
Abstract
The numbers and diversity of microbes in ecosystems within and around us is unmatched, yet most of these microorganisms remain recalcitrant to in vitro cultivation. Various high-throughput molecular techniques, collectively termed multi-omics, provide insights into the genomic structure and metabolic potential as well as activity of complex microbial communities. Nonetheless, pure or defined cultures are needed to (1) decipher microbial physiology and thus test multi-omics-based ecological hypotheses, (2) curate and improve database annotations and (3) realize novel applications in biotechnology. Cultivation thus provides context. In turn, we here argue that multi-omics information awaits integration into the development of novel cultivation strategies. This can build the foundation for a new era of omics information-guided microbial cultivation technology and reduce the inherent trial-and-error search space. This review discusses how information that can be extracted from multi-omics data can be applied for the cultivation of hitherto uncultured microorganisms. Furthermore, we summarize groundbreaking studies that successfully translated information derived from multi-omics into specific media formulations, screening techniques and selective enrichments in order to obtain novel targeted microbial isolates. By integrating these examples, we conclude with a proposed workflow to facilitate future omics-aided cultivation strategies that are inspired by the microbial complexity of the environment.
Introduction: the importance of cultivation in the age of multi-omics
The diversity and ubiquity of the microbial world that is observed in environmental samples through recent advances in high-throughput sequencing technologies is astounding. The multi-omics revolution dramatically reshaped the field of microbial ecology and led to the realization that in most ecosystems, the existing microorganisms outnumber those that are accessible through cultivation by orders of magnitude. Only a minor part of the microorganisms observed in an environmental sample can be grown and maintained axenically or in defined communities under laboratory conditions. This phenomenon, termed “the great plate count anomaly” (Staley Citation1985; Nichols Citation2007) has resulted in the development of numerous innovative cultivation techniques using advanced technologies like microfluidics (Ma et al. Citation2014; Boitard et al. Citation2015), cultivation chips (Ingham et al. Citation2007; Hesselman et al. Citation2012; Gao et al. Citation2013), manipulation of single cells (Ben-Dov et al. Citation2009; Park et al. Citation2011) and high-throughput cultivation termed “culturomics” (Lagier et al. Citation2012). These techniques immensely expanded the number of novel species brought into culture, as reviewed in several recent publications (Alain & Querellou Citation2009; Zengler Citation2009; Overmann Citation2010; Dewi Puspita et al. Citation2012; Dini-Andreote et al. Citation2012; Stewart Citation2012; Pham & Kim Citation2012; Allen-Vercoe Citation2013; Harwani Citation2013; Narihiro & Kamagata Citation2013). To date, we count approximately 11,000 isolated species distributed over 30 bacterial and five archaeal phyla that have been validly classified (List of Prokaryotic Names with Standing in Nomenclature (LPSN; http://www.bacterio.net, cited 2016 Aug 3)). In spite of the tremendous progress in cultivation technology, the “great plate count anomaly” remains in place, as the rate of discovery of microbes that are as yet uncultivable outpaces the rate of isolating novel species. During the past decades, microbiologists have extensively characterized microbial community composition based on the sequencing of universal marker genes, in most cases PCR-amplified regions of the small subunit ribosomal RNA (rRNA) gene (Lane et al. Citation1985). This led to an estimated rate of approximately 40,000 novel prokaryotic species being discovered per year, and a total of 400,000 species of bacteria and archaea are predicted to be discovered by 2017 (Yarza et al. Citation2014). To put this in perspective, the number of named bacterial phyla increased from 12 to 92 in the last four decades. Additionally, archaea were discovered as a separate domain in 1977 and since then expanded to 26 recognized phyla to date (Woese & Fox Citation1977; Baker & Dick Citation2013; Youssef et al. Citation2014; Hug et al. Citation2016). In stark contrast, less than 6% of the total number of bacterial and archaeal species included in the SILVA REF 114 database has been validly classified by physiological tests of isolates, listed in LPSN (Parte Citation2014).
Not surprisingly, sequencing instead of culturing became the trend in the field of microbial ecology after the revolution in sequencing technology. In the present review, we subsume the terms metagenomics, metatranscriptomics and metaproteomics under the term “multi-omics” (Zhang et al. Citation2010). Metabolomics is not discussed in this review because examples where this technology has successfully been embedded in novel cultivation strategies remain very scarce to date. Metagenomics is defined as the comprehensive sequence analysis of total DNA isolated from environmental samples (Handelsman et al. Citation1998; Marchesi & Ravel Citation2015). Metatranscriptomics is the analysis of total environmental RNA. It provides insights into the local and taxon-specific expression levels of genes (Frias-Lopez et al. Citation2008) at the community or even the single-cell level (Shi et al. Citation2014). Lastly, metaproteomics enables linking genotypes to phenotypes by detecting functional catalytic components of microbial communities (Wilmes et al. Citation2015). Multi-omics studies have been readily applied to characterize the diversity and metabolic potential of microbial communities in a wide range of different environments. These include soils (Dini-Andreote et al. Citation2012; Fierer et al. Citation2012), wastewater treatment bioreactors (Speth et al. Citation2016), marine sediments (Plewniak et al. Citation2013; Urich et al. Citation2014) and eukaryotic host-associated microbiomes (Erickson et al. Citation2012; Radax et al. Citation2012; Sessitsch et al. Citation2012; Segata et al. Citation2013; Fuerst Citation2014), thereby rapidly increasing our knowledge and understanding of microorganisms and their key roles in biogeochemical cycling processes and eukaryotic host functioning and health. In addition, with metagenomics, whole genomes of newly discovered uncultured species can be resolved, allowing to predict the metabolic capabilities of these microorganisms in natural or man-made ecosystems (Tyson et al. Citation2004; Siegl et al. Citation2011; Hug et al. Citation2012; Albertsen et al. Citation2013; Wilson & Piel Citation2013; McLean et al. Citation2013; Narihiro et al. Citation2014; Nielsen et al. Citation2014; Urich et al. Citation2014; Walker et al. Citation2014; Afshinnekoo et al. Citation2015; Brown et al. Citation2015).
Facing these data sets with reconstructed genomes of hundreds of microbial species and the hypotheses they give rise to, cultivation of microorganisms is more valuable than ever. Cultivation of microorganisms currently is the most reliable way to validate ecological hypotheses raised from multi-omics data. In addition, cultivation is important for the annotation and functional characterization of novel genes (Muller et al. Citation2013). With available cultures, bacterial metabolism can be studied at the biochemical level, revealing as-yet-unknown physiological traits under varying incubation conditions. Furthermore, multi-species interactions, evolutionary principles, population dynamics and pathogenicity can only be experimentally validated when isolates are available (). Lastly, stable cultures pave the way towards applications in biotechnology, for instance regarding the quest for novel bioactive compound discovery and production, bioremediation and ecosystem engineering. In fact, multi-omics and microbial cultivation studies should be acknowledged as two sides of the same coin (Leadbetter Citation2003; Overmann Citation2010). It has been suggested that – in many instances – multi-omics information can provide valuable insights for culturing additional environmental microbes (Allen-Vercoe Citation2013; Narihiro & Kamagata Citation2013); however, actual examples of attempts to link multi-omics information with cultivation technology have remained scarce.
Figure 1. A model depicting the positive feedback loop between multi-omics data generation and isolation of as yet uncultured microorganisms. The rise of multi-omics tools has led to a better understanding of microbial life in nature, the resource for novel biotechnological applications needed by our society. The tedious cultivation of microorganisms often represents the first milestone in novel biotechnological process development and facilitates testing of ecological hypotheses. Multi-omics information, curated by physiological characterization of already available microbial isolates, represents a huge pool of knowledge about the yet-uncultured microbial world. Hence, integrating multi-omics data directed at culturing novel environmental bacteria (dashed arrow) brings multi-omics data into context and has the potential to boost biotechnological innovation for the benefit of society and nature conservation.
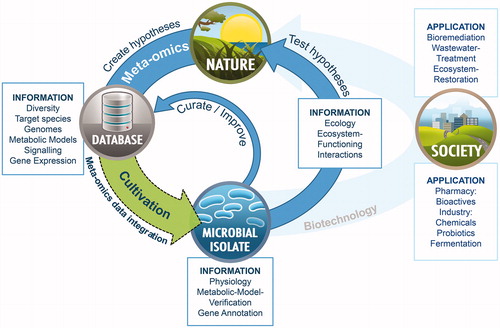
In this review, we discuss the available strategies that allow bridging the gap between current microbial cultivation and multi-omics approaches. In particular, we focus on the progress and pitfalls in the quest to integrate specific multi-omics-derived information with respect to enhancing microbial cultivation success. Secondly, we provide a literature survey, summarizing pioneering multi-omics-based cultivation experiments. Finally, we examine the current developments, extracting the experimental milestones that were achieved and propose a generalized workflow for future multi-omics inspired cultivation approaches.
