
ABSTRACT
Traumatic brain injury (TBI) is a leading cause of human death and disability with no effective therapy to fully prevent long-term neurological deficits in surviving patients. Ketone ester supplementation is protective in animal models of neurodegeneration, but its efficacy against TBI pathophysiology is unknown. Here, we assessed the neuroprotective effect of the ketone monoester, 3-hydroxybutyl-3-hydroxybutyrate, (KE) in male Sprague Dawley rats (n=32). TBI was induced using the controlled cortical impact (CCI) with Sham animals not receiving the brain impact. KE was administered daily by oral gavage (0.5 ml/kg/day) and provided ad libitum at 0.3% (v/v) in the drinking water. KE supplementation started immediately after TBI and lasted for the duration of the study. Motor and sensory deficits were assessed using the Neurobehavioral Severity Scale-Revised (NSS-R) at four weeks post-injury. The NSS-R total score in CCI + KE (1.2 ± 0.4) was significantly lower than in CCI + water (4.4 ± 0.5). Similarly, the NSS-R motor scores in CCI + KE (0.6 ± 0.7) were significantly lower than CCI + water (2.9 ± 1.5). Although the NSS-R sensory score in the CCI + KE group (0.5 ± 0.2) was significantly lower compared to CCI + water (1.8 ± 0.4), no difference was observed between CCI + water and Sham + water (1.0 ± 0.2) groups. The lesion volume was smaller in the CCI + KE (10 ± 3 mm3) compared to CCI + water (47 ± 11 mm3; p < 0.001). KE significantly decreased Iba1+ stained areas in the cortex and hippocampus, and GFAP+ stained areas in all brain regions analyzed – prefrontal cortex, hippocampus, cortex, amygdala (p < 0.01). In summary, our results indicate that KE can protect against TBI-induced morphological and functional deficits when administered immediately after an insult.
Introduction
Traumatic brain injury (TBI) is a leading cause of hospitalization, morbidity, and mortality in the United States (US) and affects both civilians and the military. It is estimated that 2.87 million people suffered TBI in 2014, which is a 53% increase from 2006 [Citation1]. Many TBI patients survive the initial incident, but frequently develop long-term sensory, motor, cognitive and emotional dysfunction, a burden on the individual, their family and their community. Current pharmacological interventions for TBI, such as anticonvulsants, antidepressants, antipsychotics and psychostimulants are ineffective in fully preventing and/or ameliorating TBI-related consequences, and contribute to unwanted side effects [Citation2,Citation3]. Therefore, new, effective and well-tolerated therapies are needed to reduce the long-term impact of TBI on individual and public health.
The pathophysiology of TBI involves both the primary injury – skull fracture, contusion, concussion, lacerations – and the consequent secondary injuries – neurochemical imbalance, excitotoxicity, oxidative stress, and mitochondrial dysfunction [Citation4]. These pathophysiological alterations directly contribute to neuronal death and also trigger neuroinflammation [Citation4,Citation5]. Microglia, astrocytes and infiltrating peripheral immune cells are attracted to the site of injury in an attempt to alleviate the injury, limit lesion progression and aid in tissue repair. However, TBI-induced inflammation frequently evolves into a chronic condition and results in sustained activation of glial cells and release of pro-inflammatory cytokines, which further contribute to cell death [Citation6].
Brain glucose metabolism also changes following TBI. A transient increase in brain glucose uptake during the acute phase of TBI is followed by a more prolonged decrease in cerebral glucose metabolism [Citation7–9]. This metabolic depression is observed in animal models and clinical TBI, and its duration varies according to age and injury severity [Citation7–13]. Cerebral blood flow is also reduced, which affects appropriate delivery of oxygen and metabolites necessary for brain function [Citation14]. Altogether, the pathophysiology of TBI is characterized by increased energy demands and reduced availability of cellular fuel, which result in an ‘energy crisis’ and contribute to further neuronal damage [Citation4].
It has been suggested that providing the brain with an alternative fuel, such as the ketone bodies d-β-hydroxybutyrate (βHB) and acetoacetate [Citation15,Citation16], may compensate for low glucose availability and prevent cell death. βHB is transported through the blood brain barrier (BBB) by the monocarboxylic acid transporters and, upon crossing the BBB, enters mitochondria to support cellular energy requirements [Citation16]. Increasing blood ketone levels by providing a ketogenic diet [Citation17,Citation18] or fasting [Citation19] appears to improve TBI pathophysiology in animal models, but such diets are restrictive and difficult to implement. Such diets also take hours, and even days, to significantly raise blood ketone levels [Citation20]. Exogenous ketones, effectively delivered as a ketone ester (KE) drink,{Clarke, 2012; Cox, 2016; Stubbs, 2017} rapidly elevate ketone blood levels without fasting or limiting carbohydrate intake and are safe for long-term use [Citation21,Citation22]. The KE has Generally Recognized As Safe (GRAS) status in the US under Food and Drug Administration (FDA) regulations, and has been sold for several years in the US as a sports drink. However, the efficacy of the KE drink as neuroprotective against TBI has not been investigated. In the present study, we assessed the protective effect of ketone-ester supplementation against TBI-induced behavioral and neuropathological alterations after controlled cortical impact (CCI) in male Sprague Dawley rats. Our hypothesis is that oral administration of ketone ester significantly increases blood levels of ketone bodies and reduces TBI-related deficit.
Materials and methods
Ethics statement
All animal experiments were conducted in accordance with guidelines from Animal Research: Reporting of In Vivo Experiments (ARRIVE) and the Uniformed Services University of the Health Sciences Institutional Animal Care and Use Committees (IACUC), which provided final approval. All efforts were made to minimize the number of animals used and any pain or distress associated with these experiments.
Animals
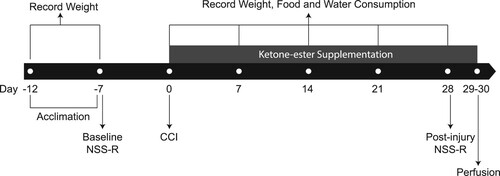
A total of 44 male Sprague Dawley rats was purchased from Taconic Farms (Rockville, MD, USA) and delivered to the Uniformed Services University of the Health Sciences animal facility at 8 weeks of age (average body weight – 313 g) – 12 rats were used to determine the dose–response curve of oral KE and 32 rats were randomly assigned to one of the four experimental groups described below to investigate the neuroprotective effect of KE. Animals were housed in pairs and allowed to acclimate to an environmentally controlled room (20–23°C, ∼44% humidity, 12-h light/12-h dark cycle [350-400 lux], lights off at 6:00 am) for four days with food (Harlan Teklad Global Diet 2018 rodent diet, 24% of calories from protein, 18% from fat, and 58% from carbohydrates; Harlan Laboratories; Indianapolis, IN) and water available ad libitum. Body weights were recorded on the day animals arrived, at the beginning of the study, and then weekly throughout the subsequent 5 weeks. Food and water consumption was recorded weekly for the duration of the study. The study timeline is summarized in .
