
Abstract
Profound depletion of the catecholamines dopamine (DA) and norepinephrine in the brain, heart, or both characterizes Lewy body diseases such as Parkinson disease, dementia with Lewy bodies, and pure autonomic failure. Although one might presume that catecholamine deficiency in these disorders results directly and solely from loss of catecholaminergic neurons, there is increasing evidence that functional abnormalities in extant residual neurons contribute to the neurotransmitter deficiencies—the “sick-but-not-dead” phenomenon. This brief review highlights two such functional abnormalities—decreased vesicular sequestration of cytoplasmic catecholamines and decreased catecholamine biosynthesis. Another abnormality, decreased activity of aldehyde dehydrogenase, may have pathogenetic significance and contribute indirectly to the loss of catecholamine stores via interactions between the autotoxic catecholaldehyde 3,4-dihydroxyphenylacetaldehyde and the protein alpha-synuclein, which is a major component of Lewy bodies. Theoretically, chronically repeated stress responses could accelerate these abnormalities, via increased exocytosis and neuronal reuptake, which indirectly shifts tissue catecholamines from vesicular stores into the cytoplasm, and via increased tyrosine hydroxylation, which augments intra-cytoplasmic DA production. The discovery of specific paths mediating the sick-but-not-dead phenomenon offers novel targets for multi-pronged therapeutic approaches.
Introduction: catecholaminergic neurodegeneration in Lewy body diseases
The movement disorder in Parkinson disease (PD) is well known to result from drastic deficiency of the catecholamine dopamine (DA) in the nigrostriatal system—especially in the putamen (Kish et al., Citation1988).
Perhaps less well known, PD and other Lewy body diseases such as dementia with Lewy bodies (DLB) and pure autonomic failure (PAF) entail equally severe loss of the closely related catecholamine norepinephrine (NE) in the heart. This has long been suspected, based on numerous cardiac sympathetic neuroimaging studies (Goldstein & Orimo, Citation2009; Jain & Goldstein, Citation2012) and on postmortem demonstrations of severely decreased immunoreactive tyrosine hydroxylase (TH), a marker of catecholamine-synthesizing neurons, in epicardial nerve trunks (Amino et al., Citation2005; Orimo et al., Citation2006); however, direct demonstration of NE deficiency by postmortem neurochemistry has come only relatively recently (Goldstein & Sharabi, Citation2019; Goldstein et al., Citation2014, Citation2019).
One might presume that the deficiencies of putamen DA and cardiac NE in Lewy body diseases straightforwardly reflect loss of nigrostriatal dopaminergic and myocardial sympathetic innervation. Several lines of evidence over the past decade have led to the view that functional abnormalities in extant residual neurons contribute to the neurotransmitter deficiencies. We call this the “sick-but-not-dead” phenomenon. There is also recent evidence that at least one dysfunction, a vesicular storage defect, is an early abnormality that can precede overt neuronal loss (Lamotte et al., Citation2019). This review highlights these recent developments and puts them in the context of pathophysiologic mechanisms and potential novel treatment or prevention strategies.
An apparent paradox
Postmortem studies of patients with PD have reported substantial loss of substantia nigra neurons (Kordower et al., Citation2013) and of immunoreactive TH in striatum (DelleDonne et al., Citation2008). The severity of these lesions has averaged about 70–80%. The extent of putamen DA deficiency, however, is substantially greater—90–99% (Goldstein, Sullivan, et al., Citation2011; Kish et al., Citation1988). Analogously, based on immunoreactive TH in epicardial nerves, there is about a 75% loss of myocardial sympathetic noradrenergic nerves in PD, in contrast with up to a 99% loss of NE (Goldstein et al., Citation2019).
This raises an apparent paradox: How can there be greater loss of the transmitter than of the neurons containing that transmitter?
