
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The same chemical reaction may be different in terms of its position of the equilibrium (i.e., thermodynamics) and its kinetics when studied in different foods. The diversity in the chemical composition of food and in its structural organization at macro-, meso-, and microscopic levels, that is, the food matrix, is responsible for this difference. In this viewpoint paper, the multiple, and interconnected ways the food matrix can affect chemical reactivity are summarized. Moreover, mechanistic and empirical approaches to explain and predict the effect of food matrix on chemical reactivity are described. Mechanistic models aim to quantify the effect of food matrix based on a detailed understanding of the chemical and physical phenomena occurring in food. Their applicability is limited at the moment to very simple food systems. Empirical modeling based on machine learning combined with data-mining techniques may represent an alternative, useful option to predict the effect of the food matrix on chemical reactivity and to identify chemical and physical properties to be further tested. In such a way the mechanistic understanding of the effect of the food matrix on chemical reactions can be improved.
Introduction
At its most basic level, food quality depends on how compounds in the food interact, via chemical and physical reactions. Such reactions are subject to basic laws from chemistry and physics, irrespective from where they occur, and they can be fully characterized in terms of thermodynamics and kinetics. However, when a chemical reaction occurs in real foods, in many cases different results are obtained as compared to the same reaction occurring in simple aqueous solutions. This happens because foods do not behave like ideal solutions. Indeed, foods are multicomponent, concentrated systems, consisting of water-insoluble materials (for instance, cell membranes and lipid droplets), complicated aqueous solutions of ionic and nonionic compounds, with various phases that can also be in different physical states. Even more importantly, however, results are likely to be different when the same reaction occurs in different foods: the food matrix changes the rate of the reaction and/or its equilibrium position. The food matrix can be defined as the whole of the chemical components of food and their molecular relationships, the chemical composition of food, and the way those components are structurally organized at micro-, meso-, and macroscopic scales. Recognizing the effect of the food matrix on chemical and enzymatic reactions means to recognize that, even starting from the same initial conditions and applying the same physical conditions to the surroundings, the same reaction would not evolve in the same way in ideal solutions as in real foods or, even more importantly, in different foods. This is equivalent to say that differences among food matrices are responsible for the apparent variability that is observed in a certain chemical behavior (for instance, the stability of a certain nutrient upon thermal treatment) within a “population” of food products.
The capacity of quantitatively describing and predicting the effect of the food matrix on chemical reactivity would be of paramount importance for food scientists because it would lead to a more rational approach to food product development and handling. Unfortunately, a fully mechanistic description of the effect of the food matrix on chemical reactivity is difficult: foods are very complex systems. Here we suggest that an empirical approach based on chemical/physical fingerprinting combined with data mining could represent a complementary strategy to predict this effect as well as to generate hypotheses to be tested with a more fundamental approach. The aim of this paper is to provide tools for understanding and predicting the effect of the food matrix on chemical reactions in food and as such, for controlling the final quality attributes of food. We will first provide two examples of the variability caused by the food matrix on thermal stability of important bioactive compounds, namely glucosinolates and vitamin C which reflects itself in different rate constants for the same reaction in different foods. Then, we will describe two approaches for the description and prediction of the food matrix effect on chemical reactivity: (1) a mechanistic approach based upon fundamental laws of chemistry and physics and (2) an empirical approach based on machine learning and data mining.
Effect of the food matrix on thermal stability of bioactive compounds: Glucosinolates
Glucosinolates (GLs, β-thioglycoside N-hydroxysulphates linked with a sulphur β-d-glucopyranose) are secondary metabolites that occur in Brassica vegetables (broccoli, cauliflower, cabbage, etc.) (Fahey et al., Citation2001). GLs are hydrolyzed by a group of endogenous β-glucosidases termed myrosinase (MYR). MYR is stored separately from glucosinolates in plants, but will come into contact with GLs upon cell damage. The hydrolysis of GLs produces isothiocyanates, nitriles, epithionitriles, or thiocyanates depending on certain conditions, such as pH, vitamin C content and the presence of epithiospecifier proteins (Hanschen et al., Citation2017). Isothiocyanates have been demonstrated to be potentially health-beneficial for humans because of their anticarcinogenic properties (Steinmetz, Citation1996; Traka, Citation2009; Miller, Citation2012; Biswa Nath Das, Citation2013).
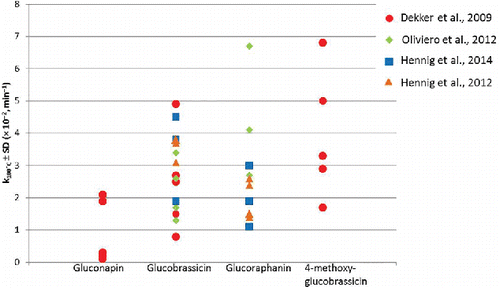
During the past 20 years, much research has been conducted on the effect of postharvest treatments on nonenzymatic GLs thermal degradation (Fahey et al., Citation2001). GLs thermal degradation has been mainly described by first-order kinetics. In , a graphical depiction of the variability in rate constants reported in the scientific literature for GLs degradation is provided. only includes a selection of rate constants from literature. A more comprehensive overview of the kinetic parameters and of the food matrices in which they have been derived is available in the Appendix. From and the Appendix, it emerges that the thermal breakdown of the same GL shows different rate constants in the different Brassica vegetables. For instance, heating ground samples of red cabbage, broccoli, Brussels sprouts, pak choi, and Chinese cabbage (previously microwaved and blended in liquid nitrogen to rule out MYR activity) at 100 ˚C resulted in different degradation rate constants for three GLs. Brussels sprouts were the vegetables in which the highest degradation rate constants for all three GLs were observed (Dekker et al., Citation2009). The authors hypothesized that the different chemical environments of the vegetables, such as differences in pH and cellular composition, can promote or inhibit GLs thermal degradation. In another study, thermal degradation of four aliphatic and four indolic GLs during boiling, steaming, and stir-frying of three Brassica oleracea varieties, the Dutch “Boerenkool” (kale), the Italian “Broccolo Lavagnino,” and “Nero di Toscana” were compared (Giambanelli et al., Citation2016). The thermal degradation rate constants were estimated by second-order kinetics, and the vegetables were only chopped (to a similar size). The results showed that for all the cooking and for all the GLs, the highest GLs degradation rates occurred in the “Nero di Toscana” variety. Such matrix effect can be detected also in different genetic lines of the same variety. Hennig and others (Citation2013) investigated the nonenzymatic GLs thermal degradation in Brassica oleracea doubled haploid populations (Hennig et al., Citation2013). The authors hypothesized that since the metabolic composition is determined by biochemical traits that are (partly) genetically regulated, the thermal degradation of GLs is genetically regulated as well and, therefore, genomic regions can be identified that influence the thermal degradation rates of GLs. GLs degradation was modeled using first-order kinetics and the rate constants were determined for five aliphatic and two indolic GLs. A threefold difference in GL degradation rate constants among the doubled haploid population was observed for most of the GLs, whereas degradation rate constants for glucoraphanin differed by ninefold. The nonenzymatic thermal degradation of GLs was also investigated in broccoli with different aw (Oliviero et al., Citation2012). It was shown that a drier matrix increased the thermal stability of GLs up to 100°C but not at 120°C. GLs degradation has been also studied in dilute aqueous solutions at different pH condition, water activity aw, and in the presence of Fe (II) and vitamin C (Hanschen et al., Citation2012). In that study it was shown that the addition of broccoli sprout powder to an aqueous model system significantly reduced thermal stability of aliphatic GLs. In line with this observation, in a more recent study it was shown that “dilution” of the broccoli matrix with ground potato, corn starch, lentil protein, or onion increases the thermal stability of GLs, in particular in broccoli-onion mixtures (Giambanelli et al., Citation2015). The authors hypothesized that the protective effect may have been caused by flavonoids of which onions are among the richest sources (Slimestad et al., Citation2007), but could not find a correlation between different flavonoid concentrations (from three onion varieties) and GLs thermal stability.
Figure 1. Selection of rate constants for thermal degradation of glucosinolates published in the scientific literature. Measures of uncertainties have not been reported for clarity of representation. RSD% was in no case > 10%. More information on the food matrices where the rate constants have been derived can be found in the appendix.