Figure 1. Study timeline. Animals acclimated for four days prior to the experiments. NSS-R test was conducted at baseline and four weeks after CCI. KE supplementation started immediately after CCI and was performed daily throughout the study. Body weight was recorded on the day animals arrived, at the beginning of the study, and then weekly for a total of 7 time points. Food and water consumption were recorded weekly at 5 time points.
Controlled cortical impact
Following acclimation, rats were subjected to CCI, which was administered according to a previously established protocol [Citation23]. Briefly, animals were anesthetized in an induction chamber with isoflurane (5%) mixed with oxygen (100%). After having their entire head shaved, they were placed in the stereotaxic frame and kept under anesthesia with isoflurane (2–3%) mixed with oxygen (100%) delivered via a nose cone. Ophthalmic ointment was applied to both eyes and core body temperature was maintained at 36–37°C by using a heating pad and the D.C. Temperature Control System (FHC, Bowdoin, ME). Surgical procedures were initiated only after verifying the depth of anesthesia, by the lack of paw-pinch reflex. The shaved area was cleaned with betadine and alcohol and a midline incision was made into the skin to expose the skull. A 5.0 mm burr hole was drilled into the skull, 3.0 mm lateral to the midline and 4.0 mm posterior to the bregma, over the left temporoparietal cortex. Injury was produced using a 3.0 mm flat steel tip to deliver an impact at a velocity of 3.5 m/sec, with a penetration depth of 2.0 mm and a dwell time of 200 ms. Following injury, the skullcap was replaced and fixed using bone wax (Ethicon, Sommerville, NJ) and the incision was closed with absorbable sutures (Stoelting, Wood Dale, IL). Sham-treated controls received the craniotomy, but no CCI injury.
Ketone-ester supplementation
The KE, (R)-3-hydroxybutyl (R)-3-hydroxybutyrate (TΔS® Ltd, Oxfordshire, UK), is a food similar to glucose with a calorie content of 4.9 kcal/g. Rat body weights were recorded and an appropriate volume of KE (0.5 ml/kg) for each rat was calculated. KE was administered by oral gavage that started immediately after surgery and then daily for the 30 days of the study. Control rats received water by gavage. A second source of KE was supplied ad libitum in drinking water at a concentration of 0.3% to ensure continuous consumption, oral gavage being performed only once a day. A group of 32 rats were randomly assigned to one of the following groups: Sham (surgery, no impact, water by gavage), Sham KE (surgery, no impact, KE by gavage), CCI (surgery, impact, water by gavage), and CCI KE (surgery, impact, KE by gavage) for a total of 8 rats per group. A dose–response curve of d-β-hydroxybutyrate blood levels following KE oral gavage was established in a separate group of 12 rats. Blood was obtained by tail prick and βHB concentrations were measured at baseline then 30, 60, 120 and 240 min after oral gavage using a handheld device (Precision Xtra Blood Glucose & Ketone Meter, SKU # 9881465; Abbott, Abbott Park, IL).
Revised neurobehavioral severity scale
The revised Neurobehavioral Severity Scale (NSS-R) was used to assess sensory and motor reflexes prior to injury (baseline) and four weeks post-injury as previously described [Citation24]. On the testing day, animals were transferred to a designated experiment room and allowed to acclimate for one hour before beginning experiments. NSS-R was administered in the morning (during rats’ dark phase) and before oral gavage in order to minimize the effects of stress on test performance. Ten tasks were administered in the order of least to most ‘stressful’, so that subsequent tasks would be minimally affected by preceding tasks. Each task was administered immediately after the preceding one, without significant interval between tasks. The order of administering the tasks was consistent for all rats and was not randomized by treatment group. Total NSS-R testing time was approximately five minutes per animal. Tasks 1–5 and tasks 6–10 were designed to assess motor and sensory reflexes, respectively. The NSS-R tasks were administered as follows: 1) General Balance Test – rats were individually placed on a balance beam (100 cm long, 2 cm wide) at a height of 29 cm and their balance was observed. 2) Landing Test – rats were dropped with their paws facing down from the height of the balance beam into the cage below. 3) Tail Raise Test – rats were gently lifted above the balance beam (∼40 cm) by the base of their tail (dorsal side up). 4) Drag Test – rats were placed on a flat surface being held by the base of their tail, and then dragged backwards at a constant speed (about 20 cm/sec) while only their forepaws were allowed to touch the surface. 5) Righting Reflex – rats were placed on their backs and observed for their ability to right themselves. 6) Ear Touch Reflex – the auditory meatus was lightly touched with the cotton end of a long Q-tip. 7) Eye Touch Reflex – both eyes (one at time) were lightly touched with the cotton end of a Q-tip. 8) Sound Reflex – a short, sharp and strong clap of hands was performed 10–15 cm away from each rat (kept facing away from hands). 9) Tail Pinch Reflex – a strong pinch was briefly applied between the middle and the base of the rat’s tail. 10) Paw Flexion Reflex – a strong pinch was briefly applied to each rat’s hind paw. NSS-R tasks were observed and scored using a three-point Likert scale, in which a normal, healthy response was assigned a ‘0’, a partial or compromised response was assigned a ‘1’, and the absence of a response was assigned a ‘2’ (). Therefore, the NSS-R has a scoring range of 0–20 and higher scores reflect a greater extent of injury. The NSS-R was scored by a trained and independent rater, who was blinded to the experimental condition of each rat and had a previously established reliability ≥ 0.9.
Table 1. Scoring criteria for the Revised Neurobehavioral Severity Scale.
