavenge free heme in plasma [Citation38], such as alpha-1-microglobulin [Citation36] and high- and low-density lipoproteins [Citation39]. The binding of alpha-1-microglobulin to heme can neutralize its pro-oxidant effects, suggesting that alpha-1-microglobulin acts as a heme scavenger in circulation. Nevertheless, it is still unclear if alpha-1-microglobulin holds a protective role against heme’s toxicity under pathophysiologic conditions.
Iron salvaging is performed by macrophages of the reticuloendothelial system, where the remaining protoporphyrin is also converted to bilirubin [Citation37]. The released ferrous iron is returned to the circulation mainly via ferroportin. Liberated iron is then oxidized to trivalent iron (by circulating ceruloplasmin), detached from transferrin and returned to maturing precursor cells of the erythroid lineage to participate in hemoglobin synthesis.
When excessive hemolysis occurs, it leads to the saturation and depletion of hemoglobin removal systems and, subsequently, to the buildup of hemoglobin and heme in plasma. The excess amounts of hemoglobin and heme iron will then be filtered into the urine [Citation40].
Reabsorption of hemoglobin dimers
As mentioned above, free circulating hemoglobin is divided into dimers, thus reducing its size by half. Free dimers with low molecular weight, are rapidly filtered by the glomerulus and reabsorbed by the epithelial cells of the proximal tubules. When inside the renal tubular cells, the iron is separated from the protoporphyrin, where it is stored as ferritin or hemosiderin.
The reabsorption of hemoglobin dimers in the proximal convoluted tubular cells is intermediated by the intracellular receptors megalin and cubilin. These receptors are expressed at the apical membrane of the proximal tubules. Megalin and cubilin are multiligand endocytic receptors and their primary function is to reabsorb small molecules that pass through the glomerular filtration barrier. As it has been previously demonstrated, hemoglobin is one of their ligands [Citation41,Citation42].
Megalin was identified in 1982. It is a member of the superfamily of low-density lipoprotein receptors LRP-2. It is a transmembrane protein, with a molecular weight of 600 kDa and consists of four repeating cysteine-rich regions, which are probably involved in the binding of small molecules [Citation43]. The cytoplasmic domain of megalin regulates receptor trafficking and endocytosis.
Cubilin is a 460-kDa glycosylated extracellular protein. It is believed that its molecular structure is responsible for the ligand binding and the interaction with other proteins with the purposes of membrane localization and endocytosis. Its intracellular circulation is complex and poorly understood. For its correct membrane localization, cubilin depends on amnionless (AMN), a 38–50-kDa single transmembrane protein. Since AMN is responsible for the correct position of cubilin in the membrane, in cases where AMN is not present, cubilin is retained in intracellular structures. Similarly, AMN is retained intracellularly if cubilin is absent. In the proximal tubule, cubulin is believed to interact with megalin, forming a multireceptor complex. Cubilin does not have an endocytosis signaling sequence. It appears that megalin is responsible for the internalization of cubilin and its ligands, in addition to internalizing its own ligands. These two receptors are structurally different, but their main function is to help small molecules to pass through the barrier of the glomerular filtration.
Hemoglobin is one of the molecules that bind to these receptors [Citation42,Citation44]. Following receptor-mediated endocytosis via apical coated pits, the complexes accumulate in lysosomes for the degradation of their proteins, whereas the receptors (including AMN) recycle to the apical plasma membrane via dense apical tubules [Citation45]. Whether megalin and cubulin receptors are constitutively associated in the plasma membrane and remain associated during recycling in dense apical tubules is not known.
Defense mechanisms against kidney damage in PNH
The exact role of the kidneys in salvaging the iron and returning it to the circulation is uncertain. Although carrier proteins, like divalent metal transporter 1 and ferroportin, have been identified in mice kidneys, their localization, function, and regulation remain unclear [Citation37].
In a case report of acute renal failure due to PNH, a marked infiltration of CD163-expressing macrophages was observed in the renal interstitium in areas with iron deposits, suggesting a possible role of this hemoglobin scavenger receptor in response to massive hemolysis. The uptake of hemoglobin by CD163 receptors not only attenuates the toxic effects of cell-free hemoglobin, but it also induces several anti-inflammatory responses, including interleukin-10 release and HO-1 synthesis [Citation15].