Effect of the food matrix on thermal stability of micronutrients: vitamin C
Vitamin C is composed of L-ascorbic acid and its oxidized form, L-dehydroascorbic acid. Vitamin C is not synthesized in humans and its deficiency causes a disease known as scurvy. The main dietary sources of vitamin C are citrus fruits, Brassica vegetables, and many other fruits and vegetables including melons, kiwi, and bell peppers. Storage and thermal treatments may drastically reduce vitamin C content in foods. The main degradation pathway for ascorbic acid is thought to be its oxidation to dehydroascorbic acid, followed by its irreversible hydrolysis to 2,3 diketogulonic acid, which possesses no vitamin C activity (Giannakourou and Taoukis, Citation2003). However, a nonoxidative pathway has been also described where ascorbic acid is degraded without producing dehydroascorbic acid (Peleg et al., Citation2016). A marked effect of the food matrix on vitamin C thermal stability is recognizable, as reported in . For instance, the degradation rate constants of vitamin C during storage were reported to be different among several citrus fruits of the same genus (Burdurlu, Citation2006). The authors stored grapefruit, tangerine, lemon, and orange juice at 28, 37, and 45 ˚C and modeled the vitamin C degradation rates by first-order kinetics. At 28 ˚C the reaction rate constant was higher in lemon juice than in orange juice, whereas at 45 ˚C the opposite was observed. The degradation rate constants of vitamin C in grapefruit and tangerine juices were similar and intermediate to those of lemon and orange juices. Stešková et al. (Citation2006) reviewed the thermal stability of vitamin C in fortified food during storage, modeling the degradation rate by first-order kinetics (Stešková, Citation2006). The fortified foods investigated were milk, evaporated milk, several types of bread, bran and potato flakes, cereals, cocoa powder, and apple chips. At the same storage temperature (25 ˚C), the highest degradation rate constants were observed in the breads, whereas in all the other foods, reaction rate constants were much lower, except for the fortified milk that showed intermediate values. The authors hypothesized that the difference in water content between the breads and the other dry foods (such as flakes and cocoa powder) can explain the different degradation rates of vitamin C. However, the fortified milk had higher water content than bread which showed the highest degradation rate constants. The same authors also reviewed the degradation of vitamin C in fortified beverages during storage. The beverages investigated were apple, cranberry, grapefruit, pineapple, tomato, and vegetable juices, but also grape and orange drinks, dry fruit drink mix and a carbonated beverage. The carbonated beverage showed highest degradation rates and the dry fruit drink mix the lowest (Stešková, Citation2006). Other studies have reported that vitamin C is more stable in dry matrices or in matrices with lower aw (Laing et al., Citation1978; Uddin et al., Citation2001) and its dependence on aw has been often used to optimize the air drying process to retain the vitamin C content ((Mishkin et al., Citation1984; Mishkin, 2006; Karim., Citation2009; Jin et al., Citation2014; Jin, Citation2014). Vitamin C stability during frozen storage was found in the order okra > green peas = green beans > spinach (Giannakourou and Taoukis, Citation2003). The different thermal stability of vitamin C in different foods can be attributed to multiple factors, for example, the different tissue structure, mechanical damage during harvesting, intrinsic enzyme (ascorbate oxidase) and sulfhydryl group content, and the presence of metal ions, such as Fe3+ and Cu2+, which act as catalysts for degradation. Last but not the least, the level of oxygen in food is also crucial since it acts as reactant in vitamin C oxidation.
Table 1. Overview of kinetic parameters reported in the scientific literature for the thermal degradation of vitamin C in different food matrices.
From and it is clear that kinetic parameters for the same reaction may differ among different food matrices (e.g., different juices or juices at different aw) and those differences depend on the particular chemical milieu wherein the reaction occurs. In the following sections we will provide some basic thermodynamic and kinetic details to understand, at least qualitatively, the effect of the food matrix and we will present two approaches to quantitatively describe and predict such effect.
Thermodynamics and kinetics of (bio)chemical reactions
Chemical reactions can be characterized in terms of their thermodynamic properties determining the position of the equilibrium (i.e., by equilibrium constants) and kinetic properties determining the rate at which equilibrium is attained (i.e., by rate constants and their temperature- and pressure-dependence). Let’s consider, for the sake of simplicity, a reversible elementary bimolecular reaction:(1)
(1)
When equilibrium is attained the following expression holds:(2)
(2) where
is the thermodynamic equilibrium constant for the reaction,
, and
are the thermodynamic activities of products C and D and reactants A and B, respectively. Thermodynamic activities are related to molar concentrations through the following relation:
(3)
(3) where
is the thermodynamic activity,
is the molar activity coefficient and
is the concentration of the chemical species in mol L−1. Under ideal conditions (gas state or very dilute solutions)
= 1 and thermodynamic activities equal concentrations. From eq. Equation2
(2)
(2) and Equation3
(3)
(3) , it follows that, in nonideal conditions, the following expression holds for experimentally observed concentrations:
(4)
(4)
In nonideal conditions, the observed equilibrium constant is different from the one that would be measured under ideal conditions
, because activities do not coincide with concentrations (
values are ≠ 1). The dependency of the equilibrium constant on temperature is expressed by the van ‘t Hoff equation:
(5)
(5) where
is the absolute temperature,
is the standard enthalpy change of the reaction and
is the gas constant.
Transition state theory has been developed to describe the effect of temperature on rate constants. According to the transition state theory, reactants are in quasi-equilibrium with the so-called transition state (or activated complex). The rate of the forward reaction in eq. Equation1(1)
(1) depends on the rate at which this transition state converts into the reaction products. This is expressed as follows:
(6)
(6)
The same holds for the reverse reaction:(7)
(7) where
is the transition state,
and
are the rate constants for the conversion of TS to C + D and to A + B, respectively.
is the equilibrium constant for the formation of the TS from reactants:
(8)
(8) where
,
, and
are the thermodynamic activities of
, A, and B, respectively.
is the equilibrium constant for the formation of the TS from products:
(9)
(9) where
,
, and
are the thermodynamic activities of
, A, and B, respectively.
Under the representation of eq. (Equation6(6)
(6) ) and (Equation7
(7)
(7) ), based on the law of mass action, the rate of the forward reaction in eq. (Equation1
(1)
(1) ) is as follows:
(10)
(10)
Which, combined with eq. (Equation3(3)
(3) ), (Equation8
(8)
(8) ), and (Equation9
(9)
(9) ) gives the following expression:
(11)
(11)
[A], [B], [C], and [D] are concentrations of reactants. In nonideal systems, values are ≠ 1 and, if they are not known, they are, by default, mathematically incorporated in the observed rate constant. In such situation, the following relationship holds:
(12)
(12) where
is the observed rate constant for the forward reaction in eq. Equation1
(1)
(1) . Under ideal conditions,
= 1 and the observed rate constants are equal to the ideal ones. The same holds true for the reverse reaction.
The rate constant depends on the temperature of the system and its temperature dependence may be expressed in terms of the Eyring equation:(13)
(13)
is Boltzmann’s constant,
is Planck’s constant,
is the absolute temperature,
is the gas constant, and
is the standard Gibbs energy change related to the formation of the TS from reactants.
The formalism expressed in the previous formulas only holds true in solutions that are ideally mixed and in general in all those cases where the rate of encounter of reactants is not limiting. When reactants diffuse slowly in the reacting medium, their frequency of collision can be the rate limiting step of the reaction and the observed rate can be very slow, despite the fact that the chemical reaction can be very fast (very high ). In such situations, the observed rate constant is expressed by the following formula (only the forward reaction is shown):
(14)
(14) where
is the rate constant for the conversion of the reactants into products (eq. Equation6
(6)
(6) ) and
is the rate of the diffusion of the reactants. If the reaction occurs in conditions such that the diffusivity of reactants is not hampered, that is,
,
is entirely dominated by
. The reaction is said to be reaction controlled. When molecular diffusion of reactants is hampered such that
then,
is mostly determined by
and the reaction is said to be diffusion controlled.
If the reaction system is homogeneous, that is, all the physical and chemical properties are the same in any infinitesimal volume, then the rate of the reaction will be the same in any infinitesimal volume of the system and is also equal to the average rate. If the system is heterogeneous, that is, with properties different in different infinitesimal volume, then the reaction rate will vary locally and the average rate will be obtained by ideally integrating the reaction rate over the entire reaction volume.
Mechanistic modeling of the food matrix effect
When a chemical reaction occurs in food, the observed rate is, most likely, different from what would be measured under ideal conditions (a very dilute solution) and different from food to food. These observed rates are therefore “apparent” rates compared to the true rates measured under ideal conditions. The position of the reaction equilibrium can be affected also. This effect is buried within observed equilibrium and rate constants. In short, the food matrix will change thermodynamic and kinetic properties of the reaction by acting on concentrations of reactants and products (see eq. Equation2(2)
(2) and Equation11
(11)
(11) ), activity coefficients (eq. Equation4
(4)
(4) and Equation12
(12)
(12) ), and diffusivity (eq. Equation14
(14)
(14) ) of reactants and products, as well as on the temperature sensed by the reactants in each part of the system (eq. Equation5
(5)
(5) and Equation13
(13)
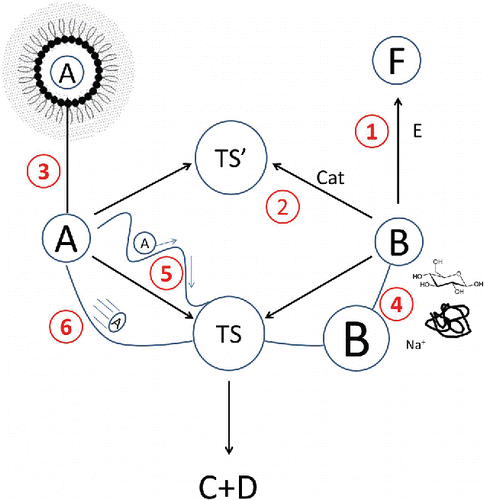
(13) ). This is summarized and visually depicted in .