Information associated with and extracted from multi-omics data
Genomes from metagenomes
Metagenomics provides information about the collective set of genes in a given community. For many purposes, it is required that these gene pools are separated and assigned to particular taxa. Depending on the complexity of the community and the depth of the analyses, sometimes even whole genomes can be assembled (Tyson et al. Citation2004; Brown et al. Citation2015). Taxonomy assignment to reads or (assembled) contigs from metagenomes can be done either based on phylogenetic affiliation (Darling et al. Citation2014) or through a process called binning. Binning can be performed using sequence composition-dependent or composition-independent methods or a combination of both (Albertsen et al. Citation2013). Composition-dependent methods use information such as GC content and tetranucleotide frequency patterns to sort reads within a metagenome into “bins” containing sequences with similar characteristics (Parks et al. Citation2011), but these can be limited by local sequence deviations within genomes. On the other hand, composition-independent methods use the assumption that reads/contigs with similar coverage profiles originate from the same microbial population and represent a proxy for its abundance. Generally, combining information on differential coverage profiles with composition-based approaches has been shown to improve binning fidelity (Imelfort et al. Citation2014).
On the basis of assembled genomes or genome fragments, predictions regarding the ecology, physiology and genetic potential of individual community members are feasible. For example, inferences concerning the metabolic pathways for nitrogen and carbon cycling, respiration mechanisms and the degradation of particular toxic compounds can be based on the relative abundance or even the presence and absence of the relevant genes (Barone et al. Citation2014; Narihiro et al. Citation2014). With the constantly improved methods for sequence generation and bioinformatic analysis, near complete genome assembly is slowly becoming a standard method (Hug et al. Citation2016). This allows insights into the metabolic potential of environmental communities with the potential to unravel the factors preventing cultivation to date, especially in combination with metabolic reconstruction.
Genome-scale reconstruction of metabolic capacities and pathways
Predicting metabolic capabilities and other phenotypical features of microorganisms based on genomic data is achieved by means of genome-scale metabolic models. These models are tools that are commonly used for linking genomic data to biochemical reaction networks controlling cellular processes (Bordbar et al. Citation2014). They can be utilized to understand the relationships between genotype and phenotype and can provide a framework for the integration of transcriptomic, proteomic and metabolomic data (Joyce & Palsson Citation2006). Such data integration thus offers an overview of in silico predicted cellular physiological and genetic responses to environmental changes in the microbial habitat. The process of genome-scale metabolic model construction has been reviewed extensively (Oberhardt et al. Citation2009; Santos et al. Citation2011; Bordbar et al. Citation2014). Briefly, it requires an initial draft genome-derived metabolic reconstruction based on gene annotation data that is coupled to information on pathways such as found in the KEGG database (Kanehisa et al. Citation2006) where genes are linked to functional categories. Following this, a model can be proposed that generates predictions about the phenotypes conferred by the analysed genomes. The models can be conceptual, but their analysis can be taken further using a mathematical representation of the bio-transformations and metabolic processes encoded within the organism’s genome. The latter is typically achieved using constraint-based methods, which impose constraints that consider network stoichiometry, thermodynamics, flux capacity and sometimes transcriptional regulation (Reed Citation2012). Multiple tools are available that either aid in the metabolic reconstruction from an assembled genome directly (The SEED) (Henry et al. Citation2010) or combine the information of multiple existing, manually curated models (Notebaart et al. Citation2006; Santos et al. Citation2011). Potentially relevant in the context of improving in vitro culturability of microorganisms, Carr and Borenstein (Citation2012) implemented NetSeed, a modelling tool, which predicts the compounds an organism needs to obtain from its environment. Based on NetSeed data, the Minimal ENvironment TOol (MENTO), predicts minimal nutritional requirements for the microorganisms at stake (Zarecki et al. Citation2014), making this a potentially useful tool in designing culture media. Currently, genome-scale metabolic models exist almost exclusively for targeted, single species and their respective genomes. However, methods for metabolic reconstruction of complex communities start to appear for well-studied ecosystems like the human gut (Magnúsdóttir et al. Citation2016), a development that is worth persuing since the input data for such analyses are accumulating rapidly in databases. Metabolic models based on genome‐scale reconstructions can be seen as collections of hypotheses, which can be systematically identified, tested and resolved to provide feedback for model refinements (Oberhardt et al. Citation2009). Therefore, for example, models of interacting species can be used to predict cross-feeding phenotypes, which would require simultaneous cultivation for growth. An interactive and iterative approach, including experiments and further model development, is expected to improve the accuracy of the predictions, in turn allowing to refine media for the cultivation of yet uncultured target microorganisms ().
Active metabolic functions at community level: metatranscriptomics and metaproteomics
Metatranscriptomics provides a snapshot of gene expression levels in a community. Application of metatranscriptomics-derived information provides another piece of the puzzle in the quest to establish robust cultivation conditions, as it allows to distinguish – under given environmental conditions – the active and passive community members and their expressed metabolic pathways (Frias-Lopez et al. Citation2008). It can even point at gene categories that are apparently required for growth and have not (yet) been highlighted in concurrent metagenomics-based studies (Radax et al. Citation2012). Furthermore, comparison of metatranscriptomes derived from samples subjected to different cultivation or environmental conditions can provide correlations between gene expression and environmental variables (Bomar et al. Citation2011). Thus, metatranscriptomics, and also metaproteomics, can assist us in understanding the abundance and function of the expressed genes and corresponding proteins in microbial habitats of interest (Keller & Hettich Citation2009). Responses to certain stress levels, alternating metabolic strategies (e.g. aerobic or anaerobic respiration vs. fermentation), defence mechanisms like antimicrobial compound production patterns or metabolite export and uptake can be differentiated. For example, information about active growth versus sole biomass maintenance can be obtained from a given community (Belnap et al. Citation2010; Erickson et al. Citation2012). This can provide detailed insights into the metabolic status of microbial communities and their adaptations towards differential conditions, representing valuable information for improving the cultivability of specific community members.
Cooperative and antagonistic interactions within microbial communities
Within microbial communities, distinct microorganisms often compete for limited resources. However, many processes occur in a cooperative manner since individual species often lack the ability to produce all essential components needed for survival. These needs are met by other microorganisms, enabling such interdependent microbes to live efficiently by clipping down the required number of encoded and expressed genes within individual community members in nutrient-limited environments (Morris et al. Citation2012). Consequently, microorganisms communicate by trading metabolites and/or signalling molecules. Processes driven by signalling molecules include, for example, biofilm formation, virulence factor secretion, bioluminescence, antibiotic production and exoenzyme production (West et al. Citation2007). Bacteria perform these actions aided by a process of cell-to-cell communication, like quorum sensing, where bacteria synchronously control gene expression in response to changes in cell density (Ng & Bassler Citation2009). Typically, intra-species interactions among bacteria include but are not limited to, signalling molecules such as N-acyl-homoserine lactones (N-AHLs), autoinducer-2 (AI-2) and antimicrobial compounds including peptides (Camilli & Bassler Citation2006; Yim et al. Citation2006). Through genomic information, it has become apparent that quorum sensing-related mechanisms are widespread in the bacterial world. For instance, the gene responsible for the production of AI-2 (LuxS) is present ubiquitously across the bacterial domain and found in over half of all sequenced bacterial genomes (Federle Citation2009; Pereira et al. Citation2013).
Hence, in cultivation attempts, the absence of metabolites or signalling molecules that are usually provided by other community members can have pronounced effects on the growth of target organisms and thus cultivation success (Camilli & Bassler Citation2006). This will particularly affect host-associated bacteria, which have highly specialized genomes (McCutcheon & Moran Citation2011). Such complex relationships are difficult to reproduce in traditional microbial cultivation approaches where cells are physically separated from each other and inter- and intra-species exchange of metabolites and/or signalling compounds is disrupted during the first stage of isolation.
To this end, the aforementioned genome-based model predictions of auxotrophies, that is, dependencies on external supply of specific compounds, may indicate which metabolic pathways have to be complemented in order to allow for in vitro growth of the targeted organism. Moreover, the discovery of phylogenetically and structurally novel signalling molecules, that provide the cues for metabolic activity, is common in microbial multi-omics data (Kimura Citation2014), potentially leading to success when integrated in cultivation methods (Bruns et al. Citation2002; Nichols et al. Citation2008; Vartoukian et al. Citation2010; Sipkema et al. Citation2011).