Decreased vesicular sequestration
A potential resolution would be decreased ability to sequester cytoplasmic catecholamines in vesicles. For instance, if there were an 80% loss of putamen dopaminergic terminals (20% remaining) and an 80% decrease in the ability to take up cytoplasmic DA into vesicles (20% of normal efficiency) then the stores of DA in the tissue would be 0.20 × 0.20 = 0.04 times normal, for a decrease in tissue DA content of 96%.
The first evidence for an intra-neuronal vesicular storage defect in catecholaminergic neurons of patients with Lewy body diseases came from a study that combined cardiac sympathetic neuroimaging with catechol neurochemistry in arterial plasma after intravenous injection of 18F-DA (Goldstein, Holmes, et al., 2011). If there were sympathetic noradrenergic denervation alone, then myocardial 18F-DA-derived radioactivity detected by positron emission tomographic (PET) scanning and arterial plasma levels of the neuronal 18F-DA metabolite 18F-3,4-dihydroxyphenylacetic acid (18F-DOPAC) would be decreased proportionately equally. If there were an isolated vesicular storage defect without a loss of sympathetic noradrenergic nerves, there would be normal 18F-DA uptake and increased arterial 18F-DOPAC levels; and if there were both a loss of sympathetic nerves and a vesicular storage defect in the residual nerves, then 18F-DA-derived radioactivity would be decreased, and arterial 18F-DOPAC levels would be decreased to a smaller extent. Based on these considerations, a vesicular uptake index was devised, and evidence for a vesicular storage defect was obtained in Lewy body diseases.
Another approach for examining vesicular storage, which has been applied both in the putamen and heart, is based on the kinetics of radioactivity after neuronal uptake of PET imaging agents. Accelerated loss of 18F-DOPA-derived radioactivity in the putamen or of 18F-DA-derived radioactivity in the left ventricular myocardium cannot be explained by loss of catecholaminergic terminals and indicates increased turnover of vesicular stores. Under resting conditions, most of tissue catecholamine turnover reflects net leakage from vesicles into the cytoplasm and enzymatic deamination of the cytoplasmic catecholamines, with rapid exit of the metabolite DOPAC (from DA) and 3,4-dihydroxyphenylglycol (DHPG, from NE) (Eisenhofer et al., Citation2004). Accordingly, the percent decline in putamen 18F-DOPA-derived radioactivity from its peak value provides an inverse index of vesicular sequestration in nigrostriatal dopaminergic terminals; and analogously the slope of decline in myocardial 18F-DA-derived radioactivity provides an inverse index of vesicular sequestration in cardiac sympathetic nerves. In PD and PAF, there is accelerated loss of myocardial 18F-DA-derived radioactivity, and in PD there is also accelerated loss of putamen 18F-DOPA-derived radioactivity (PAF does not involve a putamen dopaminergic lesion) (Goldstein, Holmes, et al., Citation2015).
Postmortem catechol neurochemistry has confirmed the results using these in vivo methods (Goldstein et al., Citation2014). We devised, validated, and applied five neurochemical indices of the sequestration–deamination shift—concentration ratios of DOPAC/DA, DA/NE, DHPG/NE, DOPAC/NE, and DHPG/DOPAC—and used a kinetic model to estimate the extent of the vesicular storage defect (Goldstein et al., Citation2014). The majority of patients with PD had severe NE depletion (mean 2% of control), and in this subgroup all five indices of a sequestration–deamination shift were found to be increased compared to controls.
A postmortem study of isolated vesicles from PD patients has helped pin-point the mechanistic bases of the vesicular storage defect (Pifl et al., Citation2014). Vesicular uptake of DA was reduced by 71–80% and binding of the type 2 vesicular monoamine transporter (VMAT2) ligand 3H-dihydrotetrabenazine by 87–90%. After correcting for loss of dopaminergic terminals, DA uptake per VMAT2 transport site was reduced in PD putamen by 55%. The results indicate that the vesicular storage defect results at least partly from abnormalities of the VMAT2 protein itself.