Figure 2. The multiple effect of a food matrix on the rate and the equilibrium of a generic reaction. To be able to convert into products C and D, reactants A and B must come into contact to form an intermediate transition state TS. This, in turn, decomposes into products. The food matrix can affect the rate of this reaction in multiple ways: (1) Parallel reactions may deplete one of the reactants or both, thus changing concentrations of reactants. A special case is the effect of pH which changes the concentration of the reactive form of the reactant. (2) In the presence of catalysts the reaction may proceed through a different path which leads to a different transition state (TS’). (3) Partitioning of (one of) the reactants between different phase (e.g., oil and water phase) changes the concentrations of reactants in each phase. (4) Short-range and long-range interactions with ionic and nonionic solutes may alter reactants and transition state chemical potential (here the change in chemical potential is represented by reactant B being of different size which has obviously nothing to do with its molecular size). (5) The presence of obstacles (surrounding molecules, pores, etc.) or changes in viscosity may augment the effective path which reactant molecules must go through before they can collide, or may slow down reactant molecules. (6) Different thermal properties may produce a thermal profile within the food during nonequilibrium conditions, which changes the energy of reactants kinetic and, thus, frequency of collisions.
Mechanistic modeling the effect of food matrix on chemical reactivity means to be able to predict its effect on reactivity based on fundamental laws of chemistry and physics. In other words, it means to disentangle the effect of food matrix from the observed rate constant by quantifying its effect on concentrations of reactants (and intermediates), activities, diffusivities, as well as on temperature of the food systems. In the following subsections, theoretical frameworks for the quantification of each single effect are described.
Reactants concentration
Reactants can be consumed (or formed) through parallel reactions with (or from) other food components which would change their actual concentrations in the system. For instance, the condensation between a reducing sugar and a particular amino group will proceed at a slower rate when the sugar is simultaneously consumed in a parallel reaction with another amino group, or due to sugar isomerization. When parallel reactions involving reactants have been identified, their effect on the rate of the reaction of interest can be accounted for by reformulating the expression of the rate law. For instance, in the simpler case where reactant A in eq. Equation1(1)
(1) is also involved in a parallel irreversible reaction:
(15)
(15)
The rate law of eq. Equation11(11)
(11) would be reformulated as follows:
(16)
(16) where
is the rate constant for the parallel reaction. Of course, much more complex reaction networks may occur in food (for example also including parallel reactions where reactants are generated). In such situation the deployment of more sophisticated modeling approaches like multiresponse modeling are necessary (Van Boekel, Citation2008).
Reactants concentration can also change because of changes in the moisture content of food. If moisture loss is homogeneous throughout food, change in reactant concentration is inversely proportional to the change in moisture. However, in transient, nonequilibrium situations where the moisture content varies in time and throughout the food, mathematical models are needed to describe the diffusion of water and thus the actual moisture distribution throughout the food. This is important, for example, during high temperature processing of solid food, like cooking and frying. Changes in moisture content also result in changes in food structure and rheology and thus in the diffusivity of reactants (see next sections).
Catalysis
The presence of a catalyst changes the kinetics of a chemical reaction by providing an alternative route for the reaction to occur. The kinetics of the reaction in such a situation can be dealt with in the same way as described above (see eq. Equation16(16)
(16) ), that is, by reformulating the rate law as a function of two parallel reactions, one catalyzed and the other noncatalyzed, each characterized by its own reaction rate. Suppose a simple irreversible unimolecular conversion:
(17)
(17)
The rate of this reaction is expressed as follows:(18)
(18)
The alternative catalyzed route is:(19)
(19) where C is the catalyst. The rate of the catalyzed reaction is:
(20)
(20)
The overall rate is therefore:(21)
(21)
Thus, the rate constant of the noncatalyzed reaction and the rate constant of the catalyzed reaction both contribute to the observed rate constant. In food systems, acids and bases and metal ions represent the most common catalyst. Acids and bases can catalyze a reaction through the undissociated acid or the undissociated base or through generation of H+, OH− ions, that is, through changes in pH. Each of the catalyst (undissociated acid, undissociated base, H+, OH−) provides an alternative route to the uncatalyzed reaction. Each route is characterized by its own reaction rate and each rate is proportional to the catalyst concentration. The overall reaction rate can be expressed as a function of multiple parallel reactions, as described in eq. Equation21(21)
(21) . On top of that, pH can change the ratio between charged and neutral species of a reactant or product, that is, their pH-dependent species distribution (of course for ionisable reactants and products). This ratio can be calculated from the well-known Henderson-Hasselbalch equation. For instance, the relative distribution of the dissociated and undissociated forms of an acidic group is linked to pH by the following expression:
(22)
(22) where
is the acid dissociation constant of the acidic group, and
and
are the thermodynamic activities of the conjugated base and acid, respectively. By applying the above equation the actual activities of the reactive species (
and
) can be calculated at any pH and thus, the effect on reaction rate can be predicted.
Partitioning of reactants among different phases
In multiphase foods, reactants can partition between different phases, which changes their concentrations in each phase. A simple example is the distribution of a reactant between an aqueous phase and a micellar phase, the so-called micellar effect (van Boekel, 2009). When amphipathic compounds like mono- and diglycerides or phospholipids are present in solutions above the critical micelle concentration, a micellar phase can form which is in equilibrium with amphipathic molecules in solution. The distribution of reactants between the aqueous and the micellar phase can result in two distinct “reactors” wherein the same reaction occurs at two different rates because of differences in reactant concentrations. An example is the catalysis of the reaction between sorbic acid and thiols by surfactants (Wedzicha and Zeb, Citation1990) or bovine serum albumin (Wedzicha and Zeb, Citation1991). Another relevant example is the partitioning of reactants in emulsions. In such systems, reactants and intermediates can partition between the aqueous phase, the lipid phase or the interphase depending on their polarity and surface activity. The condensation between a reducing sugar and an amino group occurs with a different rate in oil in water nano-emulsions compared to aqueous solutions because of the pH-dependent partitioning of the amino acid between the lipid and the aqueous phases (Troise et al., Citation2016).
Partition of solutes in heterogeneous, multiphase systems can be quantitatively described by partition coefficients. A partition coefficient, , is defined as:
(23)
(23) where
and
are the thermodynamic activities of the solute in phase 1 and phase 2, respectively, when the equilibrium between the two phases has been attained. Under ideal conditions, thermodynamic activities can be replaced by concentrations in eq. Equation23
(23)
(23) . A partition coefficient depends on the solute, on the phases and also on temperature. The interfacial region between two phases represents an additional phase (even though it is not a true phase). This partition, together with the volume fraction of each phase in the system determines the molar concentration of each compound in each phase. The overall reaction rate is the sum of the contribution to the reaction from each phase. An example is provided for emulsions by (Romsted and Bravo-Diaz, Citation2013) but it can be formally extended to other systems made up of different phases or pseudophases (e.g., a micellar system). If we consider the bimolecular reaction in eq. Equation1
(1)
(1) , as long as the rate of partitioning between phases is much faster than the reaction itself (i.e., equilibrium has been attained for the reactants between the phases), the contributions to the overall reaction rate of the three separate reaction pathways in the continuous oil, interfacial, and aqueous regions are proportional to the concentrations of A and B in each region. This leads to the general equation:
(24)
(24) where
is the second-order kinetics rate constant;
, and
are the rate constants for the reaction in each phase; [A] and [B] the reactant concentrations and φ are the volume fractions with φW + φO + φI = 1. Subscripts W, O, and I represent the water, oil, and interface phases, respectively. Eq. Equation24
(24)
(24) reduces to only one of the three terms displayed on the right-hand side if the reaction occurs entirely in one phase, that is, the reactants are soluble only in one phase. If the partition coefficients happen to be known, the amount of reactants in each phase can be calculated and eq. Equation24
(24)
(24) can be used to predict the overall rate. However, it must be stressed that rate constants may be different in different phases (i.e.,
, and
in eq. Equation24
(24)
(24) may be different) because reactants are exposed to different milieus or because of differential properties of the solvent, for example, its dielectric constant. Partition coefficients must be determined experimentally, even though partition between oil and water can be also predicted by empirical and mechanistic models (Moln r et al., 2004; Molnár et al., Citation2004; Cheng et al., Citation2007).
