
ABSTRACT
Objectives: Serum chitotriosidase activity (CHIT1) is a biomarker of macrophage activation with an important role in inflammation-induced tissue remodeling and fibrosis. Macrophages have been described to play a crucial role in regulating pathological erythropoiesis in polycythemia vera (PV). The aim of this study was to evaluate CHIT1 in patients diagnosed with Philadelphia-negative myeloproliferative neoplasms (MPNs).
Methods: Using fluorometric assay, we measured CHIT1 in 28 PV, 27 essential thrombocythemia (ET), 17 primary myelofibrosis (PMF), 19 patients with secondary myelofibrosis and in 25 healthy controls.
Results: CHIT1 was significantly higher in PV (p < .001) and post-PV myelofibrosis (MF) transformation (post-PV MF) (p = .020), but not in ET (p = .080), post-ET MF transformation (p = .086), and PMF patients (p = .287), when compared to healthy controls. CHIT1 in PV was positively correlated with hemoglobin (p = .026), hematocrit (p = .012), absolute basophil count (p = .030) and the presence of reticulin fibrosis in the bone marrow (p = .023).
Discussion: A positive correlation between CHIT1 and these distinct laboratory PV features might imply macrophages closely related to clonal erythropoiesis as cells of CHIT1 origin. In addition, a positive association between CHIT1 and reticulin fibrosis might indicate its potential role in PV progression.
Conclusion: CHIT1 might be considered as a circulating biomarker in PV. Additional studies are needed to clarify the role of CHIT1 in promoting disease progression and bone marrow fibrosis in PV.
Introduction
Classical Philadelphia-negative myeloproliferative neoplasms (MPNs) encompass polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). These neoplasms are characterized by overproduction of erythroid, megakaryocytic and granulocytic cells, bone marrow hypercellularity and fibrosis, frequent splenomegaly and an increased risk of bleeding and thrombosis. Constitutive activation of the JAK-STAT signaling pathway is a cardinal feature in these neoplasms. Three types of mutually exclusive driver mutations are associated with MPNs: activating mutations in the Janus kinase 2 (JAK2) [Citation1–3] or thrombopoietin receptor (MPL) genes [Citation4,Citation5], or alterations in the calreticulin (CALR) gene [Citation6,Citation7]. Activating mutations in the JAK2 gene are most frequent and present in all MPN subtypes, while activating mutations in the MPL gene and alterations of CALR are detected only in ET and PMF. So-called ‘triple negative’ cases, which carry neither of the mutations, account for a small percentage of MPN patients [Citation8].
MPNs are accompanied by chronic inflammation [Citation9,Citation10] that not only causes many of the debilitating symptoms associated with the disease [Citation11,Citation12] but also drives the selective expansion of the neoplastic clone [Citation13]. MPN patients show elevated serum levels of various proinflammatory cytokines including interleukin (IL)-1, IL-6, IL-8, IL-11, IL-17, tumor necrosis factor-alpha (TNF-α) and transforming growth factor-beta (TGF-β) [Citation14–16]. Inflammation might also be directly responsible for some pathologic features of the disease, such as bone marrow fibrosis [Citation17]. Moreover, JAK inhibitors were shown to efficiently decrease the production and signaling of major inflammatory cytokines, leading to significant reduction of spleen size and other inflammation-linked clinical symptoms in PMF [Citation18–21].
A potentially fatal disease complication in PV and ET is transformation to secondary myelofibrosis (SMF): post-polycythemia vera myelofibrosis (post-PV MF) and post-essential thrombocythemia myelofibrosis (post-ET MF), respectively. The underlying mechanism for transformation to MF remains unclear and is likely multifactorial. Bone marrow fibrosis is the result of a complex and poorly understood interaction between megakaryocytes, fibroblasts, endothelial cells, inflammatory cytokines and marrow stroma. Preclinical studies support a pathologenetic role of TGF-β. This protein is overexpressed by megakaryocytes in MF and the TGF-β signature is upregulated and has been targeted in MF animal models [Citation17,Citation22].
Macrophages have been confirmed to play a crucial role in regulating normal and pathological erythropoiesis. Erythroid cells develop in a specialized niche, the ‘erythroblastic island’, composed of a central macrophage surrounded by erythroblasts. Resident bone marrow macrophages are important for ‘erythroblastic island’ formation and have been shown to regulate erythropoiesis by promoting proliferation and survival of mature erythroblasts. These effects are dependent on erythroblast-macrophage contact. Once the erythrocytes have matured, they stay in the circulation for about 120 days. At the end of their lifespan, they are cleared by macrophages residing in the spleen and liver [Citation23–29].
Interesting results came from a study that investigated whether macrophages might be involved in PV pathophysiology using transgenic JAK2-positive mice. These animals exhibit PV-like phenotype, including elevated hematocrit levels, erythrocytosis and retikulocytosis. Clodronate-induced depletion of macrophages in these mice reversed those PV features, validating their role in contributing to the pathophysiology of the disease. Furthermore, proliferation of erythroid lineage cells from human PV patients was increased when they were co-cultured with macrophages compared to erythroid precursors cultured alone. Authors therefore suggested a new model for PV progression, where the JAK2 mutation functions as a primer for the PV phenotype, but the bone marrow microenvironment and the macrophage niche is required for the full manifestation of the disease in vivo [Citation30].