Antimicrobial compounds play a major role in the environment, not only as defence mechanisms against competing organisms, but also as intra-species signalling molecules (Goh et al. Citation2002; Yim et al. Citation2006; Yim et al. Citation2007; Voolaid et al. Citation2012). Therefore, antibiotic resistance genes are also widespread; they can be extracted from multi-omics data (Medema & Fischbach Citation2015) and have recently been shown to be expressed in a broad range of different natural environments (Versluis et al. Citation2015). Often, antibiotics show poor activity against oligotrophic and slow-growing organisms (Lewis Citation2007), many of which are potential targets for cultivation studies. Thus, antibiotics and their production and resistance loci detectable in multi-omics data can be used as selection criteria for the isolation of target organisms. This may include the prevention of overgrowth by fast-growing microorganisms (Sizova et al. Citation2012; Hames-Kocabas & Uzel Citation2012; Rettedal et al. Citation2014; Keren et al. Citation2015) or selection for specific phenotypic traits such as those characteristic for Gram-positive bacteria or production of antibiotic-resistant spores.
Habitat complexity and current multi-omics-based cultivation studies
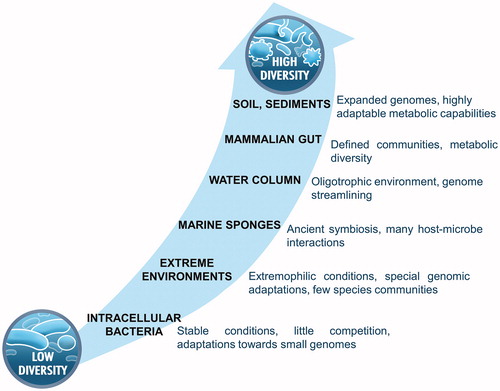
Different habitats require different strategies to obtain meaningful multi-omics data. Here, we categorize microbial habitats based on their complexity in order to interpret available literature data on the use of multi-omics data leading to cultivation successes. The complexity of a microbial habitat may include aspects of species diversity, fluctuations in environmental conditions and interactions among the community members. Temporal and spatial instability of environmental parameters favour the evolution of large genomes in a community (Guieysse & Wuertz Citation2012; Bentkowski et al. Citation2015). The presence of large genomes in a sample make it more difficult to resolve individual genomes using a given multi-omics strategy and reduce the strength of resulting hypotheses that lay the foundation for cultivation experiments. However, both environmental instability and the intricacy of interactions amongst microbes in an environment have been scarcely addressed in the multi-omics literature. Therefore, we use the parameter species diversity as a proxy for microbial habitat complexity (). Low-diversity environments are characterized by the predominance of just one or a few microbial species. These microbes may be host-associated or free-living in environments with conditions tolerated by only a handful of species, such as in acid mine drainage biofilms or hot springs. Currently, the majority of the successful –omics information based cultivation examples are from such low-diversity environments, since according multi-omics data can be analysed more thoroughly with currently available bioinformatic tools. On the contrary, most environments support the growth of a myriad of species resulting in a high microbial diversity, such as in soils, seawater or animal intestines (Torsvik Citation2002). In the following section, we elaborate how multi-omics information can be used as a basis for cultivation of targeted microorganisms from an environment with a given complexity, supported with key examples from literature.
Figure 2. Schematic depiction of selected environments discussed in this review according to the diversity of the residing microbial community.
Low diversity microbial habitats
Host associated
This category contains examples of both intra- and extracellular host-dependent or host-favoured microbes that share their habitat with only few or no other species. From intracellularly occurring microbes, genomic information can be readily obtained after purifying the microbes from host cell material by, for example, cell disruption and differential centrifugation as was done for the Q (query) fever-causing obligate intracellular pathogen Coxiella burnetti (Cockrell et al. Citation2008). In general, evolution favours the reduction of genome sizes, and hence, these microbes often have only a limited set of specialized metabolic pathways that support the host associations (Dutta & Paul Citation2012). Media and cultivation conditions have to be carefully adapted to the microbes’ demands, and multi-omics data can be instrumental in identifying nutritional requirements.
An early example of genome-inspired medium design was the development of a defined medium for Xylella fastidiosa, a slow-growing plant pathogen inhabiting the xylem of citrus plants (Lemos et al. Citation2003; Almeida et al. Citation2004; Janse & Obradovic Citation2010). Even though the organism had been isolated with empirical methods before (Wells et al. Citation1987), and its genome was sequenced in 2000 (Setubal et al. Citation2000), Lemos et al. (Citation2003) revealed that a relatively simple medium composition supported growth. They prepared five minimal media that differed in amino acid composition and concentration based on the presence or absence of genes for amino acid biosynthetic pathways in the X. fastidiosa genome. Some enzymes required for the biosynthesis of serine, cysteine and methionine were missing, and the addition of these amino acids resulted in faster growth of X. fastidiosa. Furthermore, myo-inositol, a specific precursor of plant cell wall polysaccharides, was added since it was hypothesized that X. fastidiosa metabolizes this compound based on the presence of the enzyme inositol monophosphatase. However, not all metabolic reactions the organism was performing were predicted from the genome. For example, despite a predicted serine auxotrophy, growth occurred in serine-free media. Potential reasons for this incongruence include incorrect gene annotation, multifunctional enzymes, unknown metabolic pathways or enzymes encoded by analogous genes (Lemos et al. Citation2003). This example shows how a variety of genome-tailored minimal medium designs can result in improved growth rates, but advocates at the same time that currently not all metabolic capabilities of the target organisms can be predicted correctly from genomic data.
Another example is provided by the first axenic culture of Tropheryma whipplei, the causative agent of Whipple’s disease. This obligate intracellular pathogen had resisted axenic cultivation for almost a century and was growing only in association with eukaryotic cells until its 0.9 Mb genome, obtained by differential centrifugation, was published in 2003 (Bentley et al. Citation2003). Analysing metabolic models based on the genome revealed the (partial or complete) absence of biosynthetic pathways for 16 amino acids, which suggested that T. whipplei obtains these from its host. As a follow-up, Renesto et al. (Citation2003) designed a medium providing the 16 amino acids, inoculated it with supernatant of T. whipplei-infected fibroblasts and established an axenic culture of T. whipplei.
The Q (query) fever-causing obligate intracellular pathogen Coxiella burnetti was recently liberated from its host into axenic culture (Omsland et al. Citation2009). In this case, information derived from multiple sources was used to optimize the medium that supported growth: Replicating niche characteristics (low pH, salt concentrations) and incorporating the genome-predicted metabolic capacities (amino acid uptake and metabolism) resulted in the formulation of the initial growth medium. A subsequent comparison of the transcriptomes from C. burnetti incubated in suboptimal medium and C. burnetti growing in Vero cells suggested a deficiency in amino acids for the bacteria growing in the designed medium. The addition of casamino acids and L-cysteine to the initial medium yielded an approximately 13-fold increase in protein synthesis, but not substantial growth. Further genome inspections revealed that terminal oxidases associated with both aerobic and microaerobic respiration was encoded, suggesting that C. burnetti could respire in low-oxygen environments. Incubations under different oxygen tensions showed an increased ability of C. burnetti to oxidize essential substrates under microaerophilic conditions. Finally, C. burnetti was incubated in amino acid-supplemented growth medium under 2.5% oxygen tension conditions, where three logs of growth were observed after 6 days of incubation. Thus, fine tuning growth conditions concurrently with designing media based on predicted metabolic capabilities led to the successful axenic cultivation of C. burnetti.
Bomar et al. (Citation2011) analysed the metatranscriptomes of two extracellular bacterial symbionts that make up the entire gut microbiota of the leech Hirudo verbana. The high expression levels of genes encoding mucin (and glycan) degrading enzymes suggested that mucins constitute the main carbon and energy source for the one as-yet-uncultured Rikenella-like leech symbiont. Replacement of glucose by mucin in the culture medium resulted in an isolate that was identical to the target bacterium from the leech gut. Hence, by identifying genes that are highly expressed in their original environment, key physiological properties of the target organisms can be predicted and used in targeted isolation approaches.
Extreme environments
Slightly more complex but still considered low-diversity environments are habitats that are characterized by extreme conditions. Hot springs, acid mine drainage biofilms or hypersaline lakes allow only a few highly adapted species to thrive. Such extremophilic organisms harbour a high potential for industrial processes and compounds. Furthermore, fundamental research interests in evolution, abiotic to biotic element cycling and possibilities for extraterrestrial life have made such extreme habitats popular study sites for decades. The relatively simple communities have led to many ground-breaking results, also in the field of molecular microbial ecology.