The results of a study of forearm vein biopsy specimens from patients with PAF were interpreted in terms of a vesicular storage defect in extant sympathetic noradrenergic nerves (Guo et al., Citation2018). The patients had normal abundance of cell membrane norepinephrine transporter (NET) protein and reduced VMAT2 protein expression compared with controls.
A recent study has provided evidence that decreased vesicular sequestration is an early finding that can precede overt neuronal loss (Lamotte et al., Citation2019). Interventricular septal 18F-DA-derived radioactivity at 8 min after tracer injection (8′ radioactivity) was taken to be an index of myocardial sympathetic innervation, and the slope of mono-exponential decline of radioactivity between 8 and 25 min (k8′–25′) was taken to be an index of intra-neuronal vesicular storage. Upon initial evaluation patients with PD or PAF had both low mean 8′ radioactivity and elevated k8′–25′ values compared to healthy volunteers. In PD, 8′ radioactivity decreased by a median of 4% per year, whereas k8′–25′ values did not change during follow-up. Neuroimaging evidence of decreased vesicular uptake in cardiac sympathetic nerves therefore seems to be present upon initial evaluation of patients with Lewy body diseases and may provide an early biomarker of sympathetic neurodegeneration (“sick-before-dead”).
Catecholamine biosynthesis
DA biosynthesis depends on two enzymes in series—TH, which converts the amino acid tyrosine to the catechol amino acid 3,4-dihydroxyphenylalanine (DOPA), and l-aromatic-amino-acid decarboxylase (LAAAD), which converts DOPA to DA. Postmortem neurochemistry has indicated decreased putamen LAAAD activity in PD (Goldstein et al., Citation2017) and decreased TH and LAAAD activity in myocardial sympathetic nerves (Goldstein et al., Citation2019).
NE biosynthesis depends also on vesicular uptake, since DA-beta-hydroxylase (DBH), the enzyme catalyzing the conversion of DA to NE, is localized to the vesicles. This means that decreased TH activity, decreased LAAAD activity, and decreased vesicular sequestration of cytoplasmic catecholamines would be expected to result in severely decreased NE stores. Perhaps surprisingly, DBH activity in cardiac sympathetic nerves seems to be normal in Lewy body diseases (Goldstein et al., Citation2019).
Figure 1. Overview of the catecholaldehyde hypothesis. Dopamine (DA) is synthesized from 3,4-dihydroxyphenylalanine (DOPA) by l-aromatic-amino-acid decarboxylase (LAAAD), after DOPA is produced from tyrosine via tyrosine hydroxylase (TH). DOPAL is formed from the action of monoamine oxidase-A (MAO-A) on cytoplasmic dopamine (DA). DOPAL is detoxified by aldehyde dehydrogenase (ALDH), which converts DOPAL to 3,4-dihydroxyphenylacetic acid (DOPAC). DOPAC rapidly exits the cell. According to the catecholaldehyde hypothesis, interactions of DOPAL and the protein alpha-synuclein set the stage for vicious cycles that challenge homeostasis in catecholaminergic neurons. DOPAL oxidizes spontaneously to DOPAL-quinone (DOPAL-Q). DOPAL reacts with hydrogen peroxide and divalent metal cations to form hydroxyl radicals, which peroxidate membrane lipids. The lipid peroxidation products 4-hydroxynonenal (4-HNE) and malondialdehyde (MD) inhibit ALDH. DOPAL, probably via oxidation to DOPAL-Q, oligomerizes and forms quinoprotein adducts with (“quinonizes”) AS. DOPAL-induced synuclein oligomers impede vesicular functions, evoking destabilizing positive feedback loop.