Chitotriosidase enzyme (CHIT1 or chitinase-1), belongs to a family of various chitinase and chitinase-like proteins, and is one of the most abundantly secreted proteins, being mainly produced by activated macrophages, granulocytes and epithelial cells. Its natural substrate chitin is the second most abundant polysaccharide in nature after cellulose. Chitin is a polymer of N-acetylglucosamine and the main component of the cell walls of fungi and protozoa, egg shells of helminthes, and the exoskeletons of arthropods and insects. Although mammals do not contain chitin, CHIT1 plays a pivotal role in the defense against chitin-containing microorganisms such as fungi, insects and some bacteria [Citation31–37]. Macrophages release CHIT1 after toll-like receptor stimulation and after stimulation with lipopolysaccharide, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-gamma and TNF-α [Citation38,Citation39].
In everyday clinical practice, CHIT1 is used as a circulating biomarker for monitoring the efficacy of various therapeutic approaches in Gaucher’s disease where is considered to be a surrogate serum biomarker of the tissue lipid-laden macrophages known as Gaucher cells [Citation40,Citation41]. Elevated CHIT1 was also observed in various other disorders characterized by nonspecific macrophage activation and expansion, such as β-thalassemia [Citation42,Citation43], Plasmodium falciparum malaria [Citation44], sarcoidosis [Citation45–47] and Wegener’s granulomatosis [Citation48].
On the other hand, it has been proposed that CHIT1, through inducing TGF-β signaling, might also act as a promoter of tissue remodeling and fibrosis, suggesting a role in inflammation-mediated tissue repair processes. CHIT1 released by activated Kupffer cells (liver macrophages) was shown to induce hepatic fibrosis and cirrhosis [Citation49]. Similarly, CHIT1 overexpression from alveolar macrophages in chronic obstructive pulmonary disease contributes to airway obstruction, tissue remodeling and was implicated in disease onset and progression [Citation50]. CHIT1 was also involved in the induction of fibrosis in the murine model of interstitial lung disease where bleomycin-induced pulmonary fibrosis was significantly reduced in CHIT1 -/- mice when compared to CHIT1 overexpressing transgenic mice [Citation51]. More recently, increased serum levels of CHIT1 were described in atherosclerosis, where CHIT1 produced by macrophages might enhance atherosclerotic plaque formation and subsequent thrombosis [Citation52–54].
These studies suggest that CHIT1 has a dual function; it is both an innate immune mediator and a regulator of tissue remodeling. Therefore, CHIT1 might have organ- as well as cell-specific effects in the context of infectious diseases and inflammatory disorders. As MPNs are characterized by a state of chronic inflammation, which is proposed to be directly involved in disease progression and bone marrow fibrosis, we found it relevant and timely to investigate CHIT1 in MPN patients.
Subjects and methods
Consecutive patients with PV, ET and PMF diagnosed according to World Health Organisation (WHO) 2008 criteria [Citation55] and post-ET MF and post-PV MF diagnosed according to International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) 2008 criteria [Citation56], were enrolled in the study between July 2014 and February 2017. Healthy blood donors served as controls. Excluded from participation were pregnant women, subjects younger than 18 years of age, subjects with acute infections, known autoimmune disorders or concomitant solid tumors.
Bone marrow biopsy was performed in all patients due to clinical reasons and at physician’s discretion, not as a requirement for participation in this study. Only patients whose blood samples were taken within 6 months from the bone marrow biopsy were included in this study.
This was a case-controlled study conducted at two hematological centers in Croatia (the Department of Internal medicine at the General Hospital of Sibenik-Knin County and the Division of Hematology at the Department of Internal Medicine, University Hospital Center Zagreb).
Data regarding the patients’ diagnosis, sex, age, JAK2 and CALR mutational status, presence of constitutive symptoms (fever ≥ 37.5C, night sweats or weight loss ≥ 10% in the preceding 6 months), palpable hepatosplenomegaly and treatment modalities (phlebotomy, acetylsalicylic acid and / or hydroxycarbamide therapy) were collected at the time of study enrollment. Bone marrow fibrosis was graded by hematopathologist according to the current European consensus [Citation57].
Ethics
This study was performed in accordance to the Declaration of Helsinki and approved by the General Hospital of Sibenik-Knin County and University Hospital Centre Zagreb Ethics Committees. Prior to enrollment all participants signed the informed consent.
Laboratory assays
Blood samples for measurement of blood counts, serum lactate dehydrogenase (LDH) and CHIT1 were taken at the time of study enrollment.
Samples for CHIT1 measurement were left to clot, and serum was separated from cellular fragments by centrifugation (3500 rpm, 10 minutes). All serum samples were stored at −80 °C until the analysis period. CHIT1 was determined using a fluorometric enzyme assay. Specifically, 5 μL of serum was incubated with 100 μL of the 22 μM fluorogenic substrate 4-methylumbelliferyl-β-N,N,N-triacetylchitotrioside (Sigma-Aldrich, USA) in McIlvaine buffer (pH 5.2) for 60 min at 37°C. The reaction was stopped by adding 600 μL of 0.2 M glycine–NaOH buffer (pH 10.4). The enzymatic hydrolysis product 4-methylumbelliferone was measured at specified excitation (365 nm) and emission (450 nm) wavelengths on Cary Eclipse Fluorimeter (Agilent Technologies, USA). CHIT1 was expressed as nanomoles of substrate hydrolyzed per hour per milliliter of incubated serum (μmol/L/h).