Tyson et al. (Citation2005) established one of the first breakthroughs in a metagenome-derived cultivation approach by reconstructing the genome of an as-yet-uncultured member of the Nitrospirae from a metagenome of an acid mine drainage biofilm. The reconstructed genome revealed a single nitrogen fixation operon. Based on this information, the authors developed a nitrogen-free medium. The cultivation conditions were further set up to match prevailing environmental conditions, that is, high metal concentrations, pH 0.8 and 37 °C. They successfully obtained axenic cultures of a Leptospirillum group III member by means of repeated sequential batch dilution series. This iron-oxidizing Nitrospirae isolate was described as Leptospirillum ferrodiazotrophum sp. nov. (Tyson et al. Citation2005). In a follow-up study, the entire acid mine drainage biofilm was targeted, including its complete metabolic functions (Belnap et al. Citation2010). Hence, a cultivation system was designed that maintained and propagated the biofilm in vitro. Metabolic labelling-based proteomic analysis after 12 days of growth confirmed also the presence and activity of low-abundance community members. Additionally, autotrophic primary production and stress responses were monitored. Modifying the cultivation conditions led to enhanced growth and decreased the abundance of stress response proteins as monitored by metaproteomics (Belnap et al. Citation2010). Thus, metaproteomics data as monitoring tool enabled the customization of cultivation conditions towards the metabolic demands of the targeted communities. To our knowledge, this is the only example of the use of metaproteomics for improving culturability, which might be due to the inherent complexity of community metaproteomes and the current analytical limitations of this technology (Wilmes et al. Citation2015).
Nanoarchaeota have first been described as obligate extracellular symbionts of Crenarchaeota from submarine hydrothermal vents (Huber et al. Citation2002). Using specific primers, Nanoarchaeota have since then been detected in many environments including terrestrial hot springs and hypersaline lakes with culture-independent methods (Casanueva et al. Citation2008). Single-cell sorting and whole-genome amplification of antibody-labelled archaea from the Obsidian Pool in Yellow Stone National park revealed 16 S rRNA gene sequences of a novel nanoarchaeal organism and sequences from its putative crenarchaeal host, an uncultured member of the Sulfolobales (Podar et al. Citation2013). Both nanoarchaeal and crenarchaeal whole-genome amplification products were assembled into near-complete genomes and their metabolic capacities were reconstructed. The nanoarchaeon’s fragmented, extremely small genome lacked many essential biosynthetic pathways which indicated that the nanoarchaeon cannot live autonomously and hence depends on the presence of a crenarchaeal associate. Potential glycolysis and gluconeogenesis pathways, however, are retained in the nanoarchaeon, suggesting the use of peptides or complex sugars as energy source (Podar et al. Citation2013; Wurch et al. Citation2016). On this basis, Wurch et al. (Citation2016) established enrichment cultures containing yeast extract, casamino acids and sucrose or glycogen in anoxic Brock medium with low pH and 80–85 °C and obtained stable communities with increasing relative abundance of nanoarchaeota, as monitored by qPCR. After two rounds of dilution to extinction, they transferred a single crenarchaeal cell carrying a nanoarchaeon into liquid medium using optical tweezers, thereby obtaining a pure co-culture of the crenarchaeon host (Acidilobus sp. 7 A) and its associated nanoarchaeon (strain N7A). This first isolated geothermal nanoarchaeon, proposed as “Candidatus Nanopusillus acidilobi” represents the smallest cultured organism to date (100–300 nm cell size), and available cultures now allow experimentation to reveal its particular metabolism and adaptation features. Many more ultra-small bacteria and archaea with similar ectosymbiotic or ectoparasitic lifestyles might await discovery and identification of their hosts, something that – apart from single cell genomics – is only feasible by direct cultivation (Delafont et al. Citation2015; He et al. Citation2015; Wurch et al. Citation2016).
We conclude that genomic information of microbes derived from low diversity environments can often be obtained relatively easily and that individual genome reconstructions are often feasible (. Analysing metabolic networks for the presence or absence of essential biosynthetic pathways, the presence/absence of specific catabolic pathways and uptake systems can reveal, which pathways have to be complemented by the culture medium, as well as help to choose specific carbon sources, electron donors and electron acceptors. Genome-inspired medium design in combination with fine-tuning cultivation conditions can assist in isolating as-yet-uncultivated, even host-associated microorganisms with reduced genomes. In cases where genomic information is inconclusive, metatranscriptomics or metaproteomics can be applied, indicating which bacteria are active in selected conditions. This will reveal the active metabolic functions and hint towards substrates that are preferably catabolized by the organisms under study.
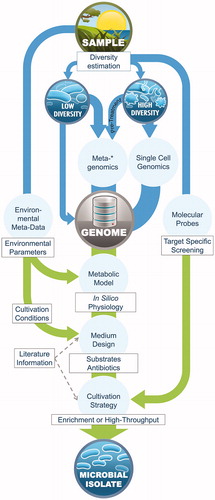
Figure 3. Proposed workflow for integrating multi-omics data into microbial cultivation. Arrows indicate the flow of information, blue for multi-omics strategies and green for microbial cultivation (in correspondence to ). *The sampling strategy prior to metagenomic or single-cell genomics differs for low- and high-diversity environments, the latter requiring a larger number of samples for enhanced genome recovery.
High diversity microbial habitats
This category contains examples of microorganisms sharing their habitat with a large variety of co-occurring species. Hence, interpreting multi-omics data represents a true challenge. Monitoring the efficiency of cultivation media through molecular and multi-omics methods for the growth of targeted species or specific microbial consortia is a common denominator for the examples in this category.
Host-associated high diversity habitats
Tian et al. (Citation2010) used DGGE (denaturing gradient gel electrophoresis, a molecular community diversity estimation method) of PCR-amplified 16 S rRNA gene fragments to compare cultivated communities to microbial profiles of the human oral cavity. They developed a cultivation medium that propagated a diverse community, which resembled the composition of the oral community the most. This in vitro grown community also contained phylotypes of the -until then- uncultivated candidate phylum TM7 (He et al. Citation2015). Some of the phylotypes contained a mutation in their 16 S rRNA gene sequence that had been previously linked to streptomycin resistance (Hugenholtz et al. Citation2001). Indeed, addition of streptomycin to the culture medium led to an enrichment of a specific TM7 phylotype (named TM7x), which was physically associated with a previously uncultured Actinomyces species. This stable coculture enabled the complete sequencing of TM7’s highly reduced genome, providing insights into the growth conditions and lifestyle of this human-associated epibiotic organism.
In addition to oral microbial communities, gut ecosystems provide a broad range of metabolic niches that are inhabited by diverse microbial communities (Vieira-Silva & Rocha Citation2010; Fodor et al. Citation2012). Especially, the human intestinal tract has been studied intensively, and many species have been cultured (Lagier et al. Citation2012; Rajilic-Stojanovic & de Vos Citation2014). The challenges of assembling genomes from such high-diversity environments are currently being overcome (Nielsen et al. Citation2014; White et al. Citation2016), allowing metabolic networks to be established for target species as well as for whole communities (Abubucker et al. Citation2012; Magnúsdóttir et al. Citation2016). Based on metabolic network information of target species, enrichment strategies can be designed that exclusively support the metabolism of the selected bacteria.
For example, nucleotide composition-based sequence binning enabled Pope and colleagues (Pope et al. Citation2011) to assemble approximately 2 Mb (∼90%) of the genome of an as-yet-uncultured member of the Succinivibrionaceae from a wallaby foregut metagenome that comprised sequences of more than 500 different species. The assembled genome was used to partially reconstruct the metabolic pathways of the bacterium as well as to search for putative antibiotic resistances. This predicted the utilization of starch as a sole carbon source and urea as a non-protein nitrogen source. A defined medium containing starch, urea and bacitracin was then developed, which led to highly enriched and (later) axenic cultures of the targeted phylotype. Further physiological characterization was consistent with the genome-based predictions, confirming that this bacterium is dependent on CO2 to support its succinate biosynthesis and produces succinate as major fermentation end product, further explaining the basis of the low methane emissions from herbivorous marsupials.
The inclusion of selection criteria such as antibiotics or other bactericidal compounds in the isolation strategy can increase the success of isolation manifold and select for specific phenotypic traits such as sporulation. Following up on the metagenomics-derived observation that many members of the human intestinal microbiota unexpectedly possess extensive genomic sporulation capacity, a pre-treatment using ethanol enriched for spores from the faecal samples and enabled the subsequent isolation of 45 novel candidate species (Browne et al. Citation2016). Overall, their isolates represented approximately 90% of the overall relative abundance at the species level in the individuals from which they were obtained and revealed novel insights into the transmission mode of human-associated strict anaerobes (Browne et al. Citation2016).
Free-living, highly complex communities
Highly diverse environments, such as water columns or soils and sediments pose particular challenges for current multi-omics approaches for multiple reasons. Firstly, such environments support the growth of microbial species with expanded genomic repertoires allowing them to adjust to oligotrophy and varying abiotic conditions (Konstantinidis & Tiedje Citation2005). Secondly, these environments are exposed to fluctuations in temperature, light, water content or salinity in daily or seasonal cycles, creating niches for a myriad of closely related species, making it difficult to separate genomes at strain or species level. As a consequence, the amount of sequencing needed to cover genomes substantially is astronomical and existing computational power needed to resolve such complex datasets exceeds current capacities (White et al. Citation2016).