Fixation and tissue processing
After completion of NSS-R assessment on week 4 of the study, rats underwent transcardial perfusion and had their brains removed and processed for histopathological analysis. Animals were anesthetized using Fatal Plus (65–75 mg/kg of body weight, intraperitoneally). Depth of anesthesia was assessed by lack of paw-pinch response and slow and even respiration. A middle sternal thoracotomy was performed to expose the heart and rats were transcardially perfused with phosphate buffered saline (PBS, 200 ml) followed by 4% paraformaldehyde (1 ml/g of body weight). After decapitation, brains were removed, post-fixed in 4% paraformaldehyde overnight at 4°C, and then transferred to a 30% sucrose solution in PBS for 72 h for cryoprotection. Brains were then quickly frozen using dry ice and stored at −80°C until sectioning. One-in-five series of sections containing the prefrontal cortex (starting at bregma + 3.70 mm) and the rostro-caudal extension of the amygdala and hippocampus (starting at bregma – 2.90 mm) was cut at 40 μm on a sliding microtome (Leica Microsystems SM2000R) and stored in a cryoprotectant solution at −20°C. One series of free-floating sections (15 slices per animal), was collected from the cryoprotectant solution and washed in PBS three times for 5 min each. Slices were mounted on a slide, air-dried and processed for Nissl staining with cresyl violet for subsequent stereological quantification of lesion volume.
Lesion volume quantification
Whole brain slices were imaged using a Zeiss AxioScan microscope (Zeiss, Jena, Germany) under a 10X objective. Lesion volume was quantified by a researcher blind to the experimental conditions using ImageJ software (National Institute of Health, USA, version 1.52k) as previously published [Citation25]. The region of interest (ROI) outlining the CCI lesion was manually drawn in the coronal plane using ImageJ software. The area of the lesion in each of the 15 slices `1wvb gf1` was calculated and multiplied by the distance between two adjacent slices (200 μm). The volume from each segment of the lesion was then summed to calculate the total volume of the lesion.
Immunohistochemistry
A total of eight slices, three containing prefrontal cortex and five containing cortical region around the lesion, hippocampus and amygdala were randomly selected from one series of free-floating sections. The slices were washed three times for 5 min each in 0.1 M PBS, and incubated in Universal Antigen Retrieval Solution (CTS015, R&D Systems, Minneapolis, MN) at 80°C for 20 min using a water bath. After cooling down to room temperature for 1 h, slices were blocked in a solution containing 10% normal goat serum (Millipore Sigma, Burlington, MA) and 0.3% Triton X-100 in PBS for 1 h at room temperature. The sections were then incubated with mouse anti-GFAP (glial fibrillary acid protein, 1:500, ab4674, Abcam, Cambridge, MA) and anti-Iba1 (ionized calcium binding protein, 1:500, 019-19741, Wako, Richmond, VA) serum, 2% normal goat serum, 0.3% Triton X-100 in PBS overnight at 4°C. The sections were rinsed, three times for 10 min each, in 0.3% Triton X-100 in PBS followed by incubation with Alexa Fluor594-conjugated goat anti-rabbit (1:1000, Jackson ImmunoResearch, West Grove, PA) and Alexa Fluor488-conjugated donkey anti-chicken (1:1000, Jackson ImmunoResearch, West Grove, PA) antibodies, 2% normal goat serum, 0.3% Triton X-100 in PBS for 1 h at room temperature. After rinsing three times for 10 min each in 0.3% Triton X-100 in PBS, the sections were mounted on slides, air-dried, and coverslips were affixed with ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA).
Stained area quantification
Image quantification of GFAP and Iba1 stained cells was performed by a researcher blind to the experimental conditions using ImageJ software as previously described [Citation26]. Fluorescent images were acquired using a Leica AF6000 microscope under a 20X objective. A total of 12 pictures (six ipsilateral and six contralateral to CCI) of each brain region (prefrontal cortex, hippocampus, amygdala and cortex) around the CCI lesion were randomly taken for each animal. All pictures were taken using the same camera setting and exposure time for each channel and 8-bit images were used for quantification. After a fixed background subtraction (Rolling Ball Radius: 50.0 pixel), we applied a fixed threshold to all images. Finally, the labeled area was quantified and averaged for each animal.
Statistical analysis
Values are presented as means ± standard error of the mean (SEM). The effects of TBI and KE on mean differences between groups of NSS-R total, motor and sensory scores were analyzed using analysis of covariance (ANCOVA). Baseline NSS-R total, sensory and motor scores were used as covariates. The effects of TBI and KE on mean differences between groups of all other outcome variables were analyzed using two-way analysis of variance (ANOVA). Multiple comparisons were performed using ANCOVA followed by Bonferroni post-hoc test or ANOVA followed by Tukey’s post-hoc test. Mean differences in the lesion volume were compared using an independent t-test. Cohen’s d effect size was calculated to assess the standardized difference between means. The relationship between different outcome variables was determined using partial correlations after controlling for baseline NSS-R scores and multivariate regression analysis. Statistical significance was at p < 0.05 (two-tailed) for all analysis methods. Sample size (n) refers to the number of rats.
Results
Ketone ester dose–response curve
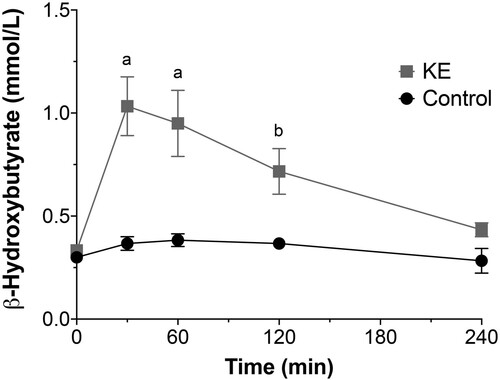
Oral gavage of KE raised blood βHB concentrations significantly higher than in control rats (): βHB concentrations were 1.03 ± 0.14 mmol/L (p < 0.0001) at 30 min, 0.96 ± 0.16 mmol/L (p < 0.0001) at 60 min, and 0.71 ± 0.11 mmol/L (p < 0.05) at 120 min after administration.
Figure 2. Dose-response curve over time of β-hydroxybutyrate blood levels following oral gavage of KE (0.5 ml/kg). KE quickly and significantly increased blood levels of β-hydroxybutyrate compared with control rats. (n = 6 rats/group; a p < 0.0001; b p < 0.05).