Statistical analysis
Categorical variables are presented with absolute and relative frequencies and are compared using chi-square test. Quantitative variables are presented as medians and ranges. After testing for normality we decided to use nonparametric tests. The Kruskal–Wallis analysis of variance was used to analyze differences in age between patients and healthy controls. The Kruskal–Wallis and the Mann–Whitney U test were used to analyze differences in CHIT1 between PV, post-PV MF, ET, post-ET, PMF and SMF patients and healthy controls. The Spearman correlation coefficients were calculated to assess correlations between CHIT1 and different clinical and laboratory variables. The Mann–Whitney U test was used to analyze differences in absolute basophil counts between patient groups. The difference in CHIT1 between bone marrow fibrosis grades was also analyzed using the Mann–Whitney U test. P-values under .05 were considered significant for all analyses. Data analysis software system IBM SPSS Statistics, version 21.0 (IBM, Armonk, NY, USA), was used.
Results
There were included 28 PV, 27 ET, 17 PMF and 19 SMF patients. Among the 19 SMF patients, there were 8 post-PV MF and 11 post-ET MF patients. There was no statistically significant difference between patients and healthy controls according to sex (p = .485) or age (p = .182). Subjects’ characteristics are shown in .
Table 1. Subject characteristics and laboratory values. PV = polycythemia vera, post-PV MF = post-polycythemia vera myelofibrosis transformation, ET = essential thrombocythemia, post-ET MF = post-essential thrombocythemia myelofibrosis transformation, PMF = primary myelofibrosis, IU/L = international units per liter.
CHIT1 was significantly higher in PV patients (median 57 μmol/L/h, range 3–458) when compared to healthy controls (median 30 μmol/L/h, range 12–78) (p < .001). PMF patients had levels similar to controls (median 33 μmol/L/h, range 8–378; p = .287) while those with ET had a trend towards higher values that failed to reach statistical significance (median 36 μmol/L/h, range 1–502; p = .080) (). The differences in CHIT1 levels between PV, ET or PMF were also not statistically significant (p = .060 and p = .109, respectively).
Figure 1. CHIT1 levels were significantly higher in PV and post-PV MF patients when compared to healthy controls. The difference between CHIT1 in ET, post-ET MF and PMF patients when compared to healthy controls were not statistically significant. PV = polycythemia vera, post-PV MF = post-polycythemia vera myelofibrosis transformation, ET = essential thrombocythemia, post-ET MF = post-essential thrombocythemia myelofibrosis transformation, PMF = primary myelofibrosis, CHIT = serum chitotriosidase activity.
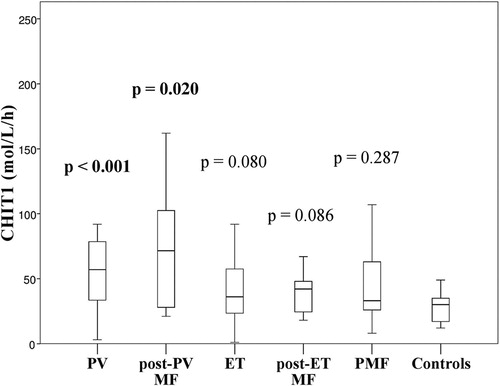
Patients with SMF had higher CHIT1 (median 44 μmol/L/h, range 18–162) than healthy controls (p = .013). However, when we divided SMF patients into those with post-PV MF and post-ET MF, only patients with post-PV MF had elevated CHIT1 levels (median 72 μmol/L/h, range 21–162; p = .020). The difference between CHIT1 in post-ET MF patients (median 42 μmol/L/h, range 18–67) and healthy controls was not statistically significant (p = .086) (), neither was the difference between post-PV MF and post-ET MF patients (p = .129).
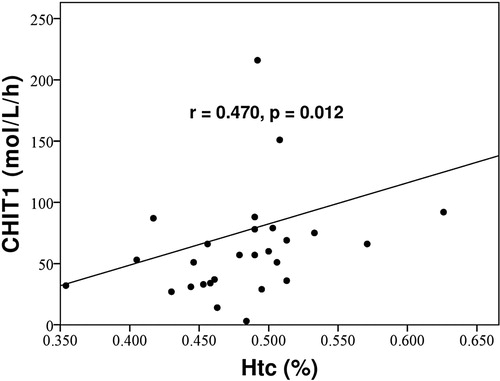
We observed a statistically significant positive correlation in PV patients between CHIT1, hemoglobin (correlation coefficient 0.420, p = .026) and hematocrit levels (correlation coefficient 0.470, p = .012) ().
Figure 2. Positive correlation in PV patients between CHIT and hematocrit level. CHIT1 = serum chitotriosidase activity, Htc = hematocrit.
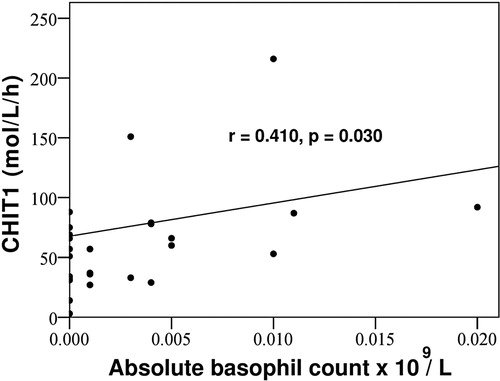
There was a statistically significant positive correlation in PV patients between CHIT1 and absolute basophil count (correlation coefficient 0.410, p = .030) (). Post-ET MF and PMF patients had higher absolute basophil count when compared to PV (p = .024 and p = .039, respectively), post-PV MF (p = .041 and p = .048, respectively) and ET patients (p = .038 and p = .047, respectively). The difference in absolute basophil count between post-ET MF and PMF patients was not statistically significant (p = .853), neither was the difference between PV, post-PV MF and ET patients (p = .758 and p = .588, respectively).
Figure 3. Positive correlation in PV patients between CHIT1 and absolute basophil count. CHIT1 = serum chitotriosidase activity.