Based on rRNA gene cloning, the SAR11 clade was found to be the most ubiquitous bacterium in ocean waters, yet recalcitrant to isolation. Hence, Rappé and colleagues (Rappé et al. Citation2002) employed a high-throughput dilution-to-extinction method to natural marine communities and inoculated a series of low-nutrient media with around 20 cells per well in microtitre plates. After 23 days of incubation, they obtained axenic cultures of 11 SAR11 strains, including the proposed “Candidatus Pelagibacter ubique”, allowing for in vitro studies with an organism of global biogeochemical significance. Further applications of this cultivation method led to the successful propagation of up to 14% of the cells of coastal waters (Connon & Giovannoni Citation2002). This empirical approach constituted a milestone in our quest to isolate the abundant bacteria in a given habitat. The analysis of two “Candidatus Pelagibacter ubique” genomes revealed an incomplete set of genes for assimilatory sulphate reduction, suggesting that the organism requires reduced sulphur compounds (e.g. methionine) for growth (Tripp et al. Citation2008). Furthermore, a fragmented glycolysis pathway and the absence of glycine and serine biosynthesis pathways suggested a metabolic dependency on low-molecular-weight organic acids as carbon sources. This information was used for the design of a defined medium, and it was shown that the addition of glycine and pyruvate as well as inorganic micro- and macronutrients and vitamins were required for robust growth of SAR11 isolates (Carini et al. Citation2012).
To our knowledge, multi-omics information-assisted cultivation approaches from highly diverse habitats such as soils and sediments remain scarce to date. The current pitfalls of multi-omics data generation and analysis are especially noticeable when it comes to multi-omics guided microbial isolation from such highly complex environments where extremely intricate community member interactions are expected (Traxler & Kolter Citation2015). However, recent breakthroughs are promising, and with the constantly improving methods for sequence generation and bioinformatics analysis, reasonably complete genome reconstruction is slowly becoming a standard method (Hug et al. Citation2016).
Pitfalls to current multi-omics methods and ways around the limitations
First, one classical problem of metagenomics and metatranscriptomics-based gene targeting is the difficulty of assigning observed functions to specific taxa (Dutilh et al. Citation2007). However, recent increases in obtainable sequence read length and assembled fragments have resulted in major improvements. Besides, computational developments are paving the way to make better use of currently available short reads. One recently developed pre-assembly method, coined latent strain analysis (LSA) (Cleary et al. Citation2015), separates the reads into “biologically informed” partitions, enabling the assembly of individual genomes from metagenomes. This is promising, since a large number of samples from high-complexity environments could enable a resolution high enough to assemble genomes of bacterial taxa present even at low abundances.
Second, a direct translation of genomics-based data to cultivation conditions and cultivation media is still difficult. For example, Lavy et al. (Citation2014) designed a medium based on genomic data of “Candidatus Poribacteria sp”. WGA-4E obtained through single-cell genomics of cells retrieved from the Red Sea sponge Theonella swinhoei (Siegl et al. Citation2011). However, the bacterium could not be brought into culture. Challenging in such endeavors is to decide on the concentrations of medium components and the combination thereof, since the (bio-) chemical composition of natural environments is often unknown despite carefully collected metadata. High concentrations of substrates can be toxic or inhibiting for environmental bacteria derived from nutrient-limited environments (Connon & Giovannoni Citation2002), or favour less-abundant, fast-growing organisms. Two different issues can be identified here: one is the isolation of community members, and another one deciphering optimal growth conditions. For the former, the concentration ranges of medium components are likely quite large, and inspiration from media that work for related microbes can be used as a proxy, although also potential competition by other, faster growing community members has to be taken into account. For the latter, further refinement of the media by factorial trial and error may be required. In fact, supplying cultivation media with ingredients that are not predicted as required according to an organism’s biosynthetic pathways can still be growth promoting (Lemos et al. Citation2003).
Third, information about the active contribution of community members to the overall nutrient cycling in an ecosystem is less frequently available as compared to their genomic potential. Bacteria can exist in a metabolically inactive dormant state, especially in nutrient-scarce environments that are subjected to regular disturbances, such as influx of toxic compounds (Epstein Citation2009; Buerger et al. Citation2012). This is gradually overcome by comparing gene expression or protein data with (meta)genomic data, which may reveal discrepancies between the most abundant and the most active organisms in a community.
Lastly, functional traits and metabolic pathways are inferred from the annotated portion of the metagenome. This fraction is a proportion of the total due to a variety of factors. First, metagenomic library construction relies on the accuracy of DNA extraction methods, which are prone to problems, such as incomplete cell extraction, cell lysis or DNA degradation (Wesolowska-Andersen et al. Citation2014). In addition, depending on the microbial diversity of the environment, approximately 30–50% of the genes found are of undetermined function altogether (Ellrott et al. Citation2010). Finally, many annotations present in databases are not accurate (Schnoes et al. Citation2009). Microbial cultivation itself is one way that can positively impact experimental validation of gene annotations, through an “ecologically validated” positive feedback loop. As a consequence, predictions of metabolic capabilities from multi-omics data can be expected to further improve. This in turn has the potential to bring a larger number of uncultured species into cultivation ().
In order to avoid some of the pitfalls mentioned above, we want to stress the importance of collecting bio- and physicochemical metadata at an appropriate temporal and spatial resolution in order to link multi-omics data to environmental cues. Microorganisms inhabit microenvironments strongly influenced by the structure of the environment, and they respond to conditions and resources at scales ranging from micrometres to a few meters (Franklin & Mills Citation2003; Cardon & Gage Citation2006). In the case of metagenomics and metatranscriptomics data, the standard of minimum information about any sequence is called MIMS (Minimum Information about a Metagenome Sequence) (Yilmaz et al. Citation2011), developed by the Genomics Standards Consortium (GSC, http://gensc.org). The metadata that describes the sampled environment usually includes collection date, specification of the environment (biome) and the location where samples were collected. We here advocate that additional parameters such as temperature, pH, oxygen concentration and biochemical data on nutrient or salt concentrations should be included as much as possible as they provide important environmental descriptors that can assist in interpreting multi-omics results and setting the appropriate cultivation conditions.
Envisioned strategies for omics-aided cultivation approaches
Even though each microbial habitat requires tailored study designs and challenges the creativity and inventiveness of individual researchers, we propose a more generic workflow based on the examples summarized in this review () as a guidance for future multi-omics based cultivation experiments.
Table 1. Summary of existing literature on the use of multi-omics for isolating as-yet uncultured microorganisms.
This workflow starts with sampling the environment of interest and collecting metadata at appropriate temporal and spatial resolution in order to link multi-omics data to environmental cues (). Estimating the species diversity of a given sample based on 16 S rRNA gene analysis, even though not a multi-omics approach sensu strictu, is helpful to determine, which of the complementary techniques of single-cell genomics and metagenomics to follow. For low diversity samples, metagenomics might enable the recovery of complete genomes of dominant species due to high copy numbers of those genomes in the samples. Applying metagenomics to high-diversity habitats may require a pre-treatment step such as cell size or cell density sorting, resulting in an enrichment of the microorganisms of interest. At the same time, the pre-treated samples can be used as pre-enriched inocula for cultivation. In addition, sequencing a large number of samples might also improve the odds for full genome recovery, given that availability of metagenomes from samples with different relative abundances aid in binning and genome reconstruction. Single-cell genomics is an alternative option to reconstruct draft genomes of the (target) microorganisms from highly complex microbial habitats.
The recovered genomes constitute a firm basis for the construction of metabolic models; however, the natural environment of the target organism and the quality of the draft genomes should be kept in consideration when examining resulting metabolic models, that is, mind the gaps! Metabolic models illustrate the targets’ genomic signatures of aerobic or anaerobic respiration, fermentation pathways, possible electron donor or acceptor molecules, carbohydrate metabolism and biosynthetic pathway deficiencies, which constitute useful information for medium design. Media formulations that were used to isolate phylogenetically related organisms represent a valuable source of inspiration for the compositional details of trace metal, salts and vitamin concentrations. We propose to aim for selective media compositions for initial isolation of the target. Identifying genetic traits that distinguish the target from other organisms such as antibiotic resistance markers or auxotrophies can represent valuable selection criteria, inhibiting undesired, fast growing organisms from the sampled community. Designing optimal growth media should only be considered after initial diversity reduction or isolation of the target since optimal growing conditions can be similar for many untargeted microbes from the environment, which may overgrow the organism of interest.
To summarize, the predicted metabolic substrates and the resulting products, in conjunction with the environmental metadata, can be translated into medium designs that can be subjected to the currently available multitude of novel cultivation strategies such as microfluidics, cultivation chips, manipulation of single cells and high throughput cultivation, mentioned in the introduction. Lastly, 16 S rRNA gene-based techniques such as fluorescent in situ hybridization (FISH) or qPCR screening enable tracking and quantification of the target organisms throughout the process of sampling and subsequent enrichment, cultivation and recovery. We surmise that, on the basis of such highly rational cultivation approaches (), a plethora of novel target species will be brought into culture.