Body weight
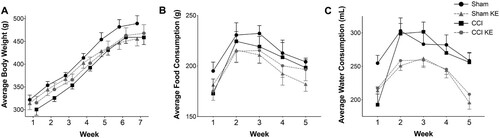
Rats in all experimental groups had similar average body weight at arrival (Sham: 322 ± 10 g; Sham KE: 315 ± 14 g; CCI: 301 ± 13 g; CCI KE: 315 ± 14 g). Body weights of animals in all four groups increased over time [F(6,168) = 187.0, p < 0.0001], but neither TBI nor KE had a significant effect on weight gain. At the end of the study, average body weight was 489 ± 17 g for Sham rats, 456 ± 16 g for Sham KE rats, 459 ± 16 g for CCI rats, and 468 ± 19 g for CCI KE rats. No significant difference between groups was observed at any time point (A).
Figure 3. Average weekly (A) body weight, (B) food and (C) water consumption throughout the study. CCI and KE did not affect weight gain (A). Rats supplemented with KE consumed less (B) food and (C) water compared to rats that received vehicle (water) by gavage. Error bars represent SEM. (n = 8 rats/group).
Food and water consumption
Food consumption varied over time [F(4,140) = 10.14, p < 0.0001; B] and no significant differences were observed between groups at any time point. However, rats supplemented with KE consumed significantly less food across the study (792.3 ± 15.6 g) compared to rats receiving water by gavage [1,028 ± 30.8 g; F(1,140) = 5.722, p < 0.05]. Water consumption also varied over time [F(4,140) = 15.54, p < 0.0001; C] with no significant difference between groups at any time point. KE supplementation also had a significant effect on water consumption [F(1,140) = 31.83, p < 0.0001], with rats who received KE displaying lower intake (1,178 ± 24 mL) throughout the study compared to rats receiving water (1,325 ± 23 mL; p < 0.0001).
Revised neurobehavioral severity scale
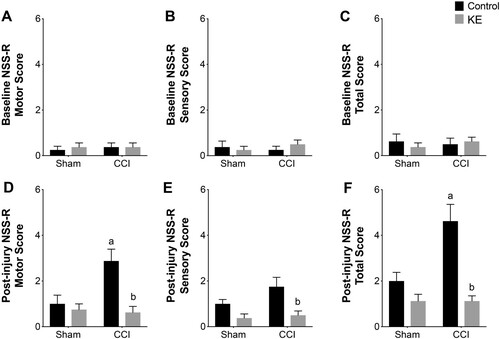
The NSS-R total, sensory and motor scores did not differ between groups at baseline, and no motor or sensory deficits were observed (A, B, C). All 32 rats were included in the study and final analysis, and their baseline NSS-R scores were used as co-variates in the ANCOVA analysis. TBI [F(1,28) = 8.29, p < 0.01], KE [F(1,28) = 22.85, p < 0.0001] and the interaction between the two factors [F(1,28) = 8.29, p < 0.01] had a significant effect on post-injury NSS-R total score variability. Multiple comparisons showed the average post-injury NSS-R total score in the CCI group (4.2 ± 0.7) was significantly higher than the Sham group (2.0 ± 0.3; p < 0.01; Cohen’s d = 1.59), and CCI KE group (1.1 ± 0.2; p < 0.01; Cohen’s d = 2.28; F). TBI [F(1,28) = 4.99, p < 0.05], KE [F(1,28) = 10.34, p < 0.05] and the interaction of both factors [F(1,28) = 7.16, p < 0.05] also had a significant effect on the variance of post-injury NSS-R motor score when controlling for baseline NSS-R motor score. The average NSS-R motor score was significantly higher in CCI rats (2.8 ± 0.5) compared to Sham rats (1.0 ± 0.3; p <0.01; Cohen’s d = 1.46) and CCI KE rats (0.6 ± 0.2; p < 0.01; Cohen’s d = 1.92; D). Finally, KE significantly affected the variability of post-injury NSS-R sensory score [F(1,28) = 14.76, p < 0.01] when baseline NSS-R sensory score was used as a covariate. Multiple comparison analysis showed that NSS-R sensory score in CCI rats (1.7 ± 0.4) was significantly higher than CCI KE rats (0.5 ± 0.1; p < 0.01; Cohen’s d = 1.37; E) but was not different from Sham rats.
Figure 4. Behavioral assessment of neurological deficits. Bar graphs of NSS-R (C, F) total, (A, D) motor, and (B, E) sensory scores at (A, B, C) baseline and (D, E, F) four weeks post-injury. Supplementation with KE significantly lowered NSS-R motor and total scores. Error bars represent SEM. (n = 8 rats/group; p < 0.05). a indicates statistical significance from Sham, Sham KE, and CCI KE. b indicates statistical significance from CCI.
Lesion volume
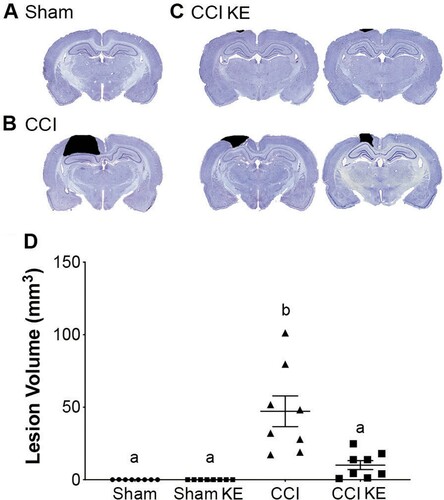
Quantification of lesion volume included only CCI and CCI KE rats, as Sham rats did not have a contusion to measure (A). TBI injury resulted in a lesion over the parietal cortex (B) with an average volume of 47.1 ± 10.6 mm3 in CCI + vehicle (water) rats (D). KE supplementation significantly reduced the lesion volume to 10.1 ± 3.1 mm3 in CCI KE rats (C, D) compared to CCI rats receiving only water (p < 0.01; Cohen’s d = 1.67; D).
Figure 5. Quantification of lesion volume. Representative photomicrographs of coronal brain sections from (A) Sham, (B) CCI, and (C) CCI KE rats processed and stained four weeks post-injury. Black shading highlights contusion area quantified in each slice for final calculation of lesion volume. (D) Scatter plot of quantified lesion volume. Supplementation with KE significantly reduced the lesion volume. Error bars represent SEM. Sham and Sham KE rats were not included in the statistical analysis as there was no lesion to be quantified. (n = 8 rats/group; p < 0.01). b indicates statistical significance from CCI rats.