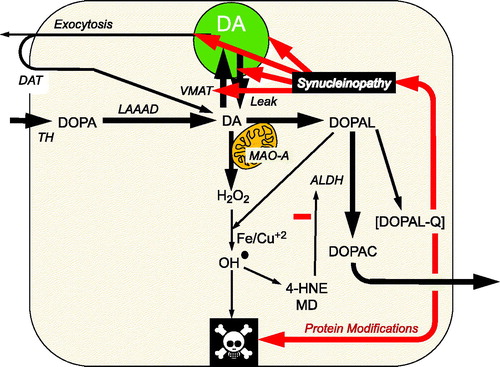
DOPAL, alpha-synuclein, and the catecholaldehyde hypothesis
Given that there is a substantial complement of catecholaminergic neurons that are sick-but-not-dead in Lewy body diseases and that the affected neurons have specific functional abnormalities that contribute importantly to neurotransmitter depletion, why are they sick? What causes the unusual susceptibility of catecholaminergic neurons in Lewy body diseases? We have proposed that the catecholaldehyde 3,4-dihydroxyphenylacetaldehyde (DOPAL), an obligate intermediate of DA metabolism, interacts with the protein alpha-synuclein (AS) to produce multiple functional abnormalities (Goldstein & Sharabi, Citation2019) (). DOPAL potently oligomerizes, aggregates, and forms quinone-protein adducts with (“quinonizes”) AS and many other intra-cellular proteins (Jinsmaa et al., Citation2018). DOPAL covalently inhibits activities of both TH (Mexas et al., Citation2011) and LAAAD (Jinsmaa et al., Citation2018). Moreover, DOPAL-induced AS oligomers impede vesicular functions (Plotegher et al., Citation2017), which could set the stage for a damaging positive feedback loop, destroying the neurons and resulting in a variety of clinical manifestations including parkinsonism and sympathetic neurocirculatory failure.
Where stress might fit in
Catecholaminergic neurons in the brain and autonomic nervous system are well known to play key roles in coordinated organismic responses to stressors. Most of the catecholamine released by exocytosis is taken back up into the neuronal cytoplasm via the cell membrane DA transporter (DAT) and cell membrane NE transporter (NET). Stress-induced exocytosis and neuronal reuptake therefore indirectly shift tissue catecholamines from vesicular stores into the cytoplasm. In the setting of a vesicular storage defect and decreased aldehyde dehydrogenase (ALDH) activity, one would predict increased cytoplasmic catecholaldehyde levels. This prediction has been confirmed in cells (Goldstein et al., Citation2012) but has not yet been tested in animals. Postmortem studies have noted both elements of this “double hit” and increased putamen DOPAL content with respect to DA content in putamen tissue from PD patients (Goldstein et al., Citation2013; Goldstein, Sullivan, et al., Citation2011) and patients with multiple system atrophy (Goldstein, Sullivan, et al., Citation2015). Chronic restraint stress reduces nigral dopaminergic and locus ceruleus (LC) noradrenergic neuronal numbers, possibly via oxidative stress (Sugama et al., Citation2016), but whether the fate of intra-neuronal catecholamines is altered in this model is unknown. A recent report focused on inclusions of the protein tau in the LC, an early neuropathologic change in Alzheimer’s disease. 3,4-Dihydroxyphenylglycolaldehyde (DOPEGAL), the catecholaldehyde produced by MAO-A acting on cytoplasmic NE, was found to elicit LC neuronal degeneration and tau pathology (Kang et al., Citation2020). Exposure to a variety of stressors increases expression and levels of TH (Fluharty et al., Citation1985; Kvetnansky et al., Citation1970; Nankova et al., Citation1994) and LAAAD (Kubovcakova et al., Citation2004), which would be expected to augment cytoplasmic DA production. In general, although chronic stress might play a role in the pathogenetic processes leading to PD (Djamshidian & Lees, Citation2014), this is an area for future research.
Therapeutic implications
The sick-but-not-dead phenomenon and catecholaldehyde hypothesis have direct treatment implications in terms of slowing or even preventing catecholaminergic neurodegeneration in Lewy body diseases. You cannot treat neurons that are dead; however, if there were a complement of dysfunctional but extant neurons, and the specific abnormalities in neurotransmitter synthesis, storage, and recycling were identified, then it might be possible in at-risk individuals to introduce multi-focused adjustments that aim to reset the underlying homeostatic machinery. Thus, increasing vesicular uptake (Lohr et al., Citation2014), decreasing MAO activity (Youdim & Weinstock, Citation2004), and augmenting ALDH activity (Chiu et al., Citation2015) tend to protect against development of parkinsonism in animal models. The catecholaldehyde hypothesis predicts that a disease-modification approach combining an MAO inhibitor (to decrease DOPAL production) with an anti-oxidant (to decrease spontaneous oxidation of DOPAL to DOPAL-quinone) might slow the progression of Lewy body diseases.