There was no correlation between CHIT1, hematocrit, hemoglobin and the absolute basophil count in post-PV MF, ET, post-ET MF, PMF and SMF patients.
CHIT1 was significantly higher in PV patients who had reticulin fibrosis (MF-1) in the bone marrow (median 76.50 μmol/L/h, range 3-458) than in those patients without reticulin fibrosis (MF-0) (median 43.50 μmol/L/h, range 14–69) (p = .023). No statistically significant difference was observed between CHIT1 and the bone marrow fibrosis grade in post-PV MF, ET, post-ET MF, PMF and SMF patients.
CHIT1 did not correlate with sex, age, erythrocyte, leukocyte, granulocyte or platelet counts, JAK2 or CALR mutational status, LDH levels, the presence of constitutive symptoms and palpable hepatosplenomegaly, treatment with acetylsalicylic and/or hydroxycarbamide therapy or the need for phlebotomy.
Discussion
To the best of our knowledge, no previous studies have examined the role of CHIT1 in MPN patients. We herein for the first time report elevated CHIT1 in PV and post-PV MF, but not in ET, post-ET MF and PMF patients.
Bone marrow environment and the macrophage niche have been shown to play a crucial role in supporting erythrocyte production, maintenance and removal. Macrophage activity stimulates signaling pathways which complement JAK2 signaling, and without their support, the proliferative potential of erythroid cells diminishes substantially. In addition, macrophages were described to play an important role in regulating pathological erythropoiesis in PV where they contribute in acquiring ‘erythroid’ phenotype. Interestingly, elevated CHIT1 in our study was observed only in PV and post-PV MF: a subset of patients with the ‘polycythemic’ phenotype. Furthermore, CHIT1 in PV was positively correlated with hematocrit and hemoglobin level, which are distinct PV features within the MPN spectrum and a ‘major’ diagnostic criteria in the WHO classification [Citation55,Citation58].
Several studies have also reported basophilia as an MPN feature. Basophils are increased in MPNs and represent a part of malignant clone and [Citation59–61]. High absolute basophil count was even shown to be an independent predictor of inferior survival in PMF [Citation62]. These cells harbor a large number of proinflammatory mediators. Among various other cytokines, they secrete GM-CSF which was shown to induce macrophage CHIT1 synthesis and secretion [Citation38,Citation39,Citation63–65]. In our study post-ET MF and PMF had higher absolute basophil counts in comparison to PV, post-PV MF and ET patients. In this context, we would expect CHIT1 to be higher in post-ET and PMF patients. On contrary, higher CHIT1 was observed in PV and post-PV MF. Furthermore, a positive correlation between CHIT1 and the absolute basophil count was noted only in PV patients. In the study of Pieri et al. [Citation61], ET and PMF patients had increased absolute basophil counts when compared to controls. However, the number of ‘activated’ basophils did not differ in comparison to controls. This was in contrast to PV, where absolute and ‘activated’ basophil counts were increased. Similarly, Lucijanic et al. [Citation62] reported increased absolute basophil count in PMF when compared to controls. In this study, high absolute basophil count positively correlated with elevated leukocyte count, LDH, massive splenomegaly and constitutive symptoms which reflects higher disease activity and proliferative potential. These observations indicate different underlying pathophysiologic processes within the MPN spectrum. In this perspective, ‘activated’ basophils in PV patients might induce CHIT1 secretion through GM-CSF-mediated signaling. Therefore, elevated CHIT1 in PV patients and a positive correlation between CHIT1 and these PV-defining features, hemoglobin, hematocrit and absolute basophil count, might imply macrophages closely related to clonal erythropoiesis as cells of CHIT1 origin.
On the other hand, elevated CHIT1 in PV patients might also result from granulocyte secretion or erythrocyte overproduction and subsequently increased spleen and liver macrophage activity due to phagocytosis of damaged erythrocytes. In our study, there was no correlation between CHIT1, the presence of palpable hepatosplenomegaly and the leukocyte, granulocyte or erythrocyte counts, which makes these hypotheses less likely. However, further studies are warranted to more precisely elucidate cell(s) of CHIT1 origin.
CHIT1 was also shown to be dysregulated in granulomatous and other human diseases characterized by inflammation and faulty tissue remodeling. As mentioned previously, various inflammatory cytokines are elevated in MPNs. In addition, TNF-α, a proinflammatory cytokine, was reported to be involved in inflammation-associated carcinogenesis [Citation66–70]. In the bone marrow, expression of TNF-α genes resulted in neutrophil activation and TNF-α-facilitated clonal expansion.
Macrophages release CHIT1 upon stimulation with TNF-α. In the MPN setting, TNF-α might directly stimulate CHIT1 secretion, which in turn, through inducing TGF-β signaling, might act as a promoter of tissue remodeling and fibrosis in PV. We found a positive correlation in PV patients between CHIT1 and the presence of reticulin fibers in the bone marrow and reticulin fibrosis has been cited as a risk factor for MF transformation [Citation71–73]. In addition, our study shows CHIT1 to be higher in SMF patients when compared to healthy controls, especially in post-PV MF patients, where highest CHIT1 levels were noted. Interestingly, these patients were actually anemic (median hemoglobin 93 g/L) with a higher bone marrow fibrosis grade. Furthermore, 75% of post-PV MF patients had constitutive symptoms, which is the highest percentage among all groups (). Although we found no correlations between CHIT1, hemoglobin/hematocrit levels, presence of constitutive symptoms and the bone marrow fibrosis grade in post-PV MF, highest CHIT1 levels in these patients could be explained by disease transition from the ‘hypercellular’ phase in PV to the ‘fibrotic’ phase in post-PV MF. In these patients elevated CHIT1 might primarily be a reflection of more advanced disease with a higher degree of inflammation and marrow fibrosis with subsequent anemia and constitutive symptoms. Similarly, higher median hemoglobin (155 g/L) and hematocrit (0.49%) levels in our PV patients despite phlebotomies and/or hydroxycarbamide therapy () might indicate uncontrolled disease with a high inflammatory state and elevated CHIT1. In this perspective, a steady rise of CHIT1 from ‘early’ PV to the ‘burnt-out’ post-PV MF transformation might indicate its role in PV progression. However, due to limited number of post-PV MF patients included in our study, additional studies are warranted to clarify the role of CHIT1 in promoting disease progression and bone marrow fibrosis in PV.