Recently, a hypothesis has been proposed that hepcidin may possess protective effects against iron-mediated AKI. It has been suggested that circulating filtered hepcidin (reabsorbed in the proximal tubules via megalin), along with hepcidin synthesized in the distal parts of the nephron play a role in the defense against iron-mediated kidney injury [Citation46].
It is known that excess intracellular heme is cytotoxic in the epithelial cells of the proximal convoluted tubules. Its degradation is mediated by HO. HO-1 is the inducible isoform of HO and its expression increases rapidly on exposure to heme-containing proteins. HO-1, a 32 kDa protein, was discovered in 1989. Studies have shown that the expression of HO-1 enzyme is induced by kidney cells as an adaptive response, in order to protect these cells against the toxic effects of heme. HO-1 knockout rat models have shown accelerated kidney injury upon exposure to heme proteins. In the absence of HO-1, excess free heme provokes cell damage [Citation47].
HO-1 is a unique regulator which participates in the catabolism of heme, by leading to the generation of equimolar amounts of iron, biliverdin and CO, therefore decreasing the production of reactive oxygen species (ROS). HO-1, CO and bilirubin are protective molecules with anti-oxidant and anti-inflammatory properties. Biliverdin reductase converts biliverdin to bilirubin within the biliverdin/bilirubin redox cycle, allowing the neutralization of ROS. The released iron is stored as ferritin in epithelial cells of proximal convoluted tubules and is detected as hemosiderin due to the apoptosis of tubular cells on kidney histological sections of PNH patients [Citation48].
The HO gene is located in the long arm of chromosome 22. The transcription of HO-1 gene (hmox1 gene) is controlled by the transcriptional factor Bach1. Bach1 is a heterodimer complex (in conjunction with Maf proteins), which binds to stress response elements (StRE) of the HO gene. The binding of Bach1 to StRE leads to the suppression of HO-1’s expression. Under homeostatic conditions, the hmox1 gene is suppressed by the Bach1 factor. A simple model for the induction of HO-1’s expression has been proposed: Bach1 seems to bind to heme with high affinity (at least in vitro [Citation37]), which alters Bach1’s DNA-binding properties resulting in the reduced affinity of Bach 1 for the StRE. When Bach1 factor is missing, expression of HO-1 is induced by the Nrf2 protein (which belongs in the same family of proteins with Bach1) which competes with the Bach1 protein for binding to StRE.
In PNH patients, a strong induction of HO-1 has been observed in renal tubular cells. Such induction represents an adaptive response that enables the degradation of heme. The same increase of HO-1’s expression has also been observed in an experimental rat model, in which repeated intramuscular administration of a hypertonic glycerol solution induced hemolysis [Citation49].
Heme cytotoxicity
When hemoglobin dimers are reabsorbed in the proximal convoluted tubules, their degradation starts resulting in the release of heme. The excess of free heme is highly toxic as it releases the oxidized iron. The liberated iron, not stored as ferritin, is particularly toxic since its divalent form (Fe+2) induces not only membrane lipid peroxidation, but also cytotoxicity through the release of lactate dehydrogenase (LDH). In the cytoplasm, the iron induces the production of ROS (Fenton reaction: Fe2+ + H2O2 → Fe3+ + OH· + OH−). ROS destroy lipid membranes, proteins and nucleic acids [Citation50] and induce apoptosis of epithelial tubular cells [Citation51]. Furthermore, iron causes inflammation and damage to the kidney which is manifested as fibrosis [Citation52].
Heme, being a lipophilic molecule, possesses the ability of intercalating in the cell membrane, impairing lipid bilayers and organelles (such as mitochondria and nuclei) and destabilizing the cytoskeleton. The iron catalyzes the oxidation of cell membrane components causing oxidation of the ingredients and forming peroxide radicals with toxic effects [Citation53]. Unsaturated fatty acid residues of phospholipids, which are characterized by high oxidation sensitivity, are particularly vulnerable to the effects of ROS. Due to the presence of large amounts of superoxide radicals, membrane lesions appear [Citation54].