Chemical potential
Since foods are nonideal systems, the chemical milieu around the reactants can change their chemical potential. This is a purely nonspecific effect exerted by the bystander molecules on those involved in the target reaction (in the sense that the bystander molecules do not chemically react directly with the molecules being affected). Neutral cosolutes will interact with the reactants through short-range forces, like dipole-dipole or dispersion forces. Ionic cosolutes will interact with neutral or charged reactants through long-range interactions like Coulomb forces. These interactions can be quite strong and may occur frequently in food which contains minerals but also dissociated acids and bases and also proteins and carbohydrates which carry charges (which depend on pH). The chemical potential of reactants can also be altered by a mere steric effect, that is, the physical occupancy of part of the available space by neighbor molecules. A typical example of this is the molecular crowding effect. This results in thermodynamic activity and diffusivity of reactants strongly deviating from those in ideal (aqueous) model systems (Ellis, Citation2001; Zhou et al., Citation2008). Since molecular crowding is ultimately a steric effect, this will be sensed more by high-molecular-weight compounds, like proteins and polysaccharides. However, the shape of the crowder also plays an essential role. Under crowding conditions, reaction equilibria are moved towards states that excludes the least volume to all the other macromolecules, for example, aggregation of macromolecules and folding of proteins are favored. Since protein unfolding is hindered under crowding conditions, heat stability of technologically relevant food enzymes is therefore expected to be higher in a crowded environment (Sasahara et al., Citation2003; Oliviero et al., Citation2014). As a rule of thumb, molecular crowding is expected to speed up transition-state-limited reactions which lead to a smaller free volume and to slow down diffusion-limited reactions because of the hindrance of reactants diffusion. The molecular crowding effect has been studied in the biochemistry domain for a while but its impact on kinetics and thermodynamics of food related chemical reactions has been very limitedly explored. Few notable exceptions are available in the scientific literature: the increase in egg white protein resistance to UV light in a crowded environment (Manzocco and Nicoli, Citation2012, Citation2015) and the increase in the rate of glycation of proteins in crowded systems (Perusko et al., Citation2015) (Weng et al., Citation2016).
In ideal solutions, that is, solutions where the average interactions between solute-solute, solute-solvent, solvent-solvent molecules are the same as in a pure liquid, the thermodynamic equilibria and the rate laws are expressed in term of concentrations of reactants. In nonideal solutions, the rate laws are expressed in terms of thermodynamic activity of the reactants and will vary accordingly. Once the activities of the probe molecules have been measured or estimated, the changes in equilibria and the reaction rates can be quantitatively accounted for (see eq. Equation4(4)
(4) and Equation12
(12)
(12) ). Despite the fact that the relationship between activity and concentration is very simple (see eq. Equation3
(3)
(3) ), the prediction of the activity coefficients of reacting species from fundamental laws of physics is quite difficult, especially in very complex systems like foods. Several mechanistic, empirical, and semiempirical models have been proposed to predict the value of the activity coefficients of probe molecules in nonideal systems.
Deviation from ideality may arise from short-range and long-range interactions between molecules. Long-range interactions refer to electrical potential experienced by charged molecules. Short-range interactions include, for example, dipole–dipole interactions and volume exclusion interactions. In electrolyte solutions nonideality arises from electrostatic interactions between solutes-solvents and solute-solute and those interactions are governed by Coulomb’s law, but also from short-range interactions. The first model to be proposed to estimate the activity coefficients of electrolytes was based on the Debye–Hückel theory (Hückel, Citation1924). In this theory the solution is considered as made up of charged ions of finite size surrounded by a cloud of opposite, point charges at a distance of minimum approach dispersed in a structureless medium of a determined relative permittivity. The model can only successfully predict the mean activity coefficient for dilute solutions when ion pairs are absent. To overcome this limitation, the mean spherical approximation (MSA) theory can be used (Blum, Citation1975). Although it has been developed for the description of thermodynamic properties of electrolyte solutions, those of nonionic cosolutes can be described as well. In the MSA approach, all the solute species, charged or uncharged, have finite size. The expression for the activity coefficients comprises a contribution from the electrostatic part and one for the volume exclusion effect via the hard sphere contribution, which reveals how space can be occupied by the cosolutes, which are approximated to have spherical shape. A very attractive alternative to calculate activity coefficients for electrolyte systems is represented by the electrolyte perturbed-chain statistical associating fluid theory (ePC-SAFT), which derives from the combination of SAFT-based equations with electrolytes theories. ePC-SAFT proved more accurate in predicting activity coefficients of biochemical compounds over Debye–Hückel theory (Hoffmann et al., Citation2013; Hoffmann et al., Citation2014). PC-SAFT accounts for molecular shape and size of solutes and their interactions with solvent by conceiving molecules as chains composed of spherical segments which interact through forces depending on their distance and diameter.
In solutions of neutral solutes, molecules experience only short-range interactions with neighboring particles. The UNIFAC model is one of the models that can be used to predict activity coefficients in such situations (Fredenslund et al., Citation1975). It is a modification of the UNIQUAC model. In both the models, the activity coefficients are split into two terms, a combinatorial term γC and a residual term γR. The first term quantifies the deviation from ideality due to differences in molecular shape. The latter is an enthalpy term that is based on the interactions between solutes. The difference between UNIFAC and UNIQUAC is that UNIFAC uses the functional groups present in interacting molecules to calculate the activity coefficients. In that respect, UNIFAC is a group contribution model, that is, a model that considers a whole solution as a mixture of functional groups rather than distinct molecules. Another example of group contribution model is the Savage-Wood Additivity of Groups (SWAG) model (Savage and Wood). This model is based on the assumption that each functional group X of solute i interacts with each group Y of cosolute C with a group-characteristic contribution that is independent of all other functional groups or relative positions and stereochemistry in solute i and cosolute C. The assumptions lead to an expression for the pairwise Gibbs energy interaction parameters in terms of specific group interactions. Other empirical models (Margules equation, van Laar equation, Wilson model, PC-SAFT, etc.) have been proposed for nonelectrolytes. A recent interesting development is represented by the conductor-like screening model for realistic solvation (COSMO-RS) model (Klamt and Schuurmann, Citation1993; Klamt, Citation1995; Klamt et al., Citation1998) and its subsequent reimplementations (e.g., COSMO-SAC) (Lin and Sandler, Citation2002). COSMO-RS model is not a group contribution model and does not require functional group parameters. This model uses quantum chemistry to calculate the chemical potential of molecular species in real solvents. First, COSMO equations are used to calculate the screening charge density on the surface of each molecule. From the pairwise interaction energies of surface patches, the chemical potential of a chemical species in a real solvent or mixture is predicted. The chemical potential of the solution or the mixture is approximately the weighted sum of the screen charge densities of its components. In analogy with the UNIFAC and UNIQUAC models, the COSMO-RS is a Gibbs excess free energy model and splits the chemical potential in a combinatorial and a residual contribution. However, compared to UNIFAC and UNIQUAC models (and other data interpolation models), the advantage of COSMO-RS models is that it does not need any adjustable, empirical group- or system-specific parameter, and chemical potentials are automatically predicted based on general or compound-specific properties, that is, COSMO-RS are more “mechanistic” compared to UNIFAC and UNIQUAC. In this respect, COSMO-MS has better predictive ability of thermodynamic properties of “new” compounds but is relatively more inaccurate in prediction of thermodynamic properties of compounds for which large amount of experimental data are available (this allows empirical parameters of (semi-)empirical models such as UNIFAC and UNIQUAC to be fitted to experimental data). Another important limitation of the COSMO-RS model is that it is, at the moment, inapplicable to electrolyte solutions (Klamt et al., Citation2010). The deviation from ideality due to purely volume exclusion effects can also be predicted, for instance via scaled-particle theory based on hard sphere approximation (Zhou et al., Citation2008). With the scaled-particle theory, within certain practical conditions, experimentally measured activity coefficients of proteins in crowded solutions can be estimated surprisingly accurately using simple structural models, in which rigid globular proteins are represented by hard particles having a size and shape similar to that of the protein at low resolution (such as spheres or spherocylinders), and large random coil polymers are represented by a random matrix of long rigid rods.
Outside chemical engineering, applications of thermodynamic models to predict chemical potentials of biological reactions are rare and can be found mostly in the pharmaceutical and the agrochemical areas (Klamt et al., Citation2002; Klamt et al., Citation2003; Klamt, Citation2005). A summary of the possible approaches can be found in (Held and Sadowski, Citation2016). Their application for the prediction of the activity coefficients of probe molecules in food is even more rare. Examples are the predictions of activity coefficient of aroma compounds in model juices or real juices, mainly based on the UNIFAC model (Carelli et al., Citation1991; Sancho et al., Citation1997) and those of amino acids in sugar solutions (Lakshmi and Nandi, Citation1976). More recently, the activity coefficient of calcium ions in milk-based systems enriched with disaccharides has been modeled using the MSA theory and used to describe the ion partitioning in milk and milk with added electrolytes (Gao et al., Citation2010a; Gao et al., Citation2010b). Other examples are the reaction of carmoisine, a food dye, with hydrogen sulphite ions (Wedzicha and Rumbelow, Citation1981) and the effect of ionic strength on dissociation of hydrogen sulphite, a food preservative (Wedzicha and Goddard, Citation1991). Thermodynamic activities can be measured also. To this end, equilibrium methods can be used, that is, methods based on partition of the compound between two phases such as osmotic measurements. This is because partition coefficients are based on activities rather than concentrations (see eq. Equation23(23)
(23) ). The activity of water, aw in solutions, for instance, is measured from its vapor pressure above the solution which is based on the water partition between the liquid and the vapor phase. Measurement of an electric potential by means of an ion-selective electrode is also an option for charged molecules (pH measurement is one such example).