Conclusions: the era of multi-omics-based microbial cultivation
In this decade, the huge increase in sequence data from genomes, metagenomes, metatranscriptomes and metaproteomes continues to unveil the enormous variety of as-yet-unexplored metabolic potential in nature. The massive amount of publicly available multi-omics data sets transits many environmental bacteria from the unknown-unknown to the known-unknown search space. Now, the decadal challenge is to further scrutinize such data sets and use them to serve our ecological and exploratory questions about members of microbiomes and their roles in the natural habitats we are studying. But, multi-omics methods have much more potential than just serving as explanatory tools. They provide hypotheses that await testing using advanced cultivation technologies to bring a range of previously recalcitrant extant microbes into cultivation. From targeted isolation (Huber et al. Citation1995; Davis et al. Citation2014) via multi-omics inspired medium development (Bomar et al. Citation2011; Pope et al. Citation2011) to high-throughput screening of a myriad of colonies or enriched liquid cultures (Lagier et al. Citation2012; Ma et al. Citation2014), the possibilities for data integration are plentiful. Given the fact that bacterial cultivation is time-consuming and tedious, the additional sources of information derived from high-end molecular tools provide highly practical advantages that may lead to important breakthroughs and should not be ignored. The integration of multi-omics knowledge in cultivation studies increases the chances of success and decreases the search space in the quest for new microbial isolates.
Acknowledgements
We thank Toni Clariana and A. A. Kampfraath for help and critical comments on designing and drawing. This research was supported by the People Programme (Marie Curie Actions) of the European Union Seventh Framework Programme FP7/2007–2013/ under REA grant agreement n° 607786, BluePharmTrain.
Disclosure statement
We state that this is an original research, which has not been previously published and has not been submitted for publication elsewhere while under consideration. We declare no conflict of interest.
Additional information
Funding
References
- Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. 2012. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 8:e1002358.
- Afshinnekoo E, Meydan C, Chowdhury S, Jaroudi D, Boyer C, Bernstein N, Maritz JM, Reeves D, Gandara J, Chhangawala S, et al. 2015. Geospatial resolution of human and bacterial diversity with city-scale metagenomics. Cels. 1:1–15.
- Alain K, Querellou J. 2009. Cultivating the uncultured: limits, advances and future challenges. Extremophiles. 13:583–594.
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol. 31:533–538.
- Allen-Vercoe E. 2013. Bringing the gut microbiota into focus through microbial culture: recent progress and future perspective. Curr Opin Microbiol. 16:625–629.
- Almeida RPP, Mann R, Purcell AH. 2004. Xylella fastidiosa cultivation on a minimal solid defined medium. Curr Microbiol. 48:368–372.
- Baker BJ, Dick GJ. 2013. Omic approaches in microbial ecology: charting the unknown. Microbe. 8:353–360.
- Barone R, Tedesco P, Visone M, Galati F. 2014. Marine metagenomics, a valuable tool for enzymes and bioactive compounds discovery. Front Mar Sci. 1:1–6.
- Belnap CP, Pan C, VerBerkmoes NC, Power ME, Samatova NF, Carver RL, Hettich RL, Banfield JF. 2010. Cultivation and quantitative proteomic analyses of acidophilic microbial communities. ISME J. 4:520–530.
- Ben-Dov E, Kramarsky-Winter E, Kushmaro A. 2009. An in situ method for cultivating microorganisms using a double encapsulation technique. FEMS Microbiol Ecol. 68:363–371.
- Bentkowski P, Oosterhout CV, Mock T. 2015. A model of genome size evolution for prokaryotes in stable and fluctuating environments. Genome Biol Evol. 7:2344–2351.
- Bentley SD, Maiwald M, Murphy LD, Pallen MJ, Yeats CA, Dover LG, Norbertczak HT, Besra GS, Quail MA, Harris DE, et al. 2003. Sequencing and analysis of the genome of the Whipple's disease bacterium Tropheryma whipplei. Lancet. 361:637–644.
- Boitard L, Cottinet D, Bremond N, Baudry J, Bibette J. 2015. Growing microbes in millifluidic droplets. Eng Life Sci. 15:318–326.
- Bomar L, Maltz M, Colston S, Graf J. 2011. Directed culturing of microorganisms using metatranscriptomics. MBio. 2:1–8.
- Bordbar A, Monk JM, King ZA, Palsson BØ. 2014. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 15:107–120.
- Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF. 2015. Unusual biology across a group comprising more than 15% of domain bacteria. Nature. 523:208–211.
- Browne HP, Forster SC, Anonye BO, Kumar N, Neville BA, Stares MD, Goulding D, Lawley TD. 2016. Culturing of “unculturable human microbiota reveals novel taxa and extensive sporulation”. Nature. 533:543–546.
- Bruns A, Cypionka H, Overmann J. 2002. Cyclic AMP and acyl homoserine lactones increase the cultivation efficiency of heterotrophic bacteria from the Central Baltic Sea. Appl Environ Microbiol. 68:3978–3987.
- Buerger S, Spoering A, Gavrish E, Leslin C, Ling L, Epstein SS. 2012. microbial scout hypothesis and microbial discovery. Appl Environ Microbiol. 78:3229–3233.
- Camilli A, Bassler BL. 2006. Bacterial small-molecule signaling pathways. Science. 311:1113–1116.
- Cardon ZG, Gage DJ. 2006. Resource exchange in the rhizosphere: molecular tools and the microbial perspective. Annu Rev Ecol Evol Syst. 37:459–488.
- Carini P, Steindler L, Beszteri S, Giovannoni SJ. 2012. Nutrient requirements for growth of the extreme oligotroph “Candidatus Pelagibacter ubique” HTCC1062 on a defined medium. IMSE J. 7:592–602.
- Carr R, Borenstein E. 2012. NetSeed: a network-based reverse-ecology tool for calculating the metabolic interface of an organism with its environment. Bioinformatics. 28:734–735.
- Casanueva A, Galada N, Baker GC, Grant WD, Heaphy S, Jones B, Yanhe M, Ventosa A, Blamey J, Cowan DA. 2008. Nanoarchaeal 16S rRNA gene sequences are widely dispersed in hyperthermophilic and mesophilic halophilic environments. Extremophiles. 12:651–656.
- Cleary B, Brito IL, Huang K, Gevers D, Shea T, Young S, Alm EJ. 2015. Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning. Nat Biotechnol. 33:1–10.
- Cockrell DC, Beare PA, Fischer ER, Howe D, Heinzen RA. 2008. A method for purifying obligate intracellular Coxiella burnetii that employs digitonin lysis of host cells. J Microbiol Methods. 72:321–325.
- Connon SA, Giovannoni SJ. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol. 68:3878–3885.
- Darling AE, Jospin G, Lowe E, Matsen FA, Bik HM, Eisen JA. 2014. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2:e243.
- Davis IJ, Bull C, Horsfall A, Morley I, Harris S. 2014. The Unculturables: targeted isolation of bacterial species associated with canine periodontal health or disease from dental plaque. BMC Microbiol. 14:196.
- Delafont V, Samba-Louaka A, Bouchon D, Moulin L, Héchard Y. 2015. Shedding light on microbial dark matter: A TM6 bacterium as natural endosymbiont of a free-living amoeba. Environ Microbiol Rep. 7:970–978.
- Dewi Puspita I, Kamagata Y, Tanaka M, Asano K, Nakatsu CH. 2012. Are uncultivated bacteria really uncultivable? Microb Environ. 27:356–366.
- Dini-Andreote F, Andreote FD, Araújo WL, Trevors JT, Elsas JD. 2012. Bacterial genomes: habitat specificity and uncharted organisms. Microb Ecol. 64:1–7.
- Dutilh BE, Noort V, Heijden RTJM, Boekhout T, Snel B, Huynen MA. 2007. Assessment of phylogenomic and orthology approaches for phylogenetic inference. Bioinformatics. 23:815–824.
- Dutta C, Paul S. 2012. Microbial lifestyle and genome signatures. Curr Genomics. 13:153–162.
- Ellrott K, Jaroszewski L, Li W, Wooley JC, Godzik A. 2010. Expansion of the protein repertoire in newly explored environments: Human gut microbiome specific protein families. PLoS Comput Biol. 6:1–11.
- Epstein SS. 2009. Microbial awakenings. Nature. 457:1083.
- Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, Raes J, et al. 2012. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 7:e49138.
- Federle MJ. 2009. Autoinducer-2-based chemical communication in bacteria: complexities of interspecies signaling. Contrib Microbiol. 16:18–32.
- Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. 2012. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci USA. 109:21390–21395.