Disclosure statement
There are no conflicts of interest to disclose.
Additional information
Funding
Notes on contributors
David S. Goldstein
David S. Goldstein, MD PhD, is a Senior Investigator and Chief of the Autonomic Medicine Section in the Division of Intramural Research, NINDS, NIH. He is a founder and thought leader in clinical catecholamine neurochemistry and catecholaminergic neuroimaging. His main discovery has been profound cardiac catecholamine deficiency in Parkinson disease.
References
- Amino, T., Orimo, S., Itoh, Y., Takahashi, A., Uchihara, T., & Mizusawa, H. (2005). Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathology (Zurich, Switzerland), 15(1), 29–34. https://doi.org/10.1111/j.1750-3639.2005.tb00097.x
- Chiu, C. C., Yeh, T. H., Lai, S. C., Wu-Chou, Y. H., Chen, C. H., Mochly-Rosen, D., Huang, Y. C., Chen, Y. J., Chen, C. L., Chang, Y. M., Wang, H. L., & Lu, C. S. (2015). Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Experimental Neurology, 263, 244–253. https://doi.org/10.1016/j.expneurol.2014.09.016
- DelleDonne, A., Klos, K. J., Fujishiro, H., Ahmed, Z., Parisi, J. E., Josephs, K. A., Frigerio, R., Burnett, M., Wszolek, Z. K., Uitti, R. J., Ahlskog, J. E., & Dickson, D. W. (2008). Incidental Lewy body disease and preclinical Parkinson disease. Archives of Neurology, 65(8), 1074–1080. https://doi.org/10.1001/archneur.65.8.1074
- Djamshidian, A., & Lees, A. J. (2014). Can stress trigger Parkinson's disease? Journal of Neurology, Neurosurgery, and Psychiatry, 85(8), 878–881. https://doi.org/10.1136/jnnp-2013-305911
- Eisenhofer, G., Kopin, I. J., & Goldstein, D. S. (2004). Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacological Reviews, 56(3), 331–349. https://doi.org/10.1124/pr.56.3.1
- Fluharty, S. J., Rabow, L. E., Stricker, E. M., & Zigmond, M. J. (1985). Tyrosine hydroxylase activity in the sympathoadrenal system under basal and stressful conditions: Effect of 6-hydroxydopamine. The Journal of Pharmacology and Experimental Therapeutics, 235(2), 354–360.