Free heme may also provoke inflammation and, thus, cause kidney damage. In epithelial cells of the proximal tubule, heme results in the activation of transcription factor Nuclear Factor kappa B (NF-kappa B). NF-kappa B plays a central role in controlling inflammation. As a result of NF-kappa B factor’s activation, inflammation could develop in the intermediate space between the tubules (interstitial nephritis). The degradation of I kappa B proteins may also provoke NF-kappa B factor’s activation. I kappa B proteins are a family of proteins which have an amino-terminal region, followed by six or more repeating ankyrin regions (ankyrin repeats) and a PEST [peptide sequence that is rich in proline (P), glutamic acid (E), serine (S), and threonine (T)] domain near the carboxylic end. NF-kappa B factor’s activation occurs mainly via the activation of the I kappa B kinase, which phosphorylates two serine residues. When I kappa B proteins degrade, the NF-kappa B enters the nucleus and induces the expression of specific genes mentioned above [Citation55]. Therefore, the inflammatory process is induced by the activation of NF-kappa B, the increased oxidative stress, the production of proinflammatory cytokines, adhesion molecules and chemotactic factors [Citation56].
Studies in an experimental rat model demonstrated that HO-1’s up-regulation in the injured kidney may assist in down-regulating cellular infiltration, by suppressing NF-kappa B-driven activation of the chemokine MCP-1 (Monocyte Chemoattractant Protein-1) [Citation57]. It has been shown that the MCP-1 protein leads to the release of IL-6 (Interleukin-6) and also induces the expression of the ICAM-1 (Intercellular Adhesion Molecule-1) in tubular epithelial cells via mechanisms that depend on Gi-proteins, protein kinase C and the intracellular calcium concentration.
Alongside the inflammation, endothelial cells are stimulated to form new vessels in the lesion area. Fibroblasts are activated by IL-13 and TGF-beta (Transforming Growth Factor-beta), in order to produce extracellular matrix and contribute to the healing process. Finally, epithelial cells proliferate and the regeneration of the damaged tissue is then completed [Citation58]. When the inflammatory reactions become chronic, continuous activation of the fibroblasts is observed, resulting in uncontrolled production and accumulation of extracellular matrix, which eventually leads to the formation of fibrosis.
Nitric oxide (NO) is the main regulator of vascular tone. Normal levels of NO lead to the dilatation of blood vessels and to increased blood flow [Citation59]. During intense hemolysis, erythrocytes release arginase in plasma. Arginase converts l-arginine into ornithine, thus reducing the primary source of NO production and synthesis. The accumulation of free hemoglobin in the plasma dramatically reduces the levels of NO (NO scavenging). NO reacts irreversibly with free hemoglobin and produces NO3 and methemoglobin. The reduction of NO, due to its binding with heme, leads to vasoconstriction, which is most pronounced in the kidney medulla. The above phenomenon results in a reduction of blood flow to the kidneys causing severe and prolonged hypoxia which ultimately leads to tubular necrosis [Citation60]. The reduction of NO may also result in a diminished activation of the soluble cytosolic guanylate cyclase (sGC) and decreased cGMP (cyclic Guanosine Monophosphate) levels, that disrupt the regulation of smooth muscle tone causing dystonias (with multiple subsequent symptoms), like systemic and pulmonary hypertension, dysphagia, esophageal spasm, retrosternal pain, atypical abdominal pain and erectile dysfunction (mainly in hemolytic crisis). Decreased cGMP levels can also lead to platelets activation and aggregation, promoting clot formation [Citation61].
Heme can also induce apoptosis of the proximal convoluted tubules’ epithelial cells. As mentioned above, HO-1 is a major regulator of apoptosis in several models portraying renal injury, including renal ischemia and reperfusion [Citation47].
Management of kidney injury in PNH
Successful management of kidney injury in PNH patients requires increased awareness by treating physicians. A precise definition and an accurate estimation of the staging of the kidney injury will also help health care professionals to better collaborate with consultant nephrologists when needed.