A specific case of thermodynamic activity that has a paramount importance in food is water activity, aw. aw can affect chemical reactivity in a direct way when water is a reactant (e.g., in a hydrolysis reaction) or in an indirect way, by changing concentration of reactants (but only if the change in aw is associated with change in water content) or chemical potential of reactants. Concentration of solutes in food at reduced aw also results in an increase in viscosity, volume occupancy or tortuosity and therefore, to a slower molecular diffusion.
Diffusivity
Differences in physical, structural and rheological properties of food affect the mobility of reactants within the food matrix which has a large effect on bimolecular (and higher order) reactions. Generally, molecular diffusivity can be described by two different approaches: Fick’s law of diffusion and the Maxwell–Stefan approach. Fick’s second law of diffusion can be written as follows:(25)
(25)
This equation gives the change in concentration of the molecular probe in time at different points (x) as a function of the concentration gradient. Based on Fick’s law for diffusion, the diffusive behavior of compounds is described in terms of their diffusion coefficient . The diffusion coefficient can be predicted from more fundamental laws in some simple cases. For particles or large molecules in a viscous fluid, the Stokes–Einstein equation can be applied:
(26)
(26) Here, k is the Boltzmann constant, μ is the solvent viscosity,
is the absolute temperature, and
is the radius of the diffusing particle. In a solution that is not made up of polymers, it is the viscosity of the medium that determines the molecular diffusivity; however, it must be stressed that in several food systems, molecular diffusion cannot be described as diffusion of inert spherical particles in a viscous medium. In several food systems this looks like a very strong approximation, because reactants occur in crowded environments that limit molecular mobility by confining them within discrete molecular boundaries, like pores, and complex structural properties may be present. At molecular scale, the effect on molecule diffusion depends on the number, size, and shape of the surrounding molecules. When molecules are moving in a crowded environment, the steric hindrance exerted by the impenetrable macromolecules would create effective pores which constrain the free movement of the probe. This effect is known as molecular confinement.
A typical example is the diffusivity of a molecule in a plant cell or a dried fruit. In such a situation, molecular diffusivity is severely hindered, which reflects itself in diffusion coefficients smaller than the theoretical ones and which are more properly referred to as effective diffusion coefficients, D*. Effective diffusion coefficients are not just a function of solvent viscosity and density, but some structural features have to be considered, like the volume fraction that is available to the solute, the tortuosity (the ratio between the actual path travelled by a fluid element between two points divided by the straight line path between the same two points) and the pore size compared to the size of the diffusing molecule.
In the framework of the Maxwell–Stefan (MS) approach to diffusion the rate of diffusion depends on the balance between the driving forces to diffusion and the resistance exerted by the surrounding molecules and expressed in friction coefficients between each pair of diffusing molecules. This can be expressed as follows:(27)
(27)
The left-hand side of eq. Equation25(25)
(25) expresses the “force” that causes the motion of component i compared to component j, whereas the right-hand side expresses the friction exerted by component j towards component i when the two compounds move with a relative velocity equal to
is the molar fraction of component j and
is the friction coefficient between components i and j. Compared to the Fickian approach, the MS approach can account for all the forces responsible for molecular diffusion (electrical, gravitational) apart from chemical forces (concentration gradients) by adding relevant expressions of the driving forces in the left hand side of eq. Equation25
(25)
(25) . Moreover, with this approach the thermodynamic nonideality is incorporated in the driving forces for the diffusing probe molecule that is a function of its activity (
) rather than its concentration. If the activity coefficients can be calculated (see previous section) or measured, then the effect of food matrix on diffusion can be explicitly taken into account, whereas it is concealed in the diffusion coefficients of the Fickian approach. In other words, the MS diffusion coefficients are relatively less sensitive to changes in the food matrix (through the effect of differences in the chemical potential of the diffusing molecule) compared with the Fickian diffusion coefficient. The MS approach to diffusion has been applied in the food domain only to a limited extent. In one study, a MS multicomponent approach was developed to model salt and water diffusion in cheese, considering cheese as a three-component system consisting of NaCl (component 1), water (component 2) and a matrix of protein and fat (component 3) (Payne and Morison, Citation1999). In another study the diffusion of salt and water through sausage during drying was modeled (Costa-Corredor et al., Citation2010).
Temperature
Finally, the rate of the reaction depends on the temperature that is sensed by the reacting molecules but during food processing, heat transfer is very often in the unsteady state, in which temperatures are changing and materials are warming or cooling. In such situations a temperature profile is generated in time and within food which depends on the thermal properties of food. For this reason, food with different thermal properties that is exposed to the same external temperature will experience a different thermal gradient before thermal equilibrium is reached. The change in the surface temperature of food during heating/cooling is determined by its resistance to heat transfer from the external medium, for example, air, water, steam or oil, through natural or forced convection. This transfer can be calculated with the following general formula for heat transfer:(28)
(28) where
is the quantity of heat flowing,
is the surface heat-transfer coefficient,
is the surface area,
is the bulk fluid temperature, and
is the food surface temperature. The surface heat transfer coefficient is largely independent from the food matrix but mostly from the heating/cooling fluid.
The heat is transferred from surface to interior (or the other way around) by conduction. This can be calculated by using the Fourier equation, which is the equivalent of the Fick equation for heat:(29)
(29) where
is the food thermal diffusivity, which in turn is function of food thermal conductivity (λ), specific heat (Cp) and density (ρ):
(30)
(30)
Specific heat (Cp) is defined as the amount of heat required to increase the temperature of a unit mass of the substance by a unit degree. As it happens, Cp is an additive property, that is, Cp of a composite material is almost equal to the weighted sum of Cp for its components, that is, water, proteins, fats, carbohydrates, ice, and ashes (Choi, Citation1986). Cp of each component is, in turn, a function of temperature and can be calculated at different temperature from empirical equations (Choi, Citation1986). The thermal conductivity of a material is defined as a measure of its ability to conduct heat. The dependence of thermal conductivity of food components from temperature can be predicted from empirical equations (Choi, Citation1986). Unlike Cp, λ for a composite food cannot be predicted simply from its relative composition in the major components, but structural features like the geometrical distribution of the phases must be considered. Examples are the parallel models (the components are assumed to be placed parallel to heat flow), series models (components are perpendicular to the heat flow) and the Krischer model (mixed distribution). Other models such as Maxwell–Eucken and Kopelman models have been developed for two-component food systems consisting of a continuous and a dispersed phase (Sahin and Sumnu, Citation2006). However, heat can be transferred in food also through other mechanisms like convection, that is, mixing of fluid masses at different temperature or like distillation, that is, evaporation and condensation of vapour within a dispersed gaseous phase (e.g., through bubbles in bread). An additional and obvious complication in food is that, during thermal treatments, thermal properties may change because of the chemical and structural changes occurring.
Food matrix and enzymatic reactions
The effects displayed in still hold true when an enzymatic reaction is considered. The reaction rate will be affected by the concentration of substrates for the enzyme, their chemical potential and diffusivity, which, in the case of enzymatic reactions, also include the frequency of enzyme-substrate encounters. On top of that, the effect of the food matrix on the enzyme itself should be considered. Food matrix can affect enzyme catalytic activity via modification of enzyme conformation. This may happen via a number of chemical inhibitors or activators directly binding to the enzyme or indirectly through, for example, pH. In addition, the volume exclusion effect is expected to affect enzymatic reactions through modification of enzyme conformation, the quaternary structure of enzymes as well as the formation of the enzyme-substrate complex (Minton and Wilf, Citation1981). An example is provided once again by the GLs-MYR system described above. For instance, vitamin C is known to increase the enzymatic activity of MYR (Burow et al., Citation2006), and thus the conversion of GLs into breakdown products whereas pH and the concentration in Fe2+ affect the activity of the ESP which converts the intermediate product of MYR activity into epithionitriles (Hanschen et al., Citation2017). As a result the relative ratio between GLs enzymatic breakdown products strongly depend on food matrix properties.