Another chitinase-like protein, YKL-40 or chitinase-3-like protein 1, was also studied in MPNs [Citation74,Citation75]. YKL-40 is secreted primarily by granulocytes and was described to be involved in cell proliferation, differentiation, angiogenesis, inflammation and remodeling of the extracellular matrix [Citation76,Citation77]. In the MPN setting, serum YKL-40 levels were reported to be significantly higher in PV and PMF patients when compared to ET and healthy controls. Furthermore, YKL-40 was shown to correlate with C-reactive protein, LDH, granulocytes, JAK2 allelic burden and the presence of fatigue in PV patients, which are common indicators of increased tumor burden in these neoplasms. Our observations of elevated CHIT1 in PV and post-PV MF patients might further substantiate the role of chitinases and chitinase-like proteins in the pathogenesis and progression of MPNs, particularly in inflammation-induced tissue repair processes.
Although we observed elevated CHIT1 in PV patients, no correlations were found between CHIT1 and some clinical and laboratory variables that are usually suggestive of a higher disease burden, which might limit the use of CHIT1 as a circulating biomarker of disease progression and/or tumor burden. However, targeting the CHIT1 activation cascade through the administration of specific antibodies or pan-chitinase inhibitors was shown to significantly ameliorate inflammation and fibrosis in several animal models of autoimmune diseases, most likely by suppressing the CHIT1 dependent release of different cytokines and chemokines [Citation78]; the safety of this approach in humans is not yet determined as it might increase susceptibility to fungal and protozoal infections. If proven safe in humans, the development of targeted therapies would be valuable option in the treatment of these neoplasms, particularly in PV.
In addition, there is current interest in therapeutically targeting the macrophages which provide critical support to the tumor cells, i.e. targeting them in chronic lymphocytic leukemia (CLL) has been shown to result in CLL cell death [Citation79]. As mentioned previously, studies on animal models have shown that bisphosphonates (clodronate) induce macrophage depletion with the reversal of PV phenotype in mice. However, in a prospective, multi-center, phase II randomized trial, zoledronic acid in myelofibrosis patients showed a lack of response [Citation80]. Finally, statins with their ‘pleiotropic effects’ have been shown to induce macrophage apoptosis and to reduce macrophage activation in atherosclerotic plaques, as well as the levels of proinflammatory cytokines, such as TNF-α and IL-6 [Citation81–83]. These drugs have also been described to have strong antiproliferative and proapoptotic properties with the potential to decrease clonal myeloproliferation. In addition, MPN-associated JAK kinase localization to lipid rafts and its signaling was shown to be inhibited by statins [Citation84–90]. One case report demonstrated a remarkable hematological and molecular response with the reduction of JAK2 allelic burden in a PV patient during simultaneous alendronate and simvastatin treatment [Citation91]. However, further experimental and clinical research is needed on the potential role of these relatively non-toxic drugs in the treatment of MPNs. We can only speculate that potentially greater response could be seen in the ‘early’ disease, such as PV, than in the ‘fibrotic’ setting.
Even though our study shows CHIT1 to be significantly higher in PV and post-PV MF patients in comparison to healthy controls, it has several limitations. The number of patients included was limited and they were tested at various time-points in their disease course after having been diagnosed with MPN. Hydroxycarbamide therapy was given to approximately 50% of PV and ET patients and to 25–35% of post-PV MF, post-ET MF and PMF patients almost certainly influenced the blood cell counts (). To minimize the effect of therapy or general patient condition, we included only patients that were clinically well, treated as out-patients and on stable hydroxycarbamide dose and/or phlebotomy schedule, i.e. patients with a stable disease who achieved an equilibrium between cell proliferation and destruction.
Conclusion
CHIT1 could be a considered as a circulating biomarker in PV, which might, through inducing TGF-β signaling, have a role in promoting tissue remodeling and bone marrow fibrosis. This observation provides an additional degree of complexity to these neoplasms and further highlights the role of the tumor microenvironment in pathogenesis and the clinical presentation of MPNs, which might help in recognizing new therapeutic targets in these neoplasms. However, studies with larger series of PV and post-PV MF patients are required to more precisely elucidate the role of CHIT1 in promoting disease progression and bone marrow fibrosis.