Neuroinflammation
Immunofluorescence staining followed by quantification of the stained area was used to assess the effect of CCI and KE on reactive astrocytes and microglia in the prefrontal cortex, cortex around the site of the lesion, hippocampus and amygdala. Two-way ANOVA revealed that TBI induced significant changes in the mean Iba1+ stained area in all four brain regions (). TBI also had a significant effect on the total variance of GFAP+ stained area in the prefrontal cortex, cortex and hippocampus (). KE had a significant effect on Iba1+ stained area in the cortex and hippocampus, and GFAP+ stained area in all brain regions analyzed (). There was a significant interaction between CCI and KE on the variance of Iba1+ stained area in the hippocampus, and on the GFAP+ stained area in all four brain regions (). Post hoc analysis revealed that rats exposed to CCI had significantly larger GFAP+ and Iba1+ stained areas in all brain regions compared to Sham rats ( and ; ). KE supplementation reduced GFAP+ stained area in all four brain regions, and Iba1+ stained area in the hippocampus of rats following CCI ( and ; ). Analyses of GFAP+ and Iba1+ stained areas in the contralateral brain structures (prefrontal cortex, cortex, hippocampus, and amygdala) did not reveal significant differences between groups (data not shown).
Figure 6. Representative photomicrographs of GFAP+ stained areas (in green). Rat brains were perfused and processed for immunohistochemistry four weeks post-injury. Prefrontal cortex from (A) Sham, (B) Sham KE, (C) CCI, (D) CCI KE rats. (E) Graphical representation of the brain slice with ipsilateral prefrontal cortex highlighted in gray. Cortex around the lesion from (F) Sham, (G) Sham KE, (H) CCI, (I) CCI KE rats. (J) Graphical representation of the brain slice with cortex around the contusion site highlighted in gray. Hippocampus from (K) Sham, (L) Sham KE, (M) CCI, (N) CCI KE rats. (O) Graphical representation of the brain slice with ipsilateral hippocampus highlighted in gray. Amygdala from (P) Sham, (Q) Sham KE, (R) CCI, (S) CCI KE rats. (T) Graphical representation of the brain slice with ipsilateral amygdala highlighted in gray. Scale bar: 50 µm.
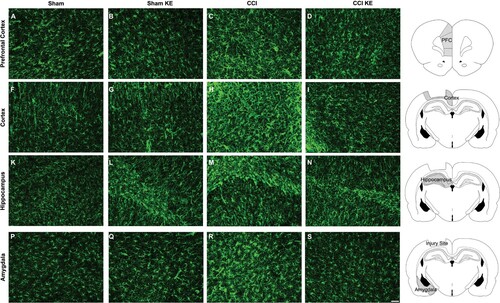
Figure 7. Representative photomicrographs of Iba1+ stained areas (in red). Rat brains were perfused and processed for immunohistochemistry four weeks post-injury. Prefrontal cortex from (A) Sham, (B) Sham KE, (C) CCI, (D) CCI KE rats. (E) Graphical representation of the brain slice with ipsilateral prefrontal cortex highlighted in gray. Cortex around the lesion from (F) Sham, (G) Sham KE, (H) CCI, (I) CCI KE rats. (J) Graphical representation of the brain slice with cortex around the contusion site highlighted in gray. Hippocampus from (K) Sham, (L) Sham KE, (M) CCI, (N) CCI KE rats. (O) Graphical representation of the brain slice with ipsilateral hippocampus highlighted in gray. Amygdala from (P) Sham, (Q) Sham KE, (R) CCI, (S) CCI KE rats. (T) Graphical representation of the brain slice with ipsilateral amygdala highlighted in gray. Scale bar: 50 µm
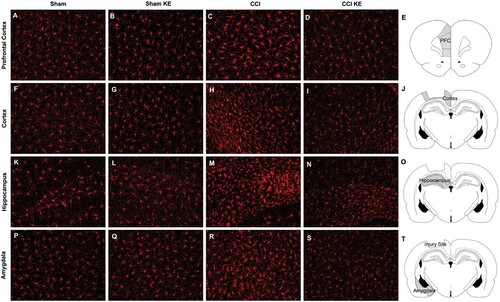
Table 2. Two-way ANOVA of GFAP and Iba1 stained area followed by Tukey’s post-hoc test and multiple comparison of groups.
Correlation and multiple regression analyses
Correlation analysis was used to assess the relationship between functional and morphological outcomes in the study subjects (). The NSS-R total score was positively associated with lesion volume, GFAP+ stained area in the cortex, prefrontal cortex and amygdala, and Iba1+ stained area in the cortex and hippocampus. Lastly, multiple regression analysis revealed that the NSS-R total score can be predicted from morphological outcomes and baseline NSSR total score when all predictors are forced into the model (R2 = 0.656; Adjusted R2 = 0.453; SSM = 67.22; p = 0.009).
Table 3. Partial correlation between post-injury NSS-R total score and morphological outcomes controlling for baseline NSS-R total score.
Discussion
Research into new neuroprotective strategies for TBI is important given the high prevalence of TBI among civilians and the military, and the lack of effective interventions to prevent and/or ameliorate the long-term consequences of a head injury. In the present study, we assessed the neuroprotective effect of KE on functional and morphological outcomes following CCI in male Sprague Dawley rats. Our results indicate that KE improved motor and sensory reflexes, and reduced lesion volume, astrogliosis and microgliosis when administered immediately after the injury. Performance in the NSS-R test positively correlated with lesion volume, GFAP+ stained area in the cortex, prefrontal cortex and amygdala, and Iba1+ stained area in the cortex and hippocampus. Multiple regression analysis revealed that morphological parameters are strong predictors of neurological function after TBI. To the best of our knowledge, we report for the first time that chronic KE, started early after TBI, is an effective and safe nutrition-based strategy to mitigate the development of TBI-related functional and morphological damage.
Although using a KE drink is a novel approach to achieve ketosis and protection against TBI, elevating blood ketone levels through a ketogenic diet (KD) and fasting have been associated with a reduction in cortical contusion volume after CCI [Citation18,Citation19]. Prins et al. [Citation18] demonstrated that contusion volumes were significantly reduced by 58% and 39% in rats at postnatal days (PND) 35 and 45, respectively when fed a KD for 7 days after CCI exposure whereas no volume reductions were seen in rats at PND 17 and 65. However, these age-related differences in neuroprotection were not associated with alterations in ketone availability. Regardless of the age, rats fed a KD displayed blood levels of βHB between 1 and 2 mmol/L (based on visual inspection of graph provided)[Citation18]. Although we did not precisely track the postnatal age of rats in the present study, they were adults and over 8 weeks old when subjected to CCI and KE: we observed a 78% reduction in the lesion volume in those supplemented with KE. Another study using adult rats (250 - 300 g) reported a significant increase in cortical tissue-sparing in rats subjected to a 24-hour fast after CCI, which was associated with increased blood levels of ketone bodies – blood levels of βHB were 0.57 ± 0.5 mmol/L [Citation19]. Our data showed that a single dose of KE administered by gavage elevated blood levels of βHB for at least two hours and that daily administration of KE for four weeks reduced the lesion volume in CCI rats. Inducing ketosis, regardless of the method used, is an effective way to achieve robust reduction in CCI-induced cortical contusion volume. Importantly, KE supplementation offers the advantage of being a faster approach in that it does not require any dietary restriction.