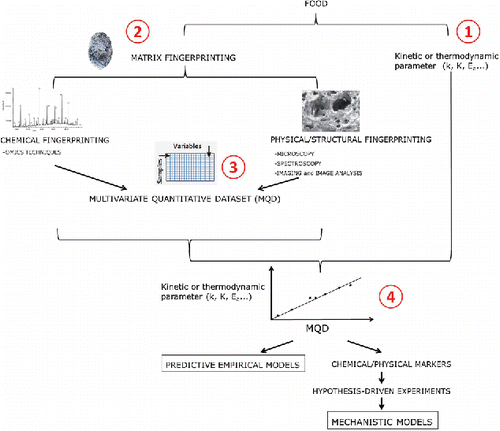
Empirical modeling of the food matrix based on fingerprinting
The approach described so far, aims at predicting the effect of the food matrix on chemical reactivity based on the knowledge of the underlying mechanisms affecting reactivity in biological systems. A fundamentally different approach for modeling food matrix effects on chemical reactivity is represented by empirical black-box modeling based on machine learning and data mining techniques. In this approach, the parameter of interest is not predicted through mechanistic models, but through statistical correlations between the parameter of interest and a set of food matrix characteristics/properties. This hypothesis-free data-driven approach is briefly described in . The first step is the quantification of the quality attribute of interest. This can be, for instance, the thermal stability of a target nutrient, measured by the rate constant for its degradation at a certain temperature. The second step is the extensive quantitative description of the chemical composition and the physical structure of several food matrices. We can name this comprehensive description of the food matrix its fingerprint. Chemical fingerprints can be obtained through a wide array of analytical techniques, such as liquid or gas chromatography (combined or not with mass spectrometry), spectroscopy, and electrophoresis. For example, chemical fingerprints can be constituted by the concentrations of a set of compounds, in the form of normalized intensities in a mass spectrum or areas of peaks in a chromatogram. It is important to stress that the identification of each single chemical descriptor is not formally necessary as long as each single variable is quantified with accuracy and precision. The physical structure of the system can also be described by combining outputs from microscopy, spectroscopy or imaging techniques. Spectroscopic data are inherently multivariate because the absorbance (or transmittance) of the sample is recorded at different wavelengths. When imaging techniques are used, these have to be translated into quantitative datasets, which is not straightforward. The authors believe that such translation of microscopy and other imaging techniques, along with the conversion of images into numerical data, would lead ultimately to empirical models that are able to predict complex quality attributes from the comprehensive numerical representation of the food matrix composition and structure. This scenario can be deemed realistic due to the fast improvement of calculation computer power as well as suitable data processing techniques. Hyperspectral imaging is a powerful technology for remotely inferring the material properties of foods. Hyperspectral images may be thought of as made up of a series of images of the same object taken at different wavelength or as a collection of pixels each represented by a spectrum. Taken as a whole, a hyperspectral image is simultaneously a chemical and structural fingerprint of the test material (Dale et al., Citation2013). Classical vibrational spectroscopy techniques, such as fluorescence spectroscopy and infrared spectroscopy also give information on the physical structure of test samples. Those techniques are typically used to describe the chemical composition of a test sample, but structural information can also be isolated with proper data processing. Other instrumental approaches to obtain quantitative information on the food matrix are, for instance, magnetic resonance imaging (MRI) and X-ray tomography combined with image analysis. Quantitative measures of physical/structural properties of the sample, such as porosity and tortuosity of a solid material, can be derived from these techniques (Schoeman et al., Citation2016).
Figure 3. Empirical modeling of the food matrix based on fingerprinting. This holistic hypothesis-free data-driven approach would consist in obtaining the values of a thermodynamic or kinetic parameters for several food matrices (Equation1(1)
(1) ) along with chemical and physical/structural fingerprint of each of them (Equation2
(2)
(2) ) and translate this fingerprint into a multivariate quantitative dataset (MQD, 3). Chemical fingerprinting can be obtained through many analytical techniques like, chromatography. The physical structure of the system can also be quantitatively described by combining outputs from microscopy, spectroscopy and imaging techniques combined, for example, with image analysis. Once MQDs are obtained for several matrices multivariate statistical techniques are applied to calibrate the MQD against the measured thermodynamic or kinetic property (Equation4
(4)
(4) ). This empirical model allows (1) to predict the thermodynamic of kinetic behavior of future samples once their fingerprinting is obtained (2) to identify a set of chemical and physical markers that can then be used to set off new hypothesis-driven experiments in order to develop proper mechanistic models.
Once the quality attribute of interest and the fingerprints have been obtained for several food matrices, a calibration model is developed that correlates the property of interest, that is, the dependent variable, to the multivariate quantitative description of the food matrix, that is, the independent variables. The derivation of the calibration model from a multivariate independent variable requires the use of special modeling tools. Examples of such modeling techniques include: basic multivariate regression algorithms, such as principal component regression (PCR) and partial least square regression (PLSR); and also statistical learning algorithms inspired by biological neural networks such as artificial neural network (ANN); support vector machine (SVM); back propagation (BP) methods; self-organizing maps (SOM); or genetic algorithms (GA). These pattern recognition techniques are also said to be supervised because the models are trained against a desired output, that is the prediction of the property of interest in a new sample. The learning algorithms inspired by biological neural networks have the advantage over traditional MLR, PLSR, and PCR to be able to deal better with highly nonlinear systems.
Once the calibration model has been developed, this can be used to predict the property of interest in a new food, once its fingerprint is obtained. The advantage of using this modeling approach to analyze a complex problem is accompanied by the drawback that it only allows the prediction of the behavior of a certain system, but does not provide any knowledge on the food system. Application of machine learning combined with data mining is not a completely new approach in food science. This type of approach has been, for instance, used for authenticity assessment and fraud detection (Capuano and van Ruth, Citation2012; Cozzolino, Citation2014; Nunes, Citation2014), food spoilage detection (Ellis et al., Citation2004; Nicolaou and Goodacre, Citation2008). More recently, an interesting use of such approach has been proposed to discriminate between different processing methods, storage conditions or food quality classes (Vervoort et al., Citation2012; Kebede et al., Citation2013; Vervoort et al., Citation2013; Grauwet et al., Citation2014). Here we propose that this approach can also be used to predict the effect of the food matrix on chemical reactivity, for example, which is the expected rate constant or equilibrium constant for a certain reaction in a certain food. In addition to regression (where a quantitative output is predicted), classification can be also applied to allocate food matrices in broad qualitative categories, for instance more or less “protective” towards a certain nutrient. Along with prediction or classification, this approach can be used to select discriminant features that can be, in turn, tested in hypothesis-driven mechanistic models. This can be done by selecting single or patterns of independent variables that are most correlated to the dependent variable. The only example of such a use of this empirical approach reported in the scientific literature so far can be found in (Hennig et al., Citation2013) where an untargeted metabolomics approach, followed by random forest regression, was applied to identify metabolites associated to thermal GL degradation in a segregating Brassica oleracea population.
Conclusions and future perspectives
Foods are very complex systems from a chemical and structural point of view. There are, therefore, several food-related factors that can affect the evolution of a chemical reaction. We have named the totality of these effects the food matrix effect. The food matrix effect is responsible for the biological variation we observe in the thermodynamics and kinetics of the same reaction occurring in different foods. The striking differences that may be observed between reactions occurring in a food, compared to an ideal aqueous system, should also alert food scientists about the proper choice of model systems when the findings must be extended to real food with an acceptable level of confidence. In this report we have presented two different approaches to describe the effect of the food matrix on reactivity. The first is a mechanistic approach based on fundamental laws of chemistry and physics. This approach should permit to predict the behavior of a food system based on the detailed knowledge of its composition and structure. Two considerations are evident with respect to this approach. The first is that the definition of mechanistic is somewhat arbitrary, since at a certain point empirical or semiempirical models must be employed. For instance, the effect of a cosolute on rate constants can be incorporated into kinetics via a fundamental law of chemistry, that is the relationship between concentration and chemical activity, but the prediction of an activity coefficient is sometimes based on (semi-)empirical models, though much progress has been made in this respect. The second consideration is that, if it is true that each single effect can be quantitatively described based on mechanistic models, the simultaneous occurrence of several effects makes the theoretical prediction of the behavior of a real food system still very far to reach. For instance, the change in one ingredient in a food formulation can simultaneously change the food pH, the hydration properties, the chemical potential, the reaction pathway, and also the diffusion behavior of reactants and intermediates. And those changes may occur in different directions so that the behavior of the system may be perceived as casual or nonlinear. In this respect, a holistic or “system” approach, which would take into account the complexity of the food system and the potentially nonlinear relationship between certain chemical or physical properties and the quality attribute of interest, would be useful. System dynamics modeling is just one of those “system” approaches. The second approach to predict the effect of food matrix is a purely empirical, correlative approach, based on an extensive characterization of the chemical and structural properties of the food matrix and a correlation of the obtained matrix fingerprint to chemical reactivity. It is clear that this approach would require reliable analytical techniques capable of producing large amount of precise and accurate data, as well as intensive computational power, which is not expected to be a limiting factor in the near future. This approach may be used for accurate predictions of the effect of food matrix on reactivity and, by the identification of key characteristics of interest of the food matrix, can be the starting point for further mechanistic investigations. In such a way, this inductive, top-down, hypothesis-free approach will proceed hand in hand with the deductive, hypothesis-based, bottom-up approach in generating knowledge about the effect of food matrix on chemical reactivity.