- Fodor AA, DeSantis TZ, Wylie KM, Badger JH, Ye Y, Hepburn T, Hu P, Sodergren E, Liolios K, Huot-Creasy H, et al. 2012. The “most wanted" taxa from the human microbiome for whole genome sequencing”. PLoS One. 7:e41294.
- Franklin RB, Mills AL. 2003. Multi-scale variation in spatial heterogeneity for microbial community structure in an eastern Virginia agricultural field. FEMS Microbiol Ecol. 44:335–346.
- Frias-Lopez J, Shi Y, Tyson G, Shi Y, Coleman M, Tyson GW, Coleman ML, Schuster S, Chrisholm SW, Delong EF, et al. 2008. Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA. 105:3805–3810.
- Fuerst JA. 2014. Diversity and biotechnological potential of microorganisms associated with marine sponges. Appl Microbiol Biotechnol. 98:7331–7347.
- Gao W, Navarroli D, Naimark J, Zhang W, Chao SH, Meldrum DR. 2013. Microbe observation and cultivation array (MOCA) for cultivating and analyzing environmental microbiota. Microbiome. 1:4.
- Goh EB, Yim G, Tsui W, McClure J, Surette MG, Davies J. 2002. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc Natl Acad Sci USA. 99:17025–17030.
- Guieysse B, Wuertz S. 2012. Metabolically versatile large-genome prokaryotes. Curr Opin Biotechnol. 23:467–473.
- Hames-Kocabas EE, Uzel A. 2012. Isolation strategies of marine-derived actinomycetes from sponge and sediment samples. J Microbiol Methods. 88:342–347.
- Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. 1998. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 5:R245–R249.
- Harwani D. 2013. Recent advances in culturing the unculturable bacteria. Int J Recent Sci Res. 4:1488–1491.
- He X, McLean JS, Edlund A, Yooseph S, Hall AP, Liu SY, Dorrestein PC, Esquenazi E, Hunter RC, Cheng G, et al. 2015. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc Natl Acad Sci USA. 112:244–249.
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. 2010. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 28:977–982.
- Hesselman MC, Odoni DI, Ryback BM, de Groot S, van Heck RGA, Keijsers J, Kolkman P, Nieuwenhuijse D, van Nuland YM, Sebus E, et al. 2012. A multi-platform flow device for microbial (co-) cultivation and microscopic analysis. PLoS One. 7:e36982.
- Huber H, Hohn MJ, Rachel R, Fuchs T, Wimmer VC, Stetter KO. 2002. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature. 417:63–67.
- Huber R, Burggraf S, Mayer T, Barns SM, Rossnagel P, Stetter KO. 1995. Isolation of a hyperthermophilic archaeum predicted by in situ RNA analysis. Nature. 376:57–58.
- Hug LA, Beiko RG, Rowe AR, Richardson RE, Edwards EA. 2012. Comparative metagenomics of three Dehalococcoides-containing enrichment cultures: the role of the non-dechlorinating community. BMC Genomics. 13:327.
- Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, et al. 2016. A new view of the tree of life. Nat Microbiol. 2016:48.
- Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol. 67:411–419.
- Imelfort M, Parks D, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW. 2014. GroopM: An automated tool for the recovery of population genomes from related metagenomes. PeerJ. 2:e603.
- Ingham CJ, Sprenkels A, Bomer J, Molenaar D, van den Berg A, van Hylckama Vlieg JET, de Vos WM. 2007. The micro-petri dish, a million-well growth chip for the culture and high-throughput screening of microorganisms. Proc Natl Acad Sci USA. 104:18217–18222.
- Janse JD, Obradovic A. 2010. Xylella Fastidiosa: Its biology, diagnosis, control and risks. J Plant Pathol. 92:35–48.
- Joyce AR, Palsson BO. 2006. The model organism as a system: integrating “omics” data sets. Nat Rev Mol Cell Biol. 7:198–210.
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita K, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34:D354–D357.
- Keller M, Hettich R. 2009. Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol Mol Biol Rev. 73:62–70.
- Keren R, Lavy A, Mayzel B, Ilan M. 2015. Culturable associated-bacteria of the sponge Theonella swinhoei show tolerance to high arsenic concentrations. Front Microbiol. 6:1–11.
- Kimura N. 2014. Metagenomic approaches to understanding phylogenetic diversity in quorum sensing. Virulence. 5:433–442.
- Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 102:2567–2572.
- Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C, Bittar F, Fournous G, Gimenez G, Maraninchi M, et al. 2012. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 18:1185–1193.
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogint ML, Pace NR. 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. 82:6955–6959.
- Lavy A, Keren R, Haber M, Schwartz I, Ilan M. 2014. Implementing sponge physiological and genomic information to enhance the diversity of its culturable associated bacteria. FEMS Microbiol Ecol. 87:486–502.
- Leadbetter JR. 2003. Cultivation of recalcitrant microbes: cells are alive, well and revealing their secrets in the 21st century laboratory. Curr Opin Microbiol. 6:274–281.
- Lemos EGDM, Alves LMC, Campanharo JC. 2003. Genomics-based design of defined growth media for the plant pathogen Xylella fastidiosa. FEMS Microbiol Lett. 219:39–45.
- Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat Rev Microbiol. 5:48–56.
- Ma L, Kim J, Hatzenpichler R, Karymov MA, Hubert N, Hanan IM, Chang EB, Ismagilov RF. 2014. Gene-targeted microfluidic cultivation validated by isolation of a gut bacterium listed in human microbiome project's most wanted taxa. Proc Natl Acad Sci USA. 111:9768–9773.
- Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, Greenhalgh K, Jäger C, Baginska J, Wilmes P, et al. 2016. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 35.
- Marchesi JR, Ravel J. 2015. The vocabulary of microbiome research: a proposal. Microbiome. 3:31.
- McCutcheon JP, Moran NA. 2011. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 10:13–26.
- McLean JS, Lombardo MJ, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, Vyahhi N, Hall AP, Yang Y, Dupont CL, et al. 2013. Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phyum. Proc Natl Acad Sci USA. 110:E2390–E2399.
- Medema MH, Fischbach MA. 2015. Computational approaches to natural product discovery. Nat Chem Biol. 11:639–648.
- Morris JJ, Lenski RE, Zinser ER. 2012. The Black Queen Hypothesis: evolution of dependencies through adaptive gene loss. MBio. 3:1–7.
- Muller EEL, Glaab E, May P, Vlassis N, Wilmes P. 2013. Condensing the omics fog of microbial communities. Trends Microbiol. 21:325–333.
- Narihiro T, Kamagata Y. 2013. Cultivating yet-to-be cultivated microbes: the challenge continues. Microbes Environ. 28:163–165.
- Narihiro T, Suzuki A, Yoshimune K, Hori T, Hoshino T, Yumoto I, Yokota A, Kimura N, Kamagata Y. 2014. The combination of functional metagenomics and an oil-fed enrichment strategy revealed the phylogenetic diversity of lipolytic bacteria overlooked by the cultivation-based method. Microbes Environ. 29:154–161.
- Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu Rev Genet. 43:197–222.
- Nichols D, Lewis K, Orjala J, Mo S, Ortenberg R, O’Connor P, Zhao C, Vouros P, Kaeberlein T, Epstein SS. 2008. Short peptide induces an “uncultivable" microorganism to grow in vitro”. Appl Environ Microbiol. 74:4889–4897.
- Nichols D. 2007. Cultivation gives context to the microbial ecologist. FEMS Microbiol Ecol. 60:351–357.
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. 2014. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 32:822–828.
- Notebaart RA, van Enckevort FHJ, Francke C, Siezen RJ, Teusink B. 2006. Accelerating the reconstruction of genome-scale metabolic networks. BMC Bioinformatics. 7:296.
- Oberhardt MA, Palsson BØ, Papin JA. 2009. Applications of genome-scale metabolic reconstructions. Mol Syst Biol. 5:1–15.
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci USA. 106:4430–4434.
- Overmann J. 2010. Novel cultivation strategies for environmentally important microorganisms. In: Barton LL, Mandl M, Loy A, editors. Geomicrobiology: molecular and environmental perspective. the Netherlands, Dordrecht: Springer; p. 69–89.
- Park J, Kerner A, Burns MA, Lin XN. 2011. Microdroplet-enabled highly parallel co-cultivation of microbial communities. PLoS One. 6:e17019.
- Parks DH, Macdonald NJ, Beiko RG. 2011. Classifying short genomic fragments from novel lineages using composition and homology. BMC Bioinformatics. 12:328.
- Parte AC. 2014. LPSN – List of prokaryotic names with standing in nomenclature. Nucl Acids Res. 42:613–616.
- Pereira CS, Thompson JA, Xavier KB. 2013. AI-2-mediated signalling in bacteria. FEMS Microbiol Rev. 37:156–181.
- Pham VHT, Kim J. 2012. Cultivation of unculturable soil bacteria. Trends Biotechnol. 30:475–484.