It is well characterized that neuroinflammation plays a critical role in the pathophysiology of TBI and contributes to secondary cell death in humans and rodents [Citation6,Citation27–31]. In the present study, quantification of astrocytes (GFAP) and microglia (Iba1), four weeks after injury, revealed that CCI triggered a chronic neuroinflammatory response, which is detrimental to cell survival. In addition, KE supplementation significantly reduced GFAP+ stained areas in all brain regions analyzed (prefrontal cortex, cortex, hippocampus and amygdala) and Iba1+ stained area in the cortex and hippocampus. Other studies have previously shown that ketone bodies reduce neuroinflammation in animal models of spinal cord injury, cuprizone-induced demyelination, kainic acid-induced seizures and stress-induced depression [Citation32–34]. A KD has been shown to reduce expression of C-X-C motif chemokine 10 (CXCL10), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), translocation of nuclear factor (NF)-κB, and activation of astrocytes and microglia [Citation32,Citation33]. Similarly, infusion of the ketone body d-βHB attenuates expression of interleukin-18 (IL-18), IL-1β and NLRP3 inflammasome [Citation34,Citation35]. However, to the best of our knowledge, the present data showed for the first time that KE supplementation reduced TBI-induced astrocyte and microglia activation.
Our data also showed that KE significantly reduced neurobehavioral deficits in rats subjected to CCI. Although different neurobehavioral tests have been used to assess functional impairment following TBI and the effectiveness of therapeutic intervention, NSS-R was selected as the main functional outcome in the present study. Assessment of motor and sensory reflexes has been extensively used to evaluate functional impairment following different models of TBI, including fluid percussion injury [Citation36–38], blast overpressure [Citation24], closed-head injury [Citation39–42], and controlled cortical impact [Citation37,Citation43–47]. Not all these studies used NSS-R; some employed its non-revised version, the Neurological Severity Score (NSS)[Citation40–44] and others selected a few specific tests to assess sensory and motor deficits [Citation37,Citation38,Citation45–47]. Nonetheless, TBI-induced sensory and motor deficits is observed in male and female mice [Citation38–40,Citation42,Citation46] and rats [Citation36,Citation37,Citation41,Citation43–45,Citation47] up to four weeks after TBI [Citation42,Citation45,Citation46]. Our data show the NSS-R total score was positively correlated with and can be predicted from morphological parameters herein investigated. Therefore, it is a valid assessment tool capable of detecting TBI-induced neurological deficits.
Although small, CCI-induced changes in NSS-R score reported in the present study are consistent with previously published data. Sharma et al. reported a mean NSS-R total score of 3.3 (SEM = 0.5) for Sham rats and 5.4 (SEM = 0.5) for injured rats five days after mild fluid percussion injury [Citation36]. Although Yarnell et al. did not include descriptive statistics in the manuscript describing NSS-R methodology, the graph provided does show a small increase in total NSS-R score after blast injury relative to baseline [Citation24]. Studies that have used the NSS (non-revised version) to assess neurobehavioral deficits following closed-head injury (CHI) and CCI in rats, also report small changes in scores from 24 h up to 28 days after injury [Citation41,Citation43]. TBI-induced neurobehavioral deficits assessed by NSS and NSS-R partially improve over time and the time points selected for behavioral assessments should be considered when comparing studies [Citation41]. This spontaneous recovery contributed to our decision to test the animals four weeks after injury. Our main objective was to determine the efficacy of a neuroprotective intervention and having a long-lasting functional impairment was desirable.
The mechanism underlying the neuroprotective properties of a dietary KE drink against TBI is largely unknown, but it has been suggested that ketone bodies confer neuroprotection by serving as an alternative fuel to glucose and providing the necessary energy to restore cerebral metabolism [Citation15,Citation16]. Investigating the mechanism underlying the neuroprotective effect of KE is beyond the scope of this study, therefore we did not conduct experiments to test this hypothesis. However, our data do show that starting KE immediately after TBI and sustaining it throughout the duration of the study effectively prevented TBI-induced impairment. This suggests that providing the brain with ketones, an alternative cellular fuel to glucose, may help ameliorate the ‘energy crisis’ and prevent multiple long-term consequences associated with TBI. In addition to its primary role as a cellular fuel, the ketone body d-βHB inhibits histone deacetylase (HDAC) enzymes and therefore can alter gene expression [Citation48,Citation49]. HDAC inhibition by βHB suppresses oxidative stress and enhances antioxidant capacity, which can contribute to neuroprotection [Citation49]. βHB also increases BDNF expression through specific inhibition of HDAC2 and HDAC3 [Citation48]. BDNF is a neurotrophic factor that supports neuronal survival and protects against injury-induced cell death [Citation50]. Finally, βHB suppresses activation of the NLRP3 inflammasome and consequent IL-1β secretion and caspase-1 activation [Citation34,Citation51]. It is likely that the metabolic and signaling properties synergistically contribute to KE neuroprotective effects.
KE supplementation is a promising nutrition-based intervention for preventing and/or reducing the development of long-term TBI-related disability. However, additional research is needed to better translate pre-clinical data into effective clinical intervention. The neuroprotective efficacy of KE needs to be tested in other animal models of TBI, such as those that replicate the rapid acceleration and rotation to the head observed in concussion, which is the most common type of head injury. Such biomechanical alterations produce diffuse injury without overt focal damage. Future studies should also include female rats, as gender differences may have a prominent role in susceptibility to TBI and KE. Gender inclusion in animal research is imperative as results showing both significant or no sex difference provide important knowledge about the etiology and treatment of various health conditions. Inclusion of aged animal is also important, given the high risk of TBI in the elderly. We also plan to investigate the neuroprotective effect of prophylactic administration of KE, particularly important to the military population who are at high risk for sustaining TBI. If pre-treatment with KE helps to ameliorate, at least in part, the negative consequences of TBI in animals, we need human studies to examine whether it may also help reduce the long-term toll of TBI in the military population. Finally, we intend to investigate the mechanism underlying the neuroprotective effect of KE after TBI and assess the efficacy of βHB, an epigenetic modulator, as an alternative cellular fuel in the recovery period after TBI.