Acknowledgment
The authors declare no conflict of interest.
References
- Biswa Nath Das, Y.-W. K. and Keum, Young-Sam (2013). Mechanisms of Nrf2/Keap1-dependent phase II cytoprotective and detoxifying gene expression and potential cellular targets of chemopreventive isothiocyanates. Oxid. Med. Cell Longev. 2013:1–7.
- Blum, L. (1975). Mean spherical model for asymmetric electrolytes. Mol. Phys. 30:1529–1535.
- Burdurlu, H. S., Koca, N. and Karadeniz, F. (2006). Degradation of vitamin C in citrus juice concentrates during storage. J. Food Eng. 74:211–216.
- Burow, M., Markert, J., Gershenzon, J. and Wittstock, U. (2006). Comparative biochemical characterization of nitrile-forming proteins from plants and insects that alter myrosinase-catalysed hydrolysis of glucosinolates. Febs J. 273:2432–2446.
- Capuano, E. and van Ruth, S. M. (2012). QA: Fraud control for foods and other biomaterials by product fingerprinting. In: Latest Research into Quality Control, pp. 111–148. Akyar, I., Ed. INTECH Open Access Publisher.
- Carelli, A. A., Crapiste, G. H. and Lozano, J. E. (1991). Activity coefficients of aroma compounds in model solutions simulating apple juice. J. Agric. Food Chem. 39:1636–1640.
- Cheng, T., Zhao, Y., Li, X., Lin, F., Xu, Y., Zhang, X., Li, Y., Wang, R. and Lai, L. (2007). Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 47:2140–2148.
- Choi, Y. (1986). Effects of temperature and composition on the thermal properties of foods. In: Food Engineering and Process Applications, Vol. 1, Transport Phenomena, pp. 93–101 Elsevier App. Sci. Pub, London, New York.
- Costa-Corredor, A., Pakowski, Z., Lenczewski, T. and Gou, P. (2010). Simulation of simultaneous water and salt diffusion in dry fermented sausages by the Stefan-Maxwell equation. J. Food Eng. 97:311–318.
- Cozzolino, D. (2014). An overview of the use of infrared spectroscopy and chemometrics in authenticity and traceability of cereals. Food Res. Int. 60:262–265.
- Dale, L. M., Thewis, A., Boudry, C., Rotar, I., Dardenne, P., Baeten, V. and Pierna, J. A. F. (2013). Hyperspectral imaging applications in agriculture and agro-food product quality and safety control: A review. Appl. Spectrosc. Rev. 48:142–159.
- Dekker, M., Hennig, K. and Verkerk, R. (2009). Differences in thermal stability of glucosinolates in five brassica vegetables. Czech J. Food Sci. 27:S85–S88.
- Ellis, D. I., Broadhurst, D. and Goodacre, R. (2004). Rapid and quantitative detection of the microbial spoilage of beef by Fourier transform infrared spectroscopy and machine learning. Anal. Chim. Acta. 514:193–201.
- Ellis, R. J. (2001). Macromolecular crowding: Obvious but underappreciated. Trends Biochem. Sci. 26:597–604.
- Fahey, J. W., Zalcmann, A. T. and Talalay, P. (2001). The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 56:5–51.
- Fredenslund, A., Jones, R. L. and Prausnitz, J. M. (1975). Group‐contribution estimation of activity coefficients in nonideal liquid mixtures. AIChE J. 21:1086–1099.
- Gao, R., van Halsema, F. E. D., Temminghoff, E. J. M., van Leeuwen, H. P., van Valenberg, H. J. F., Eisner, M. D., Giesbers, M. and van Boekel, M. A. J. S. (2010a). Modelling ion composition in simulated milk ultrafiltrate (SMUF). I: Influence of calcium phosphate precipitation. Food Chem. 122:700–709.
- Gao, R., van Halsema, F. E. D., Temminghoff, E. J. M., van Leeuwen, H. P., van Valenberg, H. J. F., Eisner, M. D. and van Boekel, M. A. J. S. (2010b). Modelling ion composition in simulated milk ultrafiltrate (SMUF) II. Influence of pH, ionic strength and polyphosphates. Food Chem. 122:710–715.
- Giambanelli, E., Verkerk, R., D’Antuono, L. F. and Oliviero, T. (2016). The kinetic of key phytochemical compounds of non-heading and heading leafy Brassica oleracea landraces as affected by traditional cooking methods. J. Sci. Food Agr. 96:4772–4784.
- Giambanelli, E., Verkerk, R., Fogliano, V., Capuano, E., D’Antuono, L. F. and Oliviero, T. (2015). Broccoli glucosinolate degradation is reduced performing thermal treatment in binary systems with other food ingredients. Rsc Adv. 5:66894–66900.
- Giannakourou, M. C. and Taoukis, P. S. (2003). Kinetic modelling of vitamin C loss in frozen green vegetables under variable storage conditions. Food Chem. 83:33–41.
- Grauwet, T., Vervoort, L., Colle, I., Van Loey, A. and Hendrickx, M. (2014). From fingerprinting to kinetics in evaluating food quality changes. Trends Biotechnol. 32:125–131.
- Hanschen, F. S., Bauer, A., Mewis, I., Keil, C., Schreiner, M., Rohn, S. and Kroh, L. W. (2012). Thermally induced degradation of aliphatic glucosinolates: identification of intermediary breakdown products and proposed degradation pathways. J. Agric. Food Chem. 60:9890–9899.
- Hanschen, F. S., Klopsch, R., Oliviero, T., Schreiner, M., Verkerk, R. and Dekker, M. (2017). Optimizing isothiocyanate formation during enzymatic glucosinolate breakdown by adjusting pH value, temperature and dilution in Brassica vegetables and Arabidopsis thaliana. Sci. Rep. 7:40807.
- Held, C. and Sadowski, G. (2016). Thermodynamics of Bioreactions. Annu. Rev. Chem. Biomol. Eng. 7:395–414.
- Hennig, K., de Vos, R. C. H., Maliepaard, C., Dekker, M., Verkerk, R. and Bonnema, G. (2014). A metabolomics approach to identify factors influencing glucosinolate thermal degradation rates in Brassica vegetables. Food Chem. 155:287–297.
- Hennig, K., Verkerk, R., Bonnema, G. and Dekker, M. (2012). Rapid estimation of glucosinolate thermal degradation rate constants in leaves of chinese kale and broccoli (Brassica oleracea) in two seasons. J. Agric. Food Chem. 60:7859–7865.
- Hennig, K., Verkerk, R., Dekker, M. and Bonnema, G. (2013). Quantitative trait loci analysis of non-enzymatic glucosinolate degradation rates in Brassica oleracea during food processing. Theor. Appl. Genet. 126:2323–2334.
- Hiwilepo-van Hal, P., Bosschaart, C., van Twisk, C., Verkerk, R. and Dekker, M. (2012). Kinetics of thermal degradation of vitamin C in marula fruit (Sclerocarya birrea subsp caffra) as compared to other selected tropical fruits. LWT - Food Sci. Technol. 49:188–191.
- Hoffmann, P., Held, C., Maskow, T. and Sadowski, G. (2014). A thermodynamic investigation of the glucose-6-phosphate isomerization. Biophys. Chem. 195:22–31.
- Hoffmann, P., Voges, M., Held, C. and Sadowski, G. (2013). The role of activity coefficients in bioreaction equilibria: thermodynamics of methyl ferulate hydrolysis. Biophys. Chem. 173-174:21–30.
- Hückel, E. (1924). Zur theorie der elektrolyte. In: Ergebnisse Der Exakten Naturwissenschaften, pp. 199–276. Springer, Berlin, Heidelberg, Berlin, Heidelberg.
- Jin, X., Oliviero, T., van der Sman, R. G. M., Verkerk, R., Dekker, M. and van Boxtel, A. J. B. (2014). Impact of different drying trajectories on degradation of nutritional compounds in broccoli (Brassica oleracea var. italica). LWT Food Sci. Technol. 59:189–195.
- Jin, X., Oliviero, T., van der Sman, R. G. M., Verkerk, R., Dekker, M. and van Boxtel, A. J. B. (2014). Impact of different drying trajectories on degradation of nutritional compounds in broccoli (Brassica oleracea var. italica). LWT Food Sci. Technol. 59:189–195.
- Karim, O. R. and Adebowale, A. A. (2009). A dynamic method for kinetic model of ascorbic acid degradation during air dehydration of pretreated pineapple slices. Int. Food Res. J. 16:555–560.
- Kebede, B. T., Grauwet, T., Tabilo-Munizaga, G., Palmers, S., Vervoort, L., Hendrickx, M. and Van Loey, A. (2013). Headspace components that discriminate between thermal and high pressure high temperature treated green vegetables: Identification and linkage to possible process-induced chemical changes. Food Chem. 141:1603–1613.