- Goldstein, D. S., Holmes, C., Kopin, I. J., & Sharabi, Y. (2011). Intra-neuronal vesicular uptake of catecholamines is decreased in patients with Lewy body diseases. The Journal of Clinical Investigation, 121(8), 3320–3330. https://doi.org/10.1172/JCI45803
- Goldstein, D. S., Holmes, C., Sullivan, P., Mash, D. C., Sidransky, E., Stefani, A., Kopin, I. J., & Sharabi, Y. (2015). Deficient vesicular storage: A common theme in catecholaminergic neurodegeneration. Parkinsonism & Related Disorders, 21(9), 1013–1022. https://doi.org/10.1016/j.parkreldis.2015.07.009
- Goldstein, D. S., & Orimo, S. (2009). Cardiac sympathetic neuroimaging: Summary of the First International Symposium. Clinical Autonomic Research, 19(3), 137–136. https://doi.org/10.1007/s10286-009-0002-9
- Goldstein, D. S., Pekker, M. J., Eisenhofer, G., & Sharabi, Y. (2019). Computational modeling reveals multiple abnormalities of myocardial noradrenergic function in Lewy body diseases. JCI Insight, 4(16):e130441. https://doi.org/10.1172/jci.insight.130441
- Goldstein, D. S., & Sharabi, Y. (2019). The heart of PD: Lewy body diseases as neurocardiologic disorders. Brain Research, 1702, 74–84. https://doi.org/10.1016/j.brainres.2017.09.033
- Goldstein, D. S., Sullivan, P., Cooney, A., Jinsmaa, Y., Sullivan, R., Gross, D. J., Holmes, C., Kopin, I. J., & Sharabi, Y. (2012). Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells: Relevance to the pathogenesis of Parkinson's disease. Journal of Neurochemistry, 123(6), 932–943. https://doi.org/10.1111/j.1471-4159.2012.07924.x
- Goldstein, D. S., Sullivan, P., Holmes, C., Kopin, I. J., Basile, M. J., & Mash, D. C. (2011). Catechols in post-mortem brain of patients with Parkinson disease. European Journal of Neurology, 18(5), 703–710. https://doi.org/10.1111/j.1468-1331.2010.03246.x
- Goldstein, D. S., Sullivan, P., Holmes, C., Kopin, I. J., Sharabi, Y., & Mash, D. C. (2015). Decreased vesicular storage and aldehyde dehydrogenase activity in multiple system atrophy. Parkinsonism & Related Disorders, 21(6), 567–572. https://doi.org/10.1016/j.parkreldis.2015.03.006
- Goldstein, D. S., Sullivan, P., Holmes, C., Mash, D. C., Kopin, I. J., & Sharabi, Y. (2017). Determinants of denervation-independent depletion of putamen dopamine in Parkinson’s disease and multiple system atrophy. Parkinsonism & Related Disorders, 35, 88–91. https://doi.org/10.1016/j.parkreldis.2016.12.011
- Goldstein, D. S., Sullivan, P., Holmes, C., Miller, G. W., Alter, S., Strong, R., Mash, D. C., Kopin, I. J., & Sharabi, Y. (2013). Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. Journal of Neurochemistry, 126(5), 591–603. https://doi.org/10.1111/jnc.12345
- Goldstein, D. S., Sullivan, P., Holmes, C., Miller, G. W., Sharabi, Y., & Kopin, I. J. (2014). A vesicular sequestration to oxidative deamination shift in myocardial sympathetic nerves in Parkinson disease. Journal of Neurochemistry, 131(2), 219–228. https://doi.org/10.1111/jnc.12766
- Guo, L., Esler, M. D., Sari, C., Phillips, S., Lambert, E. A., Straznicky, N. E., Lambert, G. W., & Corcoran, S. J. (2018). Does sympathetic dysfunction occur before denervation in pure autonomic failure? Clinical Science, 132(1), 1–16. https://doi.org/10.1042/CS20170240
- Jain, S., & Goldstein, D. S. (2012). Cardiovascular dysautonomia in Parkinson disease: From pathophysiology to pathogenesis. Neurobiology of Disease, 46(3), 572–580. https://doi.org/10.1016/j.nbd.2011.10.025
- Jinsmaa, Y., Sharabi, Y., Sullivan, P., Isonaka, R., & Goldstein, D. S. (2018). 3,4-Dihydroxyphenylacetaldehyde-induced protein modifications and their mitigation by N-acetylcysteine. The Journal of Pharmacology and Experimental Therapeutics, 366(1), 113–124. https://doi.org/10.1124/jpet.118.248492