In conclusion, the data herein presented and discussed yield valuable information on the utility of KE as an effective neuroprotective intervention against the pathophysiology of TBI. The present study is significant because it addresses the need for identifying well-tolerated and effective interventions that ameliorate the pathophysiology of TBI. Although we acknowledge the various limitations of TBI animal models in investigating therapeutic interventions for human TBI, we expect the results from this study to have a positive translational impact. The CCI model shares common pathophysiology with human TBI as well as symptomatology. KE metabolism and pharmacodynamics are similar in humans and rodents [Citation21,Citation52,Citation53]. The KE used in this study was approved in 2015 as a food for human consumption by the Food and Drug Administration (FDA) and is readily available for clinical studies. If proven to be an effective neuroprotective intervention in human TBI, KE drinks could be added to the current TBI regimen of sedatives and anti-inflammatory agents as a non-pharmacological, nutrient-based adjunct therapy. Moreover, KE could be incorporated into military rations for convenient daily supplementation in this population particularly vulnerable to TBI. In summary, our study suggests that KE offers an especially promising avenue for future clinical investigation for reducing TBI-induced impairment.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data available on request from the authors. The data that support the findings of this study are available from the corresponding author, C.P.A.-S., upon reasonable request.
Acknowledgments
We gratefully acknowledge the Center for Neuroscience and Regenerative Medicine (CNRM) for assistance with the controlled cortical impact model of TBI, and Alice Graham for technical assistance throughout the duration of these experiments.
Additional information
Funding
Notes on contributors
Camila Almeida-Suhett
P.D. acquired funding for the research; C.P.A.-S., A.N., K.C., P.D. conception and design of research; C.P.A.-S. performed experiments; C.P.A.-S. analyzed data; C.P.A.-S. interpreted results of experiments; C.P.A.-S. prepared figures; C.P.A.-S. drafted manuscript; P.D., A.N., K.C., edited and revised manuscript; C.P.A.-S., A.N., K.C., P.D. approved final version of manuscript.
Camila Almeida-Suhett is a Neuroscientist with a broad background in rodent behavioral science. Her research has focused on the etiology of neurobehavioral deficits in response to various forms of insult to the brain. She has investigated morphological and functional alterations in temporal lobe structures and their contribution to the development of cognitive deficits and anxiety disorders following traumatic brain injury. In addition, she conducted research on the impact of high-fat diet on physiological and behavioral alterations. Her long-term research goal is to identify effective interventions to prevent and/or treat such disorders and improve the lives of service members and overall population.
Aryan M. Namboodiri
Aryan M. Namboodiri is an experienced researcher in the Biochemistry and Neuroscience fields. He has led basic and preclinical studies investigating the role of N-acetylaspartate in animal models of Canavan disease. He has also conducted research to develop new neuroprotective strategies for traumatic brain injury based on acetate supplementation to the brain via the glycerol ester-based strategy.
Kieran Clarke
Kieran Clarke is Professor of Physiological Biochemistry at the University of Oxford where she has worked since 1991 on the effects of diet on energy metabolism in human heart, brain and skeletal muscle. Specifically, her interest in physical and cognitive function has led to the development of a fourth food group, a ketone ester that improves endurance exercise in athletes and could be used for the management of common metabolic diseases, such as type 2 diabetes. Before Oxford, Professor Clarke worked as a postdoctoral fellow at Harvard University Medical School and as a research scientist at the NRC in Ottawa, Canada.
Patricia A. Deuster
Patricia A. Deuster is a Professor in the Department of Military and Emergency Medicine at the Uniformed Services University of the Health Sciences in Bethesda, Maryland and Director for the Consortium for Health and Military Performance, the Defense Center of Excellence for Human Performance Optimization Translation. She has over 200 peer-reviewed papers and numerous book chapters and books relating to human performance with a focus on health, total force fitness, nutrition, dietary supplements, physical performance and exertional-related health events. In addition, she has developed multiple educational materials related to human performance and total force fitness.
References
- Center for Disease Control and Prevention. Surveillance report of traumatic brain injury-related emergency Department Visits, Hospitalizations, and Deaths—United States, 2014. Atlanta (GA): Centers for Disease Control and Prevention, US Department of Health and Human Services; 2019.
- Anghinah R, Amorim RLO, Paiva WS, et al. Traumatic brain injury pharmacological treatment: recommendations. Arq Neuropsiquiatr. 2018 Feb;76(2):100–103.
- Diaz-Arrastia R, Kochanek PM, Bergold P, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014 Jan 15;31(2):135–58.
- Prins M, Greco T, Alexander D, et al. The pathophysiology of traumatic brain injury at a glance. Dis Model Mech. 2013 Nov;6(6):1307–15.
- Wofford KL, Loane DJ, Cullen DK. Acute drivers of neuroinflammation in traumatic brain injury. Neural Regen Res. 2019 Sep;14(9):1481–1489.
- Lozano D, Gonzales-Portillo GS, Acosta S, et al. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106.
- Yoshino A, Hovda DA, Kawamata T, et al. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991 Oct 4;561(1):106–19.
- Selwyn R, Hockenbury N, Jaiswal S, et al. Mild traumatic brain injury results in depressed cerebral glucose uptake: An (18)FDG PET study. J Neurotrauma. 2013 Dec 1;30(23):1943–53.
- Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg. 1997 Feb;86(2):241–51.
- Sutton RL, Hovda DA, Adelson PD, et al. Metabolic changes following cortical contusion: relationships to edema and morphological changes. Acta Neurochir Suppl (Wien). 1994;60:446–8.
- Prins ML, Hovda DA. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J Neurotrauma. 2009 Jul;26(7):1083–93.
- Thomas S, Prins ML, Samii M, et al. Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. J Neurotrauma. 2000 Aug;17(8):649–65.
- Hattori N, Huang SC, Wu HM, et al. Correlation of regional metabolic rates of glucose with glasgow coma scale after traumatic brain injury. J Nucl Med. 2003 Nov;44(11):1709–16.