- Klamt, A. (1995). Conductor-like screening model for real solvents - a new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 99:2224–2235.
- Klamt, A. (2005). COSMO-RS: From Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design. Elsevier, Amsterdam.
- Klamt, A., Eckert, F., Diedenhofen, M. and Beck, M. E. (2003). First principles calculations of aqueous p K a values for organic and inorganic acids using COSMO− RS reveal an inconsistency in the slope of the p K a scale. J. Phys. Chem. A. 107:9380–9386.
- Klamt, A., Eckert, F. and Arlt, W. (2010). COSMO-RS: An alternative to simulation for calculating thermodynamic properties of liquid mixtures. Annu. Rev. Chem. Biomol. Eng. 1:101–122.
- Klamt, A., Eckert, F., Hornig, M., Beck, M. E. and Bürger, T. (2002). Prediction of aqueous solubility of drugs and pesticides with COSMO‐RS. J. Comput. Chem. 23:275–281.
- Klamt, A., Jonas, V., Burger, T. and Lohrenz, J. C. W. (1998). Refinement and parametrization of COSMO-RS. J. Phys. Chem.A. 102:5074–5085.
- Klamt, A. and Schuurmann, G. (1993). Cosmo - a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. II:799–805. doi:10.1039/P29930000799
- Laing, B. M., Schlueter, D. L. and Labuza, T. P. (1978). Degradation kinetics of ascorbic acid at high temperature and water activity. J. Food Sci. 43:1440–1443.
- Lakshmi, T. S. and Nandi, P. K. (1976). Effects of sugar solutions on the activity coefficients of aromatic amino acids and their N-acetyl ethyl esters. J. Phys. Chem. 80:249–252.
- Lin, S. T. and Sandler, S. I. (2002). A priori phase equilibrium prediction from a segment contribution solvation model. Ind. Eng. Chem. Res. 41:899–913.
- Manzocco, L. and Nicoli, M. C. (2012). Macromolecular crowding affects protein photosensitivity: The case of egg white immunoreactivity. Food Chem. 132:982–988.
- Manzocco, L. and Nicoli, M. C. (2015). Self Crowding as a Determinant of egg white Photostability. Food Biophys. 10:155–161.
- Miller, P. E. and Snyder, D., (2012). Phytochemicals and cancer risk: A review of the epidemiological evidence. Nutr. Clin. Pract. 27:599–612.
- Minton, A. P. and Wilf, J. (1981). Effect of macromolecular crowding upon the structure and function of an enzyme - glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 20:4821–4826.
- Mishkin, M., Saguy, I. and Karel, M. (1984). Optimization of nutrient retention during processing - ascorbic-acid in potato dehydration. J. Food Sci. 49:1262–1266.
- 7Molnár, L., Keserű, G. M., Papp, Á., Gulyás, Z. and Darvas, F. (2004). A neural network based prediction of octanol–water partition coefficients using atomic5 fragmental descriptors. Bioorg. Med. Chem. Lett. 14:851–853.
- Nicolaou, N. and Goodacre, R. (2008). Rapid and quantitative detection of the microbial spoilage in milk using Fourier transform infrared spectroscopy and chemometrics. Analyst 133:1424–1431.
- Nunes, C. A. (2014). Vibrational spectroscopy and chemometrics to assess authenticity, adulteration and intrinsic quality parameters of edible oils and fats. Food Res. Int. 60:255–261.
- Oliviero, T., Verkerk, R. and Dekker, M. (2012). Effect of water content and temperature on glucosinolate degradation kinetics in broccoli (Brassica oleracea var. italica). Food Chem. 132:2037–2045.
- Oliviero, T., Verkerk, R., Van Boekel, M. A. J. S. and Dekker, M. (2014). Effect of water content and temperature on inactivation kinetics of myrosinase in broccoli (Brassica oleracea var. italica). Food Chem. 163:197–201.
- Ozkan, M., Kirca, A. and Cemeroglu, B. (2004). Effects of hydrogen peroxide on the stability of ascorbic acid during storage in various fruit juices. Food Chem. 88:591–597.
- Payne, M. R. and Morison, K. R. (1999). A multi-component approach to salt and water diffusion in cheese. Int. Dairy J. 9:887–894.
- Peleg, M., Normand, M. D., Dixon, W. R. and Goulette, T. R. (2016). Modeling the degradation kinetics of ascorbic acid. Crit. Rev. Food Sci. Nutr. https://doi.org/10.1080/10408398.2016.1264360.
- Perusko, M., Al-Hanish, A., Velickovic, T. C. and Stanic-Vucinic, D. (2015). Macromolecular crowding conditions enhance glycation and oxidation of whey proteins in ultrasound-induced Maillard reaction. Food Chem. 177:248–257.
- Romsted, L. S. and Bravo-Diaz, C. (2013). Modeling chemical reactivity in emulsions. Curr. Opin. Colloid Interface Sci. 18:3–14.
- Sahin, S. and Sumnu, S. G. (2006). Thermal Properties of Foods. In: Physical Properties of Foods, pp. 107–155. Springer, New York.
- Sancho, M. F., Rao, M. A. and Downing, D. L. (1997). Infinite dilution activity coefficients of apple juice aroma compounds. J. Food Eng. 34:145–158.
- Sasahara, K., McPhie, P. and Minton, A. P. (2003). Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J. Mol. Biol. 326:1227–1237.
- Savage, J. J. and Wood, R. H. (1976). Enthalpy of dilution of aqueous mixtures of amides, sugars, urea, ethylene glycol, and pentaerythritol at 25°C: Enthalpy of interaction of the hydrocarbon, amide, and hydroxyl functional groups in dilute aqueous solutions. J. Solution Chem. 5:733–750.
- Schoeman, L., Williams, P., du Plessis, A. and Manley, M. (2016). X-ray micro-computed tomography (μCT) for non-destructive characterisation of food microstructure. Trends Food Sci. Technol. 47:10–24.
- Slimestad, R., Fossen, T. and Vagen, I. M. (2007). Onions: a source of unique dietary flavonoids. J. Agric. Food Chem. 55:10067–10080.
- Steinmetz, K. A. and Potter, J. D., (1996). Vegetables, fruit and cancer prevention: a review. J. Am. Diet Assoc. 96:1027–1039.
- Stešková, A., Morochovičová, M. and Lešková, E. (2006). Vitamin C degradation during storage of fortified foods. J. Food Nutr. Res. 45:55–61.
- Traka, M. and Mithen, R. (2009). Glucosinolates, isothiocyanates and human health. Phytochem. Rev. 8:269–282.
- Troise, A. D., Berton-Carabin, C. C. and Fogliano, V. (2016). Amadori products formation in emulsified systems. Food Chem. 199:51–58.
- Uddin, M. S., Hawlader, M. N. A. and Zhou, L. W. (2001). Kinetics of ascorbic acid degradation in dried kiwifruits during storage. Drying Technol. 19:437–446.
- Van Boekel, M. A. (2008). Kinetic Modeling of Reactions In Foods. CRC press, Boca Raton, FL.
- Vervoort, L., Grauwet, T., Kebede, B. T., Van der Plancken, I., Timmermans, R., Hendrickx, M. and Van Loey, A. (2012). Headspace fingerprinting as an untargeted approach to compare novel and traditional processing technologies: A case-study on orange juice pasteurisation. Food Chem. 134:2303–2312.
- Vervoort, L., Grauwet, T., Njoroge, D. M., Van der Plancken, I., Matser, A., Hendrickx, M. and Van Loey, A. (2013). Comparing thermal and high pressure processing of carrots at different processing intensities by headspace fingerprinting. Innov. Food Sci. Emerg. Technol. 18:31–42.
- Wedzicha, B. L. and Goddard, S. J. (1991). The state of sulphur dioxide at high concentration and low water activity. Food Chem. 40:119–136.
- Wedzicha, B. L. and Rumbelow, S. J. (1981). The reaction of an azo food dye with hydrogen sulphite ions. J. Sci. Food Agr. 32:699–704.
- Wedzicha, B. L. and Zeb, A. (1990). Catalysis of the reaction between sorbic acid and thiols by surfactants. Int. J. Food Sci. Tech. 25:168–179.
- Wedzicha, B. L. and Zeb, A. (1991). Catalysis of the sorbic acid–thiol reaction by bovine serum albumin. Int. J. Food Sci. Tech. 26:381–384.
- Weng, J. Y., Qi, J. R., Yin, S. W., Wang, J. M., Guo, J., Feng, J. L., Liu, Q. R., Zhu, J. H. and Yang, X. Q. (2016). Fractionation and characterization of soy beta-conglycinin-dextran conjugates via macromolecular crowding environment and dry heating. Food Chem. 196:1264–1271.
- Zhou, H. X., Rivas, G. and Minton, A. P. (2008). Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 37:375–397.