
Abstract
Pathogens have developed sophisticated strategies to evade the immune response, among which manipulation of host cellular epigenetic mechanisms plays a prominent role. In the last decade, modulation of histone acetylation in host cells has emerged as an efficient strategy of bacterial immune evasion. Virulence factors and metabolic products of pathogenic microorganisms alter expression and activity of histone acetyltransferases (HATs) and histone deacetylases (HDACs) to suppress transcription of host defense genes through epigenetic changes in histone acetylation marks. This new avenue of pathogen–host interactions is particularly important in light of introduction of HDAC inhibitors into clinical practice. Considerable effort is currently being applied to better understand the effects of HDAC inhibitors on the quality of immune responses to pathogens and to characterize the therapeutic potential of these compounds in microbial infections. In this review, we will discuss the recently discovered mechanisms utilized by bacteria to facilitate their survival within infected hosts through subversion of the host acetylation system and the effects of acetylation modulators, including HDAC inhibitors and bromodomain-containing BET protein inhibitors, on innate immune responses against microbial pathogens. Integration of these two lines of experimental evidence provides critical information on the perspectives of epigenetic therapies targeting protein acetylation in infectious diseases.
Introduction
Infectious diseases cause more than 10 million deaths per year worldwide and remain one of the main causes of morbidity and mortality, particularly in the developing world (GBD 2015 Disease and Injury Incidence and Prevalence Collaborators Citation2016). There is an urgent need to develop new treatment strategies for bacterial infections given the unchecked global rise in the incidence of antibiotic-resistant infections, coupled with an antibiotic pipeline that has produced few novel antibiotics in the last 30 years. One approach that could potentially circumvent the problem of antibiotic resistance involves host-directed therapy, which aims to promote the pathogen elimination by boosting immune responses, in particular the host defense pathways that are subverted by pathogens for their own survival advantage. While it is well established how pathogens can hijack cellular responses through disruption of intracellular signaling pathways and transcription factor activation, the observation that bacteria may exert subversive effects on host inflammatory responses by dysregulating specific epigenetic mechanisms has opened a new area in the biology of host–microbe interactions.