AKI is considered an emergency in PNH. AKI’s supportive management involves fluid resuscitation, supportive care and hemodialysis when needed [Citation1]. In cases of acute hemolysis, transfusions and glucocorticoid administrations are often necessary. In PNH patients, it is important for renal vein thrombosis to be ruled out using ultrasound scans, even though no episodes of AKI due to renal vein thrombosis have been reported so far [Citation11–18,Citation20]. In addition, it is recommended that acute thrombotic episodes should be managed with anticoagulation treatment and, in some cases, thrombolytic therapy.
In order for CRF to be prevented, hydration and a close consultation with nephrologists are essential. When complement inhibition is not available, emphasis should be given on supportive measures, such as iron or folic acid supplementation, periodic transfusions and exogenous erythropoietin administration. There are limited data on renal replacement therapy in PNH patients, while one older study reported successful renal transplantation in a PNH patient with end-stage renal disease [Citation62].
Effects of complement inhibition on renal function
Terminal complement inhibition with eculizumab is the treatment of choice for PNH patients with severe hemolytic anemia or thrombosis [Citation15,Citation16]. Eculizumab’s long-term effects on renal function have been investigated in several studies. According to available data, eculizumab administration in patients with renal dysfunction was safe and well tolerated. Hillmen et al. analyzed data from three parent clinical trials where eculizumab was administered in patients with kidney injury. Eculizumab significantly reduced intravascular hemolysis in patients with CKD stages 3–5. Similar results were found in CKD stages 1–2. Moreover, patients presented with CKD stages 1–2 at baseline were six times more likely to improve than worsen with eculizumab treatment, whereas the improvement in CKD stages 3–5 did not prove to be significant. Nevertheless, all CKD stages demonstrated an increased likelihood of improved renal function following long-term eculizumab treatment. In addition, major clinical kidney event rates were reduced to 2.10 events per 100 patient–years with eculizumab treatment compared to 4.22 events per 100 patient–years before eculizumab treatment. The analysis of a Japanese PNH population also showed that 49 out of 196 patients significantly increased their eGFR rate during eculizumab treatment, whereas the majority of patients (71.9%) maintained the same range of eGFR. In that population, however, approximately 3.5% of the patients presented with a polymorphism in C5 (c. 2654 G → A) that prevented the binding of eculizumab to C5, leading to eculizumab resistance [Citation63]. In cases of Fanconi syndrome, proximal tubular dysfunction persisted despite eculizumab treatment, suggesting that early treatment initiation is necessary. Taking into consideration the available data, potential positive effects of eculizumab seem to be time-dependent and therefore, early administration may lead to better results. In line with this, experts have suggested that meningococcal vaccination may follow initial eculizumab administration, recognizing that reduction of thrombotic and AKI incidents in PNH is an urgent clinical need [Citation1].
In terms of pathophysiology, the beneficial effects of eculizumab may be attributed to the prevention of intravascular hemolysis and, also, to the inhibition of hemosiderin deposition in the kidneys. Although eculizumab is highly effective in reducing intravascular hemolysis, breakthrough hemolysis may still occur in PNH patients following infections, surgery, pregnancy, and other conditions that increase complement activation over baseline.
Conclusion and future perspectives
Understanding the difficult but at the same time, impressive mechanisms behind PNH’s presentation remain a challenge not only for hematologists, but also for other specialists involved in PNH diagnosis and management. PNH’s pathophysiology is based on complement-mediated effects causing severe consequences in PNH patients. Kidney injury in PNH is an underdiagnosed and poorly investigated clinical feature that affects a significant portion of PNH patients. In order for those patients to be managed properly, it is essential for physicians to recognize the early signs and symptoms of acute and chronic renal insufficiency and initiate the necessary supportive therapy. It is also important for periodic re-evaluations of PNH-specific treatments to be performed in the course of the disease.
