Abstract
Modulation of the developing immune system can occur following perinatal exposure to a number of immunotoxic compounds, including polyhalogenated hydrocarbons like 2,3,7,8-tetra-chlorodibenzo-p-dioxin (TCDD; dioxin), the most toxic of the congeners. Studies in rodents have shown immunologic effects from perinatal TCDD exposure are more severe and persistent than following exposure in the adult, and include what appears to be life-long immunosuppression. Whether prenatal TCDD exposure may predispose an individual to postnatal autoimmune disease remains largely unknown. TCDD crosses the placenta and alters normal prenatal thymocyte maturation, T-cell receptor expression and expression of thymic major histocompata-bility complex Class II molecules. During the juvenile stage, mice exposed to TCDD prenatally show increased peripheral T-cells possessing “autoreactive” variable-β receptors. These data suggest that gestational exposure to TCDD may interfere with normal development of central tolerance in the thymus. In possible support of this theory, when autoimmune disease-prone mice were treated with TCDD during gestation, postnatal autoimmunity had an accelerated onset and was exacerbated. This review provides an overview of the currently available information, which appears to support a hypothesis for increased risk of postnatal autoimmune responses as a result of TCDD exposure during the sensitive time of immune system establishment.
Keywords::
TCDD exposure in humans
Halogenated organic compounds are ubiquitous contaminants of the environment. Polychlorinated dibenzofurans and polychlorinated dibenzodioxins make up a major category of over 200 related congeners. Of these congeners, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most toxic and as such has been assigned the reference toxic equivalence factor rating of 1.0. This agent is a byproduct of chemical synthesis including 2,4,5-trichlorophenol and certain chlorinated herbicides and fungicides, and is also produced by combustion of chlorinated organic compounds and chlorine-based bleaching of wood pulp (Holsapple et al., Citation1991). Due to its lipophilic nature, this compound can readily penetrate animal tissue and bioaccumulate up the food chain. Thus, animal-derived food products appear to be the primary source of human exposure to TCDD (Antignac et al., Citation2006).
As late as the early 1990s, the scientific community was, in general, satisfied with both the steps taken to reduce TCDD levels in the environment as well as determining safe levels of TCDD exposure (Rozman, Citation1989; Roberts et al., Citation1991; Collins et al., Citation1992; CitationFingerhut et al., 1992). However, in 2000, the United States Environmental Protection Agency (US EPA) issued a document describing a change in policy related to human health risks from dioxins (Scientific Reassessment of Dioxin and Dioxin-like Chemicals, http://www.aphis.usda.gov/vs/ceah/cei/health.htm). The impetus for this document was to alert the general public to new concerns regarding potential adverse effects from and safe exposure levels to dioxins. In one example, the panel predicted: “Lifetime cancer risk associated with the average person’s body burden (of dioxins) is between 1 in 1000 and 1 in 100. This estimate of risk is ten times higher than EPA’s previous estimate and represents a very significant public health concern.” The review evaluated structurally related dibenzodioxins, dibenzofurans, and polychlorinated biphenyls (PCBs), the majority of which enter the environment through industrial emissions. The EPA reassessment concluded “safe levels” of dioxin exposure are 300–600 times less than present average daily human exposure. This conclusion was based in part on rodent data suggesting TCDD may have potential to cause immune-mediated disease in humans.
TCDD and human immunity
Surprisingly, even with a large publication database on TCDD, there is still a paucity of data examining the effects of TCDD on human immunity. The few studies that do exist, point toward TCDD as a modulator of both innate and acquired immunity. TCDD has been shown to alter terminal differentiation and diminish cell number of human primary thymic epithelial cells (TEC) in culture, at concentrations as low as 0.01 nM (Riecke et al., Citation2003). Cell adhesion molecules that mediate both cell-matrix and cell-cell interactions (e.g., integrins CD49b, CD49e, CD51, CD54) displayed altered expression following the TCDD treatment. The authors reported comparable findings when TEC were also treated with 100 nM PCB 126 (3,39,4,49,5-pentachloro-biphenyl). Analogous to rodent studies, these results imply that the human thymic epithelium is sensitive to TCDD and its congeners.