Acknowledgements
The data used in this study are part of PhD thesis of Ivan Krecak.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Ivan Krecak http://orcid.org/0000-0002-8642-305X
Ivana Lapic http://orcid.org/0000-0002-0854-4526
Nadira Durakovic http://orcid.org/0000-0001-5842-0911
References
- James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023
- Ding J, Komatsu H, Wakita A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103(11):4198–4200. doi: 10.1182/blood-2003-10-3471
- Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–3476. doi: 10.1182/blood-2006-04-018879
- Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. doi: 10.1056/NEJMoa1311347
- Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. doi: 10.1056/NEJMoa1312542
- Skoda RC, Duek A, Grisouard J. Pathogenesis of myeloproliferative neoplasms. Exp Hematol. 2015;43(8):599–608. doi: 10.1016/j.exphem.2015.06.007
- Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012;119:3219–3225. doi: 10.1182/blood-2011-11-394775
- Hasselbalch HC, Bjørn ME. MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflamm. 2015;2015:102476. doi: 10.1155/2015/102476
- Geyer HL, Dueck AC, Scherber MR. Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflamm. 2015;2015:284706. doi: 10.1155/2015/284706
- Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN landmark survey. BMC Cancer. 2016;16(1):211. doi: 10.1186/s12885-016-2208-2
- Fleischman AG, Aichberger KJ, Luty SB, et al. TNF facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118(24):6392–6398. doi: 10.1182/blood-2011-04-348144
- Tefferi A, Vaidya R, Caramazza D, et al. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356–1363. doi: 10.1200/JCO.2010.32.9490
- Vaidya R, Gangat N, Jimma T, et al. Plasma cytokines in polycythemia vera: phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol. 2012;87(11):1003–1005. doi: 10.1002/ajh.23295
- Pourcelot E, Trocme C, Mondet J, et al. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp Hematol. 2014;42(5):360–368. doi: 10.1016/j.exphem.2014.01.006
- Zahr AA, Salama ME, Carreau N, et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica. 2016;101(6):660–671. doi: 10.3324/haematol.2015.141283
- Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi: 10.1056/NEJMoa1110557
- Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi: 10.1056/NEJMoa1110556
- Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. doi: 10.1056/NEJMoa1002028
- Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513–520. doi: 10.1002/cncr.28441
- Agarwal A, Morrone K, Bartenstein M, et al. Bone marrow fibrosis in primary myelofibrosis: pathogenic mechanisms and the role of TGF-β. Stem Cell Investig. 2016;3:5.
- Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112(3):470–478. doi: 10.1182/blood-2008-03-077883
- Soni S, Bala S, Gwynn B, et al. Absence of erythroblast macrophage protein (Emp) leads to failure of erythroblast nuclear extrusion. J Biol Chem. 2006;281(29):20181–20189. doi: 10.1074/jbc.M603226200
- Yoshida H, Kawane K, Koike M, et al. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature. 2005;437(7059):754–758. doi: 10.1038/nature03964
- Seki M, Shirasawa H. Role of the reticular cells during maturation process of the erythroblast. 3. The fate of phagocytized nucleus. Acta Pathol Jpn. 1965;15:387–405.
- Sadahira Y, Yasuda T, Yoshino T, et al. Impaired splenic erythropoiesis in phlebotomized mice injected with CL2MDP-liposome: an experimental model for studying the role of stromal macrophages in erythropoiesis. J Leukoc Biol. 2000;68(4):464–470.
- Hanspal M, Hanspal JS. The association of erythroblasts with macrophages promotes erythroid proliferation and maturation: a 30-kD heparin-binding protein is involved in this contact. Blood. 1994;84(10):3494–3504.
- Rhodes MM, Kopsombut P, Bondurant MC, et al. Adherence to macrophages in erythroblastic islands enhances erythroblast proliferation and increases erythrocyte production by a different mechanism than erythropoietin. Blood. 2007;111(3):1700–1708. doi: 10.1182/blood-2007-06-098178
- Ramos P, Casu C, Gardenghi S, et al. Macrophages support pathological erythropoiesis in polycythemia vera and β-thalassemia. Nat Med. 2013;19(4):437–445. doi: 10.1038/nm.3126
- Boot RG, Renkema GH, Verhock M, et al. The human chitotriosidase gene. J Biol Chem. 1998;273(40):25680–25685. doi: 10.1074/jbc.273.40.25680
- Kzhyshkowska J, Gratchev A, Goerdt S. Human chitinases and chitinase-like proteins as indicators for inflammation and cancer. Biomark Insights. 2007;2:128–146. doi: 10.1177/117727190700200023
- Malaguarnera L. Chitotriosidase: the yin and yang. Cell Mol Life Sci. 2006;63(24):3018–3029. doi: 10.1007/s00018-006-6269-2
- Van Eijk M, Van Roomen CP, Renkema GH, et al. Characterization of human phagocyte-derived chitotriosidase, a component of innate immunity. Int Immunol. 2005;17(11):1505–1512. doi: 10.1093/intimm/dxh328
- Labadaridis I, Dimitriou E, Theodorakis M, et al. Chitotriosidase in neonates with fungal and bacterial infections. Arch Dis Child Fetal Neonatal Ed. 2005;90(6):F531–F532. doi: 10.1136/adc.2004.051284
- Choi EH, Zimmerman PA, Foster CB, et al. Genetic polymorphisms in molecules of innate immunity and susceptibility to infection with Wuchereria bancrofti in south india. Genes Immun. 2001;2(5):248–253. doi: 10.1038/sj.gene.6363767