Complement inhibition with eculizumab has been reported to be safe and effective, especially when it is initiated early in the presence of renal dysfunction. Eculizumab should be administered intravenously every two weeks and must be continued indefinitely. Excluding cases of breakthrough intravascular hemolysis, approximately 20% of PNH patients on eculizumab continue to have symptomatic extravascular hemolysis due to C3 fragment accumulation [Citation1]. For those patients, novel complement inhibitors that specifically target alternative complement pathway activation may be useful. However, their efficacy and safety in patients with renal involvement remain to be investigated.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Hill A, DeZern AE, Kinoshita T, et al. Paroxysmal nocturnal hemoglobinuria. Nat Rev Dis Primers. 2017;3:17028. doi: 10.1038/nrdp.2017.28
- Hillmen P, Elebute M, Kelly R, et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010;85(8):553–559. doi: 10.1002/ajh.21757
- Nishimura J, Kanakura Y, Ware RE, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore). 2004;83(3):193–207. doi: 10.1097/01.md.0000126763.68170.46
- Ninomiya H, Obara N, Chiba S, et al. Interim analysis of post-marketing surveillance of eculizumab for paroxysmal nocturnal hemoglobinuria in Japan. Int J Hematol. 2016;104(5):548–558. doi: 10.1007/s12185-016-2065-4
- Munoz-Linares C, Ojeda E, Fores R, et al. Paroxysmal nocturnal hemoglobinuria: a single Spanish center’s experience over the last 40 yr. Eur J Hematol. 2014;93(4):309–319. doi: 10.1111/ejh.12346
- Jang JH, Kim JS, Yoon SS, et al. Predictive factors of mortality in population of patients with paroxysmal nocturnal hemoglobinuria (PNH): results from a Korean PNH registry. J Korean Med Sci. 2016;31(2):214–221. doi: 10.3346/jkms.2016.31.2.214
- Bellomo R, Ronco C, Kellum JA, et al. Acute renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the second international consensus conference of the acute dialysis quality initiative (ADQI) group. Crit Care. 2004;8(4):R204–R212. doi: 10.1186/cc2872
- Mehta RL, Kellum JA, Shah SV, et al. Acute kidney injury network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. http://ccforum.com/content/11/2/R31 doi: 10.1186/cc5713
- Group KDIGOKAKIW. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:1–138. doi: 10.1038/kisup.2012.1
- Thomas ME, Blaine C, Dawnay A, et al. The definition of acute kidney injury and its use in practice. Kidney Int. 2015;87(1):62–73. doi: 10.1038/ki.2014.328
- Nair RK, Khaira A, Sharma A, et al. Spectrum of renal involvement in paroxysmal nocturnal hemoglobinuria: report of three cases and a brief review of the literature. Int Urol Nephrol. 2008;40(2):471–475. doi: 10.1007/s11255-008-9356-5
- Balwani MR, Kute VB, Shah PR, et al. Manifestation of paroxysmal nocturnal hemoglobinuria as repeated acute kidney injury. J Nephropharmacol. 2016;5(2):116–118.
- Chen SC, Hung CC, Hsu CP, et al. Recurrent acute renal failure in a patient with aplastic anemia-paroxysmal nocturnal hemoglobinuria syndrome: a case report. Kaohsiung J Med Sci. 2007;23(11):579–583.
- Kirkizlar O, Kendir M, Karaali Z, et al. Acute renal failure in a patient with severe hemolysis. Int Urol Nephrol. 2007;39(2):651–654. doi: 10.1007/s11255-006-9096-3
- Ballarin J, Arce Y, Torra Balcells R, et al. Acute renal failure associated to paroxysmal nocturnal hemoglobinuria leads to intratubular hemosiderin accumulation and CD163 expression. Nephrol Dial Transplant. 2011;26(10):3408–3411. doi: 10.1093/ndt/gfr391
- Hussain S, Qureshi A, Kazi J. Renal involvement in paroxysmal nocturnal hemoglobinuria. Nephron Clin Pract. 2013;123(1–2):28–35. doi: 10.1159/000351345
- Chow KM, Lai FM, Wang AY, et al. Reversible renal failure in paroxysmal nocturnal hemoglobinuria. Am J Kidney Dis. 2001;37(2):E17. doi: 10.1053/ajkd.2001.21361
- Jose MD, Lynn KL. Acute renal failure in a patient with paroxysmal nocturnal hemoglobinuria. Clin Nephrol. 2001;56(2):172–174.