- Plewniak F, Koechler S, Navet B, Dugat-Bony É, Bouchez O, Peyret P, Séby F, Battaglia-Brunet F, Bertin PN. 2013. Metagenomic insights into microbial metabolism affecting arsenic dispersion in Mediterranean marine sediments. Mol Ecol. 22:4870–4883.
- Podar M, Makarova KS, Graham DE, Wolf YI, Koonin EV, Reysenbach AL. 2013. Insights into archaeal evolution and symbiosis from the genomes of a nanoarchaeon and its inferred crenarchaeal host from Obsidian Pool, Yellowstone National Park. Biol Direct. 8:9.
- Pope PB, Smith W, Denman SE, Tringe SG, Barry K, Hugenholtz P, McSweeney CS, McHardy AC, Morrison M. 2011. Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar Wallabies. Science. 333:646–648.
- Radax R, Rattei T, Lanzen A, Bayer C, Rapp HT, Urich T, Schleper C. 2012. Metatranscriptomics of the marine sponge Geodia barretti: Tackling phylogeny and function of its microbial community. Environ Microbiol. 14:1308–1324.
- Rajilic-Stojanovic M, de Vos WM. 2014. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 38:996–1047.
- Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature. 418:630–633.
- Reed JL. 2012. Shrinking the metabolic solution space using experimental datasets. PLoS Comput Biol. 8:e1002662.
- Renesto P, Crapoulet N, Ogata H, Scola BL, Vestris G, Claverie JM, Raoult D. 2003. Genome-based design of a cell-free culture medium for Tropheryma whipplei. Lancet. 362:447–449.
- Rettedal EA, Gumpert H, Sommer MOA. 2014. Cultivation-based multiplex phenotyping of human gut microbiota allows targeted recovery of previously uncultured bacteria. Nat Comms. 5:4714.
- Santos F, Boele J, Teusink B. 2011. A practical guide to genome-scale metabolic models and their analysis. In: Jameson D, Verma M, Westerhoff HV, editors. Methods in enzymology, 1st ed. Burlington: Academic Press/Elsevier Inc; p. 509–532.
- Schnoes AM, Brown SD, Dodevski I, Babbitt PC. 2009. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 5:e1000605.
- Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. 2013. Computational meta'omics for microbial community studies. Mol Syst Biol. 9:666.
- Sessitsch A, Hardoim P, Döring J, Weilharter A, Krause A, Woyke T, Mitter B, Hauberg-Lotte L, Friedrich F, Rahalkar M, et al. 2012. Functional characteristics of an endophyte community colonizing rice roots as revealed by metagenomic analysis. Mol Plant Microbe Interact. 25:28–36.
- Setubal JC, Simpson AJG, Reinach FC, Arruda P, Abreu FA, Acencio M, Alvarenga R, Alves LMC, Araya JE, et al. 2000. The genome sequence of the plant pathogen Xylella fastidiosa. Nature. 406:151–157.
- Shi X, Gao W, Wang J, Chao SH, Zhang W, Meldrum DR. 2014. Measuring gene expression in single bacterial cells: recent advances in methods and micro-devices. Crit Rev Biotechnol. 8551:1–13.
- Siegl A, Kamke J, Hochmuth T, Piel J, Richter M, Liang C, Dandekar T, Hentschel U. 2011. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5:61–70.
- Sipkema D, Schippers K, Maalcke WJ, Yang Y, Salim S, Blanch HW. 2011. Multiple approaches to enhance the cultivability of bacteria associated with the marine sponge Haliclona (gellius) sp. Appl Environ Microbiol. 77:2130–2140.
- Sizova MV, Hohmann T, Hazen A, Paster BJ, Halem SR, Murphy CM, Panikov NS, Epstein SS. 2012. New approaches for isolation of previously uncultivated oral bacteria. Appl Environ Microbiol. 78:194–203.
- Speth DR, Zandt MH, Guerrero-Cruz S, Dutilh BE, Jetten MSM. 2016. Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nat Comms. 7:11172.
- Staley JT. 1985. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 39:321–346.
- Stewart EJ. 2012. Growing unculturable bacteria. J Bacteriol. 194:4151–4160.
- Tian Y, He X, Torralba M, Yooseph S, Nelson KE, Lux R, McLean JS, Yu G, Shi W. 2010. Using DGGE profiling to develop a novel culture medium suitable for oral microbial communities. Mol Oral Microbiol. 25:357–367.
- Torsvik V. 2002. Prokaryotic diversity-magnitude, dynamics, and controlling factors. Science. 296:1064–1066.
- Traxler MF, Kolter R. 2015. Natural products in soil microbe interactions and evolution. Nat Prod Rep. 32:956–970.
- Tripp HJ, Kitner JB, Schwalbach MS, Dacey JWH, Wilhelm LJ, Giovannoni SJ. 2008. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature. 452:741–744.
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. 2004. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 428:37–43.
- Tyson GW, Lo I, Baker BJ, Allen EE, Hugenholtz P, Banfield JF. 2005. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl Environ Microbiol. 71:6319–6324.
- Urich T, Lanzén A, Stokke R, Pedersen RB, Bayer C, Thorseth IH, Schleper C, Steen IH, Øvreas L. 2014. Microbial community structure and functioning in marine sediments associated with diffuse hydrothermal venting assessed by integrated meta-omics. Environ Microbiol. 16:2699–2710.
- Vartoukian SR, Palmer RM, Wade WG. 2010. Strategies for culture of “unculturable” bacteria. FEMS Microbiol Lett. 309:1–7.
- Versluis D, D’Andrea MM, Garcia JR, Leimena MM, Hugenholtz F, Zhang J, Ozturk B, Nylund L, Sipkema D, van Schaik W, et al. 2015. Mining microbial metatranscriptomes for expression of antibiotic resistance genes under natural conditions. Sci Rep. 5:11981.
- Vieira-Silva S, Rocha EPC. 2010. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 6:e1000808.
- Voolaid V, Jõers A, Kisand V, Tenson T. 2012. Co-occurrence of resistance to different antibiotics among aquatic bacteria. BMC Microbiol. 12:225.
- Walker AW, Duncan SH, Louis P, Flint HJ. 2014. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol. 22:267–274.
- Wells JM, Raju BC, Hung HY, Weisburg WG, Mandelco-Paul L, Brenner DJ. 1987. Xylella fastidiosa gen. nov., sp. nov: Gram-negative, xylem-limited, fastidious plant bacteria related to Xanthomonas spp. Int J Syst Bacteriol. 37:136–143.
- Wesolowska-Andersen A, Bahl MI, Carvalho V, Kristiansen K, Sicheritz-Pontén T, Gupta R, Licht TR. 2014. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome. 2:19.
- West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. 2007. The social lives of microbes. Annu Rev Ecol Evol Syst. 38:53–77.
- White RA, Bottos EM, Roy Chowdhury T, Zucker JD, Brislawn CJ, Nicora CD, Fansler SJ, Glaesemann KR, Glass K, Jansson JK. 2016. Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems. 1:e00045-16.
- Wilmes P, Heintz-Buschart A, Bond PL. 2015. A decade of metaproteomics: where we stand and what the future holds. Proteomics. 15:3409–3417.
- Wilson MC, Piel J. 2013. Metagenomic approaches for exploiting uncultivated bacteria as a resource for novel biosynthetic enzymology. Chem Biol. 20:636–647.
- Woese CR, Fox GE. 1977. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 74:5088–5090.
- Wurch L, Giannone RJ, Belisle BS, Swift C, Utturkar S, Hettich RL, Reysenbach AL, Podar M. 2016. Genomics-informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nat Commun. 7:12115.
- Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R. 2014. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 12:635–645.
- Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch-Mizrachi I, Johnston A, Cochrane G, et al. 2011. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 29:415–420.
- Yim G, Huimi Wang H, Davies J. 2006. The truth about antibiotics. Int J Med Microbiol. 296:163–170.
- Yim G, Huimi Wang H, Davies J. 2007. Antibiotics as signalling molecules. Philos Trans R Soc Lond B Biol Sci. 362:1195–1200.
- Youssef NH, Couger MB, McCully AL, Criado AEG, Elshahed MS. 2014. Assessing the global phylum level diversity within the bacterial domain: a review. J Adv Res. 1:269–282.
- Zarecki R, Oberhardt MA, Reshef L, Gophna U, Ruppin E. 2014. A novel nutritional predictor links microbial fastidiousness with lowered ubiquity, growth rate, and cooperativeness. PLoS Comput Biol. 10:e1003726.
- Zengler K. 2009. Central role of the cell in microbial ecology. Microbiol Mol Biol Rev. 73:712–729.
- Zhang W, Li F, Nie L. 2010. Integrating multiple 'omics' analysis for microbial biology: application and methodologies. Microbiology (Reading, Engl). 156:287–301.