- Malaguarnera L, Simpore J, Prodi DA, et al. A 24-bp duplication in exon 10 of human chitotriosidase gene from the sub-Saharan to the Mediterranean area: role of parasitic diseases and environmental conditions. Genes Immun. 2003;4(8):570–574. doi: 10.1038/sj.gene.6364025
- Malaguarnera L, Musumeci M, Di Rosa M, et al. Interferon-gamma, tumor necrosis factor-alpha, and lipopolysaccharide promote chitotriosidase gene expression in human macrophages. J Clin Lab Anal. 2005;19(3):128–132. doi: 10.1002/jcla.20063
- Van Eijk M, Scheij SS, Van Roomen CP, et al. TLR- and NOD2-dependent regulation of human phagocyte-specific chitotriosidase. FEBS Lett. 2007;581(28):5389–5395. doi: 10.1016/j.febslet.2007.10.039
- Hollak CE, Weely SV, Oers MHV, et al. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93(3):1288–1292. doi: 10.1172/JCI117084
- Aerts JM, Hollak CE. 4 plasma and metabolic abnormalities in Gaucher’s disease. Baillieres Clin Haematol. 1997;10(4):691–709. doi: 10.1016/S0950-3536(97)80034-0
- Barone R, Di Gregorio F, Romeo MA, et al. Plasma chitotriosidase activity in patients with β-thalassemia. Blood Cells Mol Dis. 1999;25(1):1–8. doi: 10.1006/bcmd.1999.0221
- Musumeci M, Caruso V, Medulla E, et al. Serum YKL-40 levels and chitotriosidase activity in patients with beta-thalassemia major. Dis Markers. 2014;2014:965971. doi: 10.1155/2014/965971
- Barone R, Simpore J, Malaguarnera L, et al. Plasma chitotriosidase activity in acute plasmodium falciparum malaria. Clin Chim Acta. 2003;331(1–2):79–85. doi: 10.1016/S0009-8981(03)00089-5
- Bargagli E, Bianchi N, Margollicci M, et al. Chitotriosidase and soluble IL-2 receptor: comparison of two markers of sarcoidosis severity. Scand J Clin Invest. 2008;68(6):479–483. doi: 10.1080/00365510701854975
- Boot RG, Hollak CEM, Verhoek M, et al. Plasma chitotriosidase and CCL18 as surrogate markers for granulomatous macrophages in sarcoidosis. Clin Chim Acta. 2010;411(1–2):31–36. doi: 10.1016/j.cca.2009.09.034
- Popevic S, Sumarac Z, Jovanovic D, et al. Verifying sarcoidosis activity: chitotriosidase versus ACE in sarcoidosis-A case-control study-. J Med Biochem. 2016;35(4):390–400. doi: 10.1515/jomb-2016-0017
- Koening CL, Gota C, Langford CA, et al. Serum chitotriosidase activity and Wegener’s granulomatosis. Clin Biochem. 2010;43(4–5):512–514. doi: 10.1016/j.clinbiochem.2009.11.015
- Malaguarnera L, Rosa MD, Zambito AM, et al. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut. 2006;55(9):1313–1320. doi: 10.1136/gut.2005.075697
- Létuvé S, Kozhich A, Humbles A, et al. Lung chitinolytic activity and chitotriosidase are elevated in chronic obstructive pulmonary disease and contribute to lung inflammation. Am J Pathol. 2010;176(2):638–649. doi: 10.2353/ajpath.2010.090455
- Lee CG, Herzog EL, Ahangari F, et al. Chitinase 1 Is a biomarker for and therapeutic target in scleroderma-associated interstitial lung disease that augments TGF- 1 signaling. J Immunol. 2012;189(5):2635–2644. doi: 10.4049/jimmunol.1201115
- Karadag B, Kucur M, Isman FK, et al. Serum chitotriosidase activity in patients with coronary artery disease. Circ J. 2008;72(1):71–75. doi: 10.1253/circj.72.71
- Artieda M, Cenarro A, Ganan A, et al. Serum chitotriosidase activity is increased in subjects with atherosclerosis disease. Arterioscler Thromb Vasc Biol. 2003;23(9):1645–1652. doi: 10.1161/01.ATV.0000089329.09061.07
- Yildiz BS, Barutcuoglu B, Alihanoglu YI, et al. Serum chitotriosidase activity in acute coronary syndrome. J Atheroscler Thromb. 2013;20(2):134–141. doi: 10.5551/jat.13920
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the world health organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262
- Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the international working group for myelofibrosis research and treatment. Leukemia. 2008;22(2):437–438. doi: 10.1038/sj.leu.2404914
- Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128–1132.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544
- Arnalich F, Lahoz C, Larrocha C, et al. Incidence and clinical significance of peripheral and bone marrow basophilia. J Med. 1987;18(5–6):293–303.
- Agis H, Krauth M-T, Mosberger I, et al. Enumeration and immunohistochemical characterisation of bone marrow basophils in myeloproliferative disorders using the basophil specific monoclonal antibody 2D7. J Clin Pathol. 2006;59(4):396–402. doi: 10.1136/jcp.2005.029215
- Pieri L, Bogani C, Guglielmelli P, et al. The JAK2V617 mutation induces constitutive activation and agonist hypersensitivity in basophils from patients with polycythemia vera. Haematologica. 2009;94(11):1537–1545. doi: 10.3324/haematol.2009.007047
- Lucijanic M, Livun A, Stoos-Veic T, et al. High absolute basophil count is a powerful independent predictor of inferior overall survival in patients with primary myelofibrosis. Hematology. 2017;14:1–7.
- Schroeder JT, MacGlashan DW Jr, Lichtenstein LM. Human basophils: mediator release and cytokine production. Adv Immunol. 2001;77:93–122. doi: 10.1016/S0065-2776(01)77015-0
- Stone KD, Prussin C, Metcalfe DD. Ige, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S73–S80. doi: 10.1016/j.jaci.2009.11.017
- Valent P, Bettelheim P. The human basophil. Crit Rev Oncol/Hematol. 1990;10(4):327–352. doi: 10.1016/1040-8428(90)90009-H
- Suganuma M, Okabe S, Marino MW, et al. Essential role of tumor necrosis factor alpha (TNF-alpha) in tumor promotion as revealed by TNF-alpha-deficient mice. Cancer Res. 1999;59(18):4516–4518.
- Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29(11):1275–1288. doi: 10.1111/j.1745-7254.2008.00889.x
- Moore RJ, Owens DM, Stamp G, et al. Mice deficient in tumor necrosis factor-α are resistant to skin carcinogenesis. Nat Med. 1999;5(7):828–831. doi: 10.1038/10552
- Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118(2):560–570.
- Matthews N, Watkins JF. Tumour-necrosis factor from the rabbit. I. Mode of action, specificity and physicochemical properties. Br J Cancer. 1978;38(2):302–309. doi: 10.1038/bjc.1978.202
- Barbui T, Thiele J, Passamonti F, et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood. 2012;119(10):2239–2241. doi: 10.1182/blood-2011-11-393819
- Tiribelli M, Barraco D, De Marchi F, et al. Clinical factors predictive of myelofibrotic evolution in patients with polycythemia vera. Ann Hematol. 2015;94(5):873–874. doi: 10.1007/s00277-014-2257-z
- Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366. doi: 10.1038/bcj.2015.95
- Bjørn ME, Andersen CL, Jensen MK, et al. Circulating YKL-40 in myelofibrosis a potential novel biomarker of disease activity and the inflammatory state. Eur J Haematol. 2014;93(3):224–228. doi: 10.1111/ejh.12332
- Andersen CL, Bjørn ME, McMullin MF, et al. Circulating YKL-40 in patients with essential thrombocythemia and polycythemia vera treated with the novel histone deacetylase inhibitor vorinostat. Leuk Res. 2014;38(7):816–821. doi: 10.1016/j.leukres.2014.04.002
- Johansen JS, Schultz NA, Jensen BV. Plasma YKL-40: a potential new cancer biomarker? Future Oncol. 2009;5(7):1065–1082. doi: 10.2217/fon.09.66
- Kazakova MH, Sarafian VS. YKL-40--a novel biomarker in clinical practice? Folia Med Plovdiv. 2009;51(1):5–14.
- Sutherland TE, Maizels RM, Allen JE. Chitinases and chitinase-like proteins: potential therapeutic targets for the treatment of T-helper type 2 allergies. Clin Exp Allergy. 2009;39(7):943–955. doi: 10.1111/j.1365-2222.2009.03243.x
- Galleti G, Scielzo C, Barbaglio F, et al. Targeting macrophages sensitizes chronic lymphocytic leukemia to apoptosis and inhibits disease progression. Cell Rep. 2016; 23;14(7):1748–1760. doi: 10.1016/j.celrep.2016.01.042
- Delforge M, Devos T, Rowe J, et al. Lack of clinical benefit of zoledronic acid in myelofibrosis: results of a prospective multi-center phase II trial. Leuk Lymphoma. 2016;57(2):470–473. doi: 10.3109/10428194.2015.1055481
- Sakai M, Kobori S, Matsumura T, et al. HMG-CoA reductase inhibitors suppress macrophage growth induced by oxidized low density lipoprotein. Atherosclerosis. 1997;133(1):51–59. doi: 10.1016/S0021-9150(97)00118-4
- Corsini A, Mazzotti M, Raiteri M, et al. Relationship between mevalonate pathway and arterial myocyte proliferation: in vitro studies with inhibitors of HMG-CoA reductase. Atherosclerosis. 1993;101(1):117–125. doi: 10.1016/0021-9150(93)90107-6
- Croons V, De Meyer I, Houten SM, et al. Effect of statins on the viability of macrophages and smooth muscle cells. J Cardiovasc Pharmacol. 2010;55(3):269–275. doi: 10.1097/FJC.0b013e3181d0bec3
- Liang SL, Liu H, Zhou A. Lovastatin-induced apoptosis in macrophages through the Rac1/Cdc42/JNK pathway. J Immunol. 2006;177(1):651–656. doi: 10.4049/jimmunol.177.1.651
- Hasselbalch HC, Riley CH. Statins in the treatment of polycythaemia vera and allied disorders: an antithrombotic and cytoreductive potential? Leuk Res. 2006;30(10):1217–1225. doi: 10.1016/j.leukres.2005.12.018
- Newman A, Clutterbuck RD, Powles RL, et al. Selective inhibition of primary acute myeloid leukaemia cell growth by simvastatin. Leukemia. 1994;8(11):2023–2029.
- Newman A, Clutterbuck RD, Powles RL, et al. A comparison of the effect of the 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase inhibitors simvastatin, lovastatin, and pravastatin on leukaemic and normal bone marrow progenitors. Leuk Lymphoma. 1997;24(5–6):533–537. doi: 10.3109/10428199709055590
- Dimitroulakos J, Nohynek D, Backway KL, et al. Increased sensitivity of acute myeloid leukemias to lovastatin-induced apoptosis: a potential therapeutic approach. Blood. 1999;93(4):1308–1318.
- Dimitroulakos J, Thai S, Wasfy GH, et al. Lovastatin induces a pronounced differentiation response in acute myeloid leukemias. Leuk Lymphoma. 2000;40(1):167–178. doi: 10.3109/10428190009054894
- Griner LN, McGraw KL, Johnson JO, et al. JAK2-V617F-mediated signalling is dependent on lipid rafts and statins inhibit JAK2-V617F-dependent cell growth. Br J Haematol. 2013;160(2):177–187. doi: 10.1111/bjh.12103
- Sørensen AE, Kallenbach K, Hasselbalch HC. A remarkable hematological and molecular response pattern in a patient with polycythemia vera during combination therapy with simvastatin and alendronate. Leuk Res Rep. 2016;6:20–23.