- Ram R, Adiraju KP, Gudithi S, et al. Renal manifestations in paroxysmal nocturnal hemoglobinuria. Indian J Nephrol. 2017;27(4):289–293. doi: 10.4103/0971-4065.205201
- Hillmen P, Muus P, Duhrsen U, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;110(12):4123–4128. doi: 10.1182/blood-2007-06-095646
- Clark DA, Butler SA, Braren V, et al. The kidneys in paroxysmal nocturnal hemoglobinuria. Blood. 1981;57(1):83–89.
- Rimola J, Martin J, Puig J, et al. The kidney in paroxysmal nocturnal hemoglobinuria: MRI findings. Br J Radiol. 2004;77(923):953–956. doi: 10.1259/bjr/51760601
- Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825–830. doi: 10.7326/0003-4819-158-11-201306040-00007
- Riley AL, Ryan LM, Roth DA. Renal proximal tubular dysfunction and paroxysmal nocturnal hemoglobinuria. Am J Med. 1977;62(1):125–129. doi: 10.1016/0002-9343(77)90357-6
- Saito T, Furyama T, Onodera S, et al. Renal impairment in patients with paroxysmal nocturnal hemoglobinuria. Tohoku J Exp Med. 1975;116(3):267–275. doi: 10.1620/tjem.116.267
- Hsiao PJ, Wang SC, Wen MC, et al. Fanconi syndrome and CKD in a patient with paroxysmal nocturnal hemoglobinuria and hemosiderosis. Am J Kidney Dis. 2010;55(1):e1–e5. doi: 10.1053/j.ajkd.2009.07.022
- Economou M, Printza N, Teli A, et al. Renal dysfunction in patients with beta-thalassemia major receiving iron chelation therapy either with deferoxamine and deferiprone or with deferasirox. Acta Haematol. 2010;123(3):148–152. doi: 10.1159/000287238
- Vahdani K, Makrygiannis G, Kaneshyogan H, et al. Bilateral central serous retinopathy in a patient with paroxysmal nocturnal hemoglobinuria treated with deferoxamine. Eur J Ophthalmol. 2016;26(6):e152–e154. doi: 10.5301/ejo.5000840
- Wijewickrama ES, Gooneratne L, De Silva C, et al. Acute tubular necrosis in a patient with paroxysmal nocturnal hemoglobinuria. Saudi J Kidney Dis Transpl. 2013;24(1):105–108. doi: 10.4103/1319-2442.106302
- Khalighi MA, Henriksen KJ, Chang A, et al. Intratubular hemoglobin casts in hemolysis-associated acute kidney injury. Am J Kidney Dis. 2015;65(2):337–341. doi: 10.1053/j.ajkd.2014.08.020
- Zachee P, Henckens M, Van Damme B, et al. Chronic renal failure due to renal hemosiderosis in a patient with paroxysmal nocturnal hemoglobinuria. Clin Nephrol. 1993;39(1):28–31.
- Al-Harbi A, Alfurayh O, Sobh M, et al. Paroxysmal nocturnal hemoglobinuria and renal failure. Saudi J Kidney Dis Transpl. 1998;9(2):147–151.
- Melamed-Frank M, Lache O, Enav BI, et al. Structure-function analysis of the antioxidant properties of haptoglobin. Blood. 2001;98(13):3693–3698. doi: 10.1182/blood.V98.13.3693
- Nielsen MJ, Moestrup SK. Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood. 2009;114(4):764–771. doi: 10.1182/blood-2009-01-198309
- Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the hemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi: 10.1038/35051594
- Allhorn M, Berggard T, Nordberg J, et al. Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood. 2002;99(6):1894–1901. doi: 10.1182/blood.V99.6.1894
- Doig K. Introduction to increased destruction of erythrocytes. In: Keohane Elaine, Smith Larry, Walenga Jeanine, editors. Rodak’s hematology: clinical principles and applications. 5th ed. New Jersey: Elsevier Saunders; 2016. p. 353–354.