- Hlatky R, Contant CF, Diaz-Marchan P, et al. Significance of a reduced cerebral blood flow during the first 12 h after traumatic brain injury. Neurocrit Care. 2004;1(1):69–83.
- Prins ML, Matsumoto JH. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J Lipid Res. 2014 Dec;55(12):2450–7.
- White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care. 2011;15(2):219–228
- Hu ZG, Wang HD, Qiao L, et al. The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Inj. 2009 May;23(5):459–65.
- Prins ML, Fujima LS, Hovda DA. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res. 2005 Nov 1;82(3):413–20.
- Davis LM, Pauly JR, Readnower RD, et al. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res. 2008 Jun;86(8):1812–22.
- Cahill GJ, Veech RL. Ketoacids? Good medicine? Trans Am Clin Climatol Assoc. 2003;114:149–61. discussion 162-3.
- Clarke K, Tchabanenko K, Pawlosky R, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012 Aug;63(3):401–8.
- Shivva V, Cox PJ, Clarke K, et al. The population Pharmacokinetics of D-beta-hydroxybutyrate following administration of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. AAPS J. 2016 May;18(3):678–88.
- Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma. 1988;5(1):1–15.
- Yarnell AM, Barry ES, Mountney A, et al. The Revised Neurobehavioral Severity Scale (NSS-R) for rodents. Curr Protoc Neurosci. 2016 Apr 8;75:9 52 1–9 52 16.
- Anwer M, Immonen R, Hayward N, et al. Lateral fluid-percussion injury leads to pituitary atrophy in rats. Sci Rep. 2019 Aug 14;9(1):11819.
- Almeida-Suhett CP, Graham A, Chen Y, et al. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol Behav. 2017 Feb 1;169:130–140.
- Chiu CC, Liao YE, Yang LY, et al. Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods. 2016 Oct 15;272:38–49.
- Giunta B, Obregon D, Velisetty R, et al. The immunology of traumatic brain injury: a prime target for Alzheimer's disease prevention. J Neuroinflammation. 2012 Aug 1;9:185.
- Fluiter K, Opperhuizen AL, Morgan BP, et al. Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol. 2014 Mar 1;192(5):2339–48.
- Bellander BM, Singhrao SK, Ohlsson M, et al. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001 Dec;18(12):1295–311.
- Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015 Mar;72(3):355–62.
- Zhang N, Liu C, Zhang R, et al. Amelioration of clinical course and demyelination in the cuprizone mouse model in relation to ketogenic diet. Food Funct. 2020 Jun 24;11(6):5647–5663.
- Jeong EA, Jeon BT, Shin HJ, et al. Ketogenic diet-induced peroxisome proliferator-activated receptor-gamma activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp Neurol. 2011 Dec;232(2):195–202.
- Qian J, Zhu W, Lu M, et al. D-beta-hydroxybutyrate promotes functional recovery and relieves pain hypersensitivity in mice with spinal cord injury. Br J Pharmacol. 2017 Jul;174(13):1961–1971.
- Kajitani N, Iwata M, Miura A, et al. Prefrontal cortex infusion of beta-hydroxybutyrate, an endogenous NLRP3 inflammasome inhibitor, produces antidepressant-like effects in a rodent model of depression. Neuropsychopharmacol Rep. 2020 Jun;40(2):157–165.
- Sharma P, Su YA, Barry ES, et al. Mitochondrial targeted neuron focused genes in hippocampus of rats with traumatic brain injury. Int J Crit Illn Inj Sci. 2012 Sep;2(3):172–9.
- Peterson TC, Maass WR, Anderson JR, et al. A behavioral and histological comparison of fluid percussion injury and controlled cortical impact injury to the rat sensorimotor cortex. Behav Brain Res. 2015 Nov 1;294:254–63.
- Sackheim AM, Stockwell D, Villalba N, et al. Traumatic brain injury impairs sensorimotor function in mice. J Surg Res. 2017 Jun 1;213:100–109.
- Sharma A, Chandran R, Barry ES, et al. Identification of serum microRNA signatures for diagnosis of mild traumatic brain injury in a closed head injury model. PLoS One. 2014;9(11):e112019.
- Tsenter J, Beni-Adani L, Assaf Y, et al. Dynamic changes in the recovery after traumatic brain injury in mice: effect of injury severity on T2-weighted MRI abnormalities, and motor and cognitive functions. J Neurotrauma. 2008 Apr;25(4):324–33.
- Kao YJ, Lui YW, Lu CF, et al. Behavioral and Structural effects of single and Repeat closed-head injury. AJNR Am J Neuroradiol. 2019 Apr;40(4):601–608.
- Oron A, Oron U, Streeter J, et al.. low-level laser therapy applied transcranially to mice following traumatic brain injury significantly reduces long-term neurological deficits. J Neurotrauma. 2007 Apr;24(4):651–6.
- Mahmood A, Lu D, Lu M, et al. Treatment of traumatic brain injury in adult rats with intravenous administration of human bone marrow stromal cells. Neurosurgery. 2003 Sep;53(3):697–702. discussion 702-3.
- Mahmood A, Lu D, Qu C, et al. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J Neurosurg. 2006 Feb;104(2):272–7.
- Russell KL, Kutchko KM, Fowler SC, et al. Sensorimotor behavioral tests for use in a juvenile rat model of traumatic brain injury: assessment of sex differences. J Neurosci Methods. 2011 Aug 15;199(2):214–22.
- Fox GB, Fan L, Levasseur RA, et al. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma. 1998 Aug;15(8):599–614.
- Liu ZK, Ng CF, Shiu HT, et al. Neuroprotective effect of Da Chuanxiong Formula against cognitive and motor deficits in a rat controlled cortical impact model of traumatic brain injury. J Ethnopharmacol. 2018 May 10;217:11–22.
- Sleiman SF, Henry J, Al-Haddad R, et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body beta-hydroxybutyrate. Elife. 2016 Jun 2;5.
- Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013 Jan 11;339(6116):211–4.
- Zhao Z, Alam S, Oppenheim RW, et al. Overexpression of glial cell line-derived neurotrophic factor in the CNS rescues motoneurons from programmed cell death and promotes their long-term survival following axotomy. Exp Neurol. 2004 Dec;190(2):356–72.
- Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015 Mar;21(3):263–9.
- Clarke K, Tchabanenko K, Pawlosky R, et al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul Toxicol Pharmacol. 2012 Jul;63(2):196–208.
- Kesl SL, Poff AM, Ward NP, et al. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr Metab (Lond). 2016;13:9.