- Kang, S. S., Liu, X., Ahn, E. H., Xiang, J., Manfredsson, F. P., Yang, X., Luo, H. R., Liles, L. C., Weinshenker, D., & Ye, K. (2020). Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. The Journal of Clinical Investigation, 130(1), 422–437. https://doi.org/10.1172/JCI130513
- Kish, S. J., Shannak, K., & Hornykiewicz, O. (1988). Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. The New England Journal of Medicine, 318(14), 876–880. https://doi.org/10.1056/NEJM198804073181402
- Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., Halliday, G. M., & Bartus, R. T. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain, 136(Pt 8), 2419–2431. https://doi.org/10.1093/brain/awt192
- Kubovcakova, L., Krizanova, O., & Kvetnansky, R. (2004). Identification of the aromatic l-amino acid decarboxylase gene expression in various mice tissues and its modulation by immobilization stress in stellate ganglia. Neuroscience, 126(2), 375–380. https://doi.org/10.1016/j.neuroscience.2004.04.005
- Kvetnansky, R., Weise, V. K., & Kopin, I. J. (1970). Elevation of adrenal tyrosine hydroxylase and phenylethanolamine-N-methyl transferase by repeated immobilization of rats. Endocrinology, 87(4), 744–749. https://doi.org/10.1210/endo-87-4-744
- Lamotte, G., Holmes, C., Wu, T., & Goldstein, D. S. (2019). Long-term trends in myocardial sympathetic innervation and function in synucleinopathies. Parkinsonism & Related Disorders, 67, 27–33. https://doi.org/10.1016/j.parkreldis.2019.09.014
- Lohr, K. M., Bernstein, A. I., Stout, K. A., Dunn, A. R., Lazo, C. R., Alter, S. P., Wang, M., Li, Y., Fan, X., Hess, E. J., Yi, H., Vecchio, L. M., Goldstein, D. S., Guillot, T. S., Salahpour, A., & Miller, G. W. (2014). Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proceedings of the National Academy of Sciences of the United States of America, 111(27), 9977–9982. https://doi.org/10.1073/pnas.1402134111
- Mexas, L. M., Florang, V. R., & Doorn, J. A. (2011). Inhibition and covalent modification of tyrosine hydroxylase by 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Neurotoxicology, 32(4), 471–477. https://doi.org/10.1016/j.neuro.2011.03.013
- Nankova, B., Kvetnansky, R., McMahon, A., Viskupic, E., Hiremagalur, B., Frankle, G., Fukuhara, K., Kopin, I. J., & Sabban, E. L. (1994). Induction of tyrosine hydroxylase gene expression by a nonneuronal nonpituitary-mediated mechanism in immobilization stress. Proceedings of the National Academy of Sciences of the United States of America, 91(13), 5937–5941. https://doi.org/10.1073/pnas.91.13.5937
- Orimo, S., Amino, T., Takahashi, A., Kojo, T., Uchihara, T., Mori, F., Wakabayashi, K., & Takahashi, H. (2006). Cardiac sympathetic denervation in Lewy body disease. Parkinsonism & Related Disorders, 12(Suppl. 2), S99–S105. https://doi.org/10.1016/j.parkreldis.2006.05.030
- Pifl, C., Rajput, A., Reither, H., Blesa, J., Cavada, C., Obeso, J. A., Rajput, A. H., & Hornykiewicz, O. (2014). Is Parkinson's disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. The Journal of Neuroscience, 34(24), 8210–8218. https://doi.org/10.1523/JNEUROSCI.5456-13.2014
- Plotegher, N., Berti, G., Ferrari, E., Tessari, I., Zanetti, M., Lunelli, L., Greggio, E., Bisaglia, M., Veronesi, M., Girotto, S., Dalla Serra, M., Perego, C., Casella, L., & Bubacco, L. (2017). DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Scientific Reports, 7, 40699. https://doi.org/10.1038/srep40699
- Sugama, S., Sekiyama, K., Kodama, T., Takamatsu, Y., Takenouchi, T., Hashimoto, M., Bruno, C., & Kakinuma, K. (2016). Chronic restraint stress triggers dopaminergic and noradrenergic neurodegeneration: Possible role of chronic stress in the onset of Parkinson's disease. Brain, Behavior, and Immunity, 51, 39–46. https://doi.org/10.1016/j.bbi.2015.08.015
- Youdim, M. B., & Weinstock, M. (2004). Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology, 25(1–2), 243–250. https://doi.org/10.1016/S0161-813X(03)00103-7