- Bunn HF, Jandl JH. Exchange of heme among hemoglobin molecules. Proc Natl Acad Sci USA. 1966;56(3):974–978. doi: 10.1073/pnas.56.3.974
- Miller YI, Shaklai N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim Biophys Acta. 1999;1454(2):153–164. doi: 10.1016/S0925-4439(99)00027-7
- Lathem W. The renal excretion of hemoglobin: regulatory mechanisms and the differential excretion of free and protein-bound hemoglobin. J Clin Invest. 1959;38(4):652–658. doi: 10.1172/JCI103843
- Christensen EI, Birn H. Renal handling of albumin in normal rat. Kidney Int. 2000;57(3):1207–1209. doi: 10.1046/j.1523-1755.2000.00952.x
- Gburek J, Verroust PJ, Willnow TE, et al. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J Am Soc Nephrol. 2002;13(2):423–430.
- Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 2016;89(1):58–67. doi: 10.1016/j.kint.2015.11.007
- Christensen EI, Birn H. Megalin and cubilin: synergistic endocytic receptors in renal proximal tubule. Am J Physiol Renal Physiol. 2001;280(4):F562–F573. doi: 10.1152/ajprenal.2001.280.4.F562
- Christensen EI, Birn H, Storm T, et al. Endocytic receptors in the renal proximal tubule. Physiology (Bethesda). 2012;27(4):223–236.
- van Swelm RP, Wetzels JF, Verweij VG, et al. Renal handling of circulating and renal-synthesized hepcidin and its protective effects against hemoglobin-mediated kidney injury. J Am Soc Nephrol. 2016;27(9):2720–2732.
- Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol. 2007;18(2):414–420. doi: 10.1681/ASN.2006080894
- Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70(3):432–443. doi: 10.1038/sj.ki.5001565
- Miller YI, Chang MK, Funk CD, et al. 12/15-lipoxygenase translocation enhances site-specific actin polymerization in macrophages phagocytosing apoptotic cells. J Biol Chem. 2001;276(22):19431–19439. doi: 10.1074/jbc.M011276200
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202(2):199–211. doi: 10.1016/j.taap.2004.06.021
- Zager RA, Foerder CA. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J Clin Invest. 1992;89(3):989–995. doi: 10.1172/JCI115682
- Yuste C, Gutierrez E, Sevillano AM, et al. Pathogenesis of glomerular hematuria. World J Nephrol. 2015;4(2):185–195. doi: 10.5527/wjn.v4.i2.185
- Nath KA. Preservation of the kidney by carbon monoxide: a black swan phenomenon. Kidney Int. 2008;74(8):989–991. doi: 10.1038/ki.2008.423
- Marnett LJ. Lipid peroxidation-DNA damage by malondialdehyde. Mutat Res. 1999;424(1–2):83–95. doi: 10.1016/S0027-5107(99)00010-X
- Nelson DE, Ihekwaba AE, Elliott M, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306(5696):704–708. doi: 10.1126/science.1099962
- Bauge C, Beauchef G, Leclercq S, et al. NFkappab mediates IL-1beta-induced down-regulation of TbetaRII through the modulation of Sp3 expression. J Cell Mol Med. 2008;12(5A):1754–1766. doi: 10.1111/j.1582-4934.2007.00173.x
- Nath KA, Vercellotti GM, Grande JP, et al. Heme protein-induced chronic renal inflammation: suppressive effect of induced heme oxygenase-1. Kidney Int. 2001;59(1):106–117. doi: 10.1046/j.1523-1755.2001.00471.x
- Grande MT, Perez-Barriocanal F, Lopez-Novoa JM. Role of inflammation in tubulo-interstitial damage associated to obstructive nephropathy. J Inflamm (Lond). 2010;7:19. doi: 10.1186/1476-9255-7-19
- Denninger JW, Marletta MA. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim Biophys Acta. 1999;1411(2–3):334–350. doi: 10.1016/S0005-2728(99)00024-9
- Heyman SN, Fuchs S, Brezis M. The role of medullary ischemia in acute renal failure. New Horiz. 1995;3(4):597–607.
- Rother RP, Bell L, Hillmen P, et al. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. J Am Med Assoc. 2005;293(13):1653–1662. doi: 10.1001/jama.293.13.1653
- Vanwalleghem J, Zachee P, Kuypers D, et al. Renal transplantation for end-stage renal disease due to paroxysmal nocturnal hemoglobinuria. Nephrol Dial Transplant. 1998;13(12):3250–3252. doi: 10.1093/ndt/13.12.3250
- Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632–639. doi: 10.1056/NEJMoa1311084