Almost 20 years after an accidental exposure to TCDD in Seveso, Italy, adult human patients showed an inverse relationship between plasma IgG and plasma levels of TCDD (Baccarelli et al., Citation2002). An immune endpoint evaluation of this same cohort showed patients with increased antinuclear antibody titers, increased immune complex deposition and decreased numbers of natural killer cells (Sweeney and Mocarelli, Citation2000). In a study conducted in the Netherlands, children showed direct correlations between maternal dietary dioxin and PCB exposure, and altered immunity in the form of increased γ˜δ-T-cell receptor (TCR)- positive T-cells at birth, and increased total T-cells and CD8+, TCRαβ+ and TCRγδ+ T-cells at 18 months of age (Weisglas-Kuperus et al., Citation1995). Thus, collectively available human studies, while limited, suggest that dioxin and dioxin-related compounds have modulatory capability on the human immune system including post-developmental exposure.
Human/Rodent developmental exposure comparison
Recently, our laboratory observed a significant decrease in thymocyte cellularity in gestation Day 18 outbred ICR mice (a relatively non-sensitive strain to TCDD) when dosed with 0.3 μg TCDD/kg/day during gestation day 7–16 (total litter cellularity decreased from 2.1 [± 0.2] × 107 cells, to 1.6 [± 0.1] × 107 cells). Approximately 0.5% of injected TCDD crosses the mouse placenta (Weber and Birnbaum, Citation1985), indicating total TCDD exposure to these fetal ICR mice was approximately 15 ng/kg (0.3 μg/kg × 10 doses × 0.005 = 0.015 μg/kg or 15 ng/kg). For comparative purposes to humans, total infant exposure to TCDD has been estimated as about 5.5 ng/kg throughout the first 6 months of life. Briefly, TCDD exposure in the general human population occurs at about 0.2–1.0 pg/kg body weight per day (predominantly via food) and for infants, up to 30 pg/kg body weight per day via mothers milk (Neubert et al., 1990). Human infant exposure to 30 pg/kg TCDD via lactation for the first 6 months of life the child would translate to a total dose of 30 pg/kg/day × 182.5 days = 5475 pg/kg TCDD, or about 5.5 ng/kg. Neubert et al. (1990) indicated that in certain human populations, the actual TCDD exposure to the infant could be considerably higher than 5.5 ng/kg because TCDD exposure levels were 200–300-fold higher in certain occupational and geographical regions. Further, this postnatal (lactational) exposure would be additive to the transplacental TCDD exposure that would also occur but has yet to be quantitated. Based on these data, it appears likely that human infant cohorts exist where exposure to TCDD may be in the range of developmental rodent studies that demonstrated immunotoxicity. As such, concerns over the effects of TCDD exposure during critical stages of human immune development would be justified.
Immune effects of TCDD in rodents
In the mouse, thymic involution is a classical outcome of TCDD exposure (Knutson and Poland, Citation1982; Kamath et al., Citation1997). In addition, TCDD also has been shown to impair normal thymocyte differentiation during development (Blaylock et al., Citation1992; Gehrs, et al., Citation1997; Gehrs and Smialowicz, Citation1997). Using flow cytometry and fluorescent monoclonal antibodies to identify CD4 and CD8 thymocyte differentiation antigens, prenatal TCDD exposure was shown to significantly decrease the percentage of CD4+CD8+ fetal thymocytes and significantly increase the relative percentage of CD4-CD8- and CD4-CD8+ fetal thymocyte subsets (Holladay and Luster, Citation1996).
These phenotypic changes at gestation day (gd) 18 inferred that thymocyte differentiation is delayed by TCDD. However, it is not known whether these changes continue post-natally and for what duration. We recently exposed pregnant C57BL/6 mice, a high affinity Aryl hydrocarbon receptor (AhR) murine strain, to a single mid-gestation oral dose of 2.5 or 5.0 μg/kg TCDD, and then allowed the F1 generation to mature to 24-weeks-of-age. Interestingly, females but not males showed significantly altered thymus to body weight ratios and thymocyte phenotypes (). This gender bias in the females could be a result of a hormonal additive effect such as estrogen, as a number of investigators have shown estrogens to also induce thymic atrophy and affect thymoctye differentiation (Rijsinghani et al., Citation1996; Zoller and Kersh, Citation2006). This increased sensitivity of the female thymus to developmental TCDD is different from a T-cell function endpoint previously described in rats. In particular, F344 rats exposed to TCDD on gd 14 showed significant depression of the delayed-type hypersensitivity (DTH) response lasting to 4 months-of-age in females and 19 months-of-age in males (Gehrs and Smialowicz, Citation1999).
Like TCDD, cyclosporine A (CsA), a therapeutic immunosuppressant, causes a general inhibition of thymocyte differentiation (Kosugi et al., Citation1989; Holladay and Smialowicz, Citation2000). It has been proposed that this shared outcome may be due to a down-regulating effect of both compounds on thymic self-antigen presenting (MHC Class I and II) molecules (Hess et al., Citation1990; DeWall et al., Citation1992). Expression of these molecules is critical as they support normal thymocyte differentiation as well as help mediate deletion of autoreactive cells (Holladay, Citation1999; Blaylock et al., Citation2005). TCDD- and CsA-like inhibition of T-cell differentiation in the thymus has also been shown to spontaneously occur in autoimmune disease-prone murine strains (Kakkanaiah et al., Citation1990) and in mice administered monoclonal antibodies to MHC Class I and Class II molecules, in vivo (Kruisbeek et al., Citation1985). In addition to thymic epithelial MHC antigens, TCDD down-regulated expression of an MHC Class I gene (Q1b) in a mouse hepatoma cell line (Dong et al., Citation1997). These authors showed that MHC Q1b cDNA encoded for the α3 domain and transmembrane domain of the Q1b Class I protein, inferring that the MHC gene product interacts with β2-microglobulin. Thus, these authors proposed that the MHC Q1b molecule, when down-regulated by TCDD, may function similarly to the MHC Class I antigen in antigen presentation during T cell development. Given these known effects of TCDD on MHC Class I and II molecule expression levels, our laboratory hypothesized that TCDD may be capable of inducing autoimmune disease in rodents via the same mechanisms as CsA (Blaylock et al., Citation2005).
In addition to modified CD4 and CD8 antigen expression, TCDD can also alter expression of the TCR in late gestation, resulting in an increased expression of γδ TCR (CitationHolladay et al., 1991). Interestingly, this shift in γδ TCR expression has not been reported in TCDD-exposed adult animals. During T-cell differentiation in the postnatal thymus, TCR gene rearrangement to encode the TCRβ chain is required developmental step in order for the thymocytes to progress in maturation from DN or TCR-CD4-CD8+ to the αβTcRCD4+CD8+ phenotype (Takahama, Citation2006) (). As a result, positive and negative selection of αβTCR with moderate affinity to self-MHC Class I and II molecules and high affinity for self-antigen, respectively, occurs. During the time of development when such deletion of autoreactive αβTCR is occurring in the control fetal thymus, the TCR phenotype of TCDD-exposed fetal thymus is significantly shifted toward γδTcR (Holladay, Citation1999). Thus, TCDD not only has the potential to interfere with positive and negative selection through altered CD4 and CD8 surface antigen expression and reduced thymic MHC antigens, but also shifts TCR expression during the initiation of development of neonatal central tolerance (Blaylock et al., Citation2005). Additionally, developmental exposure to TCDD increases the rate of fetal thymocyte apoptosis (Camacho et al., Citation2004; Besteman et al., Citation2005), and dysregulates the cellular architecture of the thymic cortico-medulary junction (Besteman et al., Citation2005). All these observations support existing data that TCDD alters the normal cycle of fetal thymocyte deletion.
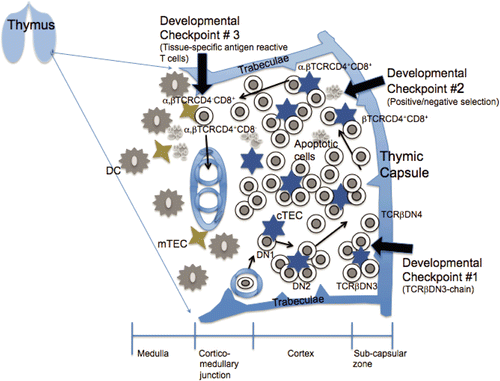
Figure 1. Thymocyte trafficking, development and selection. T-Lymphoid progenitor cells enter the thymus via the vasculature and migrate through the cortical region via chemokine signaling initially with a double negative (DN) phenotype. At developmental checkpoint #1, CD4-CD8-CD25+CD44- (double-negative 3, DN3) thymocytes are required to undergo gene rearrangement to encode TCRβ chain. These thymocytes then migrate through the cortical region and differentiate into a CD4+CD8+ (double positive; DP) phenotype under control by transforming growth factor-β (TGFβ). At developmental checkpoint #2, the highly motile unselected DP thymocytes interact with the cortical thymic epithelial cells (cTEC), and undergo positive and negative selection (central tolerance). The surviving thymocytes differentiate into CD4+ or CD8+ single positive (SP) cells. These cells migrate to the corticomedullary junction encountering the medullary thymic epithelial cells (mTEC), developmental checkpoint #3, and are screened for tissue specific antigen reactivity and deleted. Select medullary SP thymocytes also differentiate to become regulatory T-cells. Upon final maturation and differentiation the T-cells migrate out of the thymus through the blood vessels.
In addition to potential modulating effects on central tolerance, TCDD appears to increase extrathymic production of T-cells. Silverstone et al. (Citation1994) observed that TCDD induced extrathymic T-cell differentiation in the liver of young adult mice, and that such extrathymic cells expressed elevated levels of CD4+ Vβ17α TCR. Such thymocytes expressing the TCR variable β (Vβ) chains are usually deleted in the thymus by reaction with self-MHC and minor lymphocyte stimulatory antigens (Okuyama et al., Citation1992; Hanawa et al., Citation1993), and have been associated with autoimmunity in experimental mouse models (Rocha et al., Citation1992). After a single mid-gestation maternal TCDD exposure in C57BL/6 mice, we recently observed a significant increase in expression of TCR Vβ17α+ and Vβ3+ T-cells in the spleens of 24-week-old male, but not female, offspring ().
Silverstone et al. (Citation1994) suggested that the increased production of Vβ+ autoreactive T-cells following developmental TCDD exposure may promote autoimmune disease in genetically predisposed animals. In support of this hypothesis, the authors later reported accelerated onset of autoimmune nephritis in the male SNF1 mice offspring following a single maternal exposure to 10 μg/kg TCDD (Silverstone et al., Citation1998a, Citation1998b). Increases in peripherally restricted, potentially autoreactive Vβ3+ T-cells have also been detected in homogenized livers of both BALB/c and C3H adult mice (non-autoimmune strains) after exposure to TCDD (Silverstone et al., Citation1994). We recently observed focal centers of lymphocytic infiltrates in the livers of 24-week-old mice exposed to a single 5 μg/kg mid-gestation dose of TCDD, however these cells have not yet been phenotyped (Mustafa et al., 2008). Interestingly, an increase in hepatic Vβ3+ T-cells in estrogen treated mice has been related to promotion of autoimmune responses by this hormone (Okuyama et al., Citation1992). This raises an interesting question as to whether a developmental exposure to TCDD has the potential to increase the susceptibility to autoimmune disease based on sex.
Autoimmune diseases have multiple etiologies but in all cases the acquired immune system (T-cells and B-cells) is the principal mediator. The majority of available TCDD developmental immunotoxicity literature to date has focused on T-cell immunity, and indicates TCDD preferentially affects establishment of cellular immunity (e.g., DTH responses) (Miller et al., 1998; Gehrs and Smialowicz, Citation1999) However, the absence of studies investigating altered B-cell development and function after early TCDD exposure has been identified as an area lacking and worthy of further examination (Luster et al., 2003; Holsapple et al., 2005). Recent adult mouse studies suggest B-cells play a more important role in autoimmune response than previously perceived (Martin and Chan Citation2006). For example, the disruption of normal B-cell function can precipitate into autoimmunity by production of autoantibodies or potentially by altered mediation of other important immune events, including antigen presentation, cytokine production and modulation of other immune cells (Fujimoto and Sato, Citation2007). We recently detected altered B-cell lymphopoiesis and function in 24-week-old mice that were exposed to a single mid-gestation dose of 5 μg/kg TCDD. Phenotyping studies indicated that B-cell development was impaired in the bone marrow of the TCDD-treated mice (). Altered mature B-cell populations were also present in the secondary lymphoid organs of these mice, including increased spleen CD138+ plasma cells and CD24-CD21+ marginal zone (MZ) B-cells. MZ B-cells are more efficient than follicular B-cells at priming naïve T-cells, and are over-represented in lupus-prone strains, suggesting that MZ B-cells help trigger the T-cell dependent disease (Cariappa et al., Citation2001). Autoantibody titers in these mice were also increased in response to the prenatal TCDD exposure ().
Collectively, therefore, available rodent data show that perinatal TCDD dysregulates thymic deletion of autoreactive T-cells, and appears to drive T-cell differentiation into extrathymic compartments that lack the molecular capability to delete autoreactive T-cells. In addition, perinatal TCDD exposure can impair B-cell lymphopoiesis in the bone marrow, and increase splenic marginal zone B-cells and autoantibody titers in the adult. The net effect of these immune events, when TCDD exposure occurs during immune system ontogeny and establishment of neonatal tolerance, has not been fully characterized in rodents. However, it would appear that perinatal TCDD exposure is likely to increase the risk of autoimmune responses by affecting both arms of the acquired immune system.
Related Research Data
References
- Antignac, J. P., Marchand, P., Gade, C., Matayron, G., el Qannari, M., Le Bizec, B. and Andre, F. 2006. Studying variations in the PCDD/PCDF profile across various food products using multivariate statistical analysis. Anal. Bioanal. Chem. 384:271–279.
- Baccarelli, A., Mocarelli, P., D. G., Patterson, Jr., Bonzini, M., Pesatori, A. C., Caporaso, N. and Landi, M. T. 2002. Immunologic effects of dioxin: New results from Seveso and comparison with other studies. Environ. Health Perspect. 110:1169–1173.
- Baron, F., Gothot, A., Salmon, J.P., Hermanne, J. P., Pierard, G. E., Fillet, G. and Beguin, Y. 2000. Clinical course and predictive factors for cyclosporine-induced autologous graft-versus-host disease after autologous hematopoietic stem cell transplantation. Br. J. Haematol. 111:745–753.
- Besteman, E. G., Zimmerman, K. L. and Holladay, S. D. 2005. Tetrachlorodibenzo-p-dioxin (TCDD) inhibits differentiation and increases apoptotic cell death of precursor T-cells in fetal mouse thymus. J. Immunotoxicol. 2:107–114.
- Blaylock, B. L., Ahmed, S. A. and Holladay, S. D. 2005. Prenatal immunotoxicant exposure and autoimmune disease. In: Developmental Immunotoxicology, (S. D., Holladay, Ed.), Boca Raton, FL: CRC Press, pp. 215–228.
- Blaylock, B. L., Holladay, S. D., Comment, C. E., Heindel, J. J. and Luster, M. I. 1992. Exposure to tetrachlorodibenzo-p-dioxin (TCDD) alters fetal thymocyte maturation. Toxicol. Appl. Pharmacol. 112:207–213.
- Boverhoff, D. R., Tam, E., Harney, A. S., Crawford, R. B., Kaminski, N. E. and Zacharewski, R. 2004. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces suppressor of cytokine signaling 2 in murine B-cells. Mol. Pharmacol. 66:1662–1670.
- Camacho, I. A., Nagarkatti, M. and Nagarkatti, P. S. 2004. Evidence for induction of apoptosis in T-cells from murine fetal thymus following perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Sci. 78:96–106.
- Cariappa, A., Tang, M., Parng, C., Nebelitskiy, E., Carroll, M., Georgopoulos, K. and Pillai, S. 2001. The follicular versus marginal zone B-lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity 14:603–615.
- Collins, J. J., Acquavella, J. F. and Friedlander, B. R. 1992. Reconciling old and new findings in dioxin. Epidemiology 3:65–69.
- DeWaal, E. J., Schuurman, H. J., Loeber, J. G., Van Loveren, H. and Vos, J. G. 1992. Alterations in the cortical thymic epithelium of rats after in vivo exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): An (immuno)histological study. Toxicol. Appl. Pharmacol. 115:80–88.
- Dong, L., Ma, Q. and Whitlock, J. P. 1997. Down-regulation of major histocompatibility complex Q1b gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 272:29614–29619.
- Esser, C., Steinwachs, S., Herder, C., Majora, M. and Lai, Z. 2005. Effects of a single dose of 2,3,7,8-tetrachlorodibenzo-p-dioxin given at post-puberty, in senescent mice. Toxicol. Lett. 157:89–98.
- Fingerhut, M.A., Steenland, K., Sweeney, M. H., Halperin, W. E., Piacitelli, L. A. and Marlow, D. A. 1992. Old and new reflections on dioxin. Epidemiology 3:66–72.
- Fujimoto, M. and Sato, S. 2007. B-Cell signaling and autoimmune diseases: CD19/CD22 loop as a B-cell signaling device to regulate the balance of autoimmunity. J. Dematol. Sci. 46:1–9.
- Gehrs, B. C. and Smialowicz, R. J. 1999. Persistent suppression of delayed-type hypersensitivity in adult F344 rats after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology 134:79–88.
- Gehrs, B. C. and Smialowicz, R. J. 1997. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin: I. Effects on the fetus and the neonate. Toxicology 122:219–228.
- Gehrs, B. C., Riddle, M. M., Williams, W. C. and Smialowicz, R. J. 1997. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin: II. Effects on the pup and the adult. Toxicology 122:229–240.
- Hanawa, H., Tsuchida, M., Matsumoto, Y., Watanabe, H., Abo, T., Sekikawa, H., Kodama, M., Zhang, S., Izumi, T. and Shibata, A. 1993. Characterization of T-cells infiltrating the heart in rats with experimental autoimmune myocarditis. Their similarity to extrathymic T-cells in mice and the site of proliferation. J. Immunol. 150:5682–5695.
- Hess, A. D., Fischer, A. C. and Beschorner, W. E. 1990. Effector mechanisms in cyclosporine A-induced syngeneic graft-versus-host disease. Role of CD4+ and CD8+ T lymphocyte subsets. J. Immunol. 145:526–533.
- Holladay, S. D. 1999. Prenatal immunotoxicant exposure: Increased risk of postnatal autoimmune disease? Environ. Health Perspect. 107:687–691.
- Holladay, S. D. and Luster, M. I. 1996. Alterations in fetal thymic and liver hematopoietic cells as an indicator of exposure to developmental immunotoxicants. Environ. Health Perspect. 104:809–813.
- Holladay, S. D., Lindstrom, P., Blaylock, B. L., Comment, C. E., Germolec, D. R., Heindel, J. J. and Luster, M. I. 1991. Perinatal thymocyte antigen expression and postnatal immune development altered by exposure to tetrachlorodibenzo-p-dioxin (TCDD). Teratology 44:385–393.
- Holladay, S.D. and Smialowicz, R.J. 2000. Development of the immune system: Differential effects depending on time of exposure. Environ. Health Perspect 108:463–473.
- Holsapple, M. P., Snyder, N. K., Wood, S. C. and Morris, D. L. 1991. A review of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: 1991 update. Toxicology 69:219–255.
- Kakkanaiah, V. N., Pyle, R. H., Nagarkatti, M. and Nagarkatti, P. S. 1990. Evidence for major alterations in the thymocyte subpopulations in murine models of autoimmune diseases. J. Autoimmunity 3:27–28.
- Kamath, A. B., Xu, H., Nagarkatti, P. S. and Nagarkatti, M. 1997. Evidence for the induction of apoptosis in thymocytes by 2,3,7,8-tetrachlorodibenzo-p-dioxin in vivo. Toxicol. Appl. Pharmacol. 142:367–377.
- Knutson, J. C. and Poland, A. 1982. Response of murine epidermis to 2,3,7,8-tetrachlorodibenzon-p-dioxin: Interaction of the ah and hr loci. Cell 30:225–234.
- Kosugi, A., Zuniga-Pflunker, J. C., Sharrow, S. O., Kruisbeek, A. M. and Shearer, G. M. 1989. Effect of cyclosporin A on lymphopoiesis. II. Developmental effects on immature thymocytes in fetal thymus organ culture treated with cyclosporin A. J. Immunol. 143:3134–3140.
- Kruisbeek, A. M., Bridges, S., Carmen, J., Longo, D. L. and Mond, J. J. 1985. In vivo treatment of neonatal mice with anti-I-A antibodies interferes with the development of the Class I, Class II, and Mls-reactive proliferating T-cell subset. J. Immunol. 134:3597–3604.
- Martin, F. and Chan, A. C. 2006. B-Cell immunobiology in disease: Evolving concepts from the clinic. Annu. Rev. Immunol. 24:467–496.
- Mustafa, A., Holladay, S. D., Goff, M., Witonsky, S., Kerr, R., Reilly, C., Sponenberg, P. and R. M., Gogal, Jr. An enhanced postnatal autoimmune profile in 24-week-old C57BL/6 mice developmentally-exposed to TCDD. Toxicol. Appl. Pharmacol. (In press).
- Neubert, R., Jacob-Muller, U., Stahlmann, R., Helgo, H. and Neubert, D. Polyhalogenated dibenzo-p-dioxins and dibenzofurans and the immune system. 1. Effects on peripheral lymphocyte subpopulation of a non-human primate (Callithrix jacchus) after treatment with 2,3,7,8- tetrachlorodibenzo-p-dioxin (TCDD). Arch. Toxicol. 64:345–359.
- Okuyama, R., Abo, T., Seki, S., Ohteki, T., Sugiura, K., Kusumi, A. and Kumagai, K. 1992. Estrogen administration activates extrathymic T-cell differentiation in the liver. J. Exp. Med. 175:661–669.
- Reicke, K., Schmidt, A. and Stahlmann, R. 2003. Effects of 2,3,7,8-TCDD and PCB 126 on human thymic epithelial cells in vitro. Arch. Toxicol. 77:358–364.
- Rijhsinghani, A. G., Thompson, K., Bhatia, S. K. and Waldschmidt, T. J. 1996. Estrogen blocks early T-cell development in the thymus. Am. J. Reprod. Immunol. 36:269–277.
- Roberts, L. 1991. EPA moves to reassess the risk of dioxin. Science 252:911.
- Rocha, B., Bassalli, P. and Guy-Grand, D. 1992. The extrathymic T-cell development pathway. Immunol. Today 13:449–454.
- Rozman, K. 1989. A critical view of the mechanism(s) of toxicity of 2,3,7,8-tetrachlorodibenzo-p- dioxin. Implications for human safety assessment. Derm. Beruf. Umwelt. 37:81–92.
- Silverstone, A. E., D. E., Frazier, Jr., Gasiewicz, T. A. 1994. Alternate immune system targets for TCDD: Lymphocyte stem cells and extrathymic T-cell development. Exp. Clin. Immunogenet. 11:94–101.
- Silverstone, A. E., Gavalchin, J. and Gasiewicz, T. A. 1998a. TCDD, DES, and estradiol potentiate a lupus-like autoimmune nephritis in NZB × SWR (SNF1) mice. Toxicologist 42:403.
- Silverstone, A. E., Gavalchin, J., Silvin, C. A., Staples, J. E., Ames, I. H. and Shanley, P. 1998b. Effects of 2,3,7,8-tetrachlorodibenzo-p- dioxin on a murine model of a lupus-like nephritis. Chemosphere 18:11–23.
- Sweeney, M. H. and Mocarelli, P. 2000. Human health effects after exposure to 2,3,7,8-TCDD. Food Add. Contam. 17:303–316.
- Takahama, Y. 2006. Journey through the thymus: Stromal guides for T-cell development and selection. Nat. Rev. Immunol. 6:127–135.
- Weber, H. and Birnbaum, L. S. 1985. Dosing with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and with 2,3,7,8-tetrachlorodibenzo-p-furan (TCDF) in pregnant C57Bl/6N mice: Distribution and excretion. Arch. Toxicol. 57:159–162.
- Weisglas-Kuperus, N. T., Sas, C. J., Koopman-Esserboom, C., Van Der Zwan, C. W., De Ridder, M. A., Beishuizen, A., Hooijkaas, H. and Sauer, P. J. 1995. Immunologic effects of background prenatal and postnatal exposure to dioxins and polychlorinated biphenyls in Dutch infants. Pediatr. Res. 38:404–410.
- Zoller, A. L. and Kersh, G. J. 2006. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of β-selected thymocytes. J. Immunol. 176:7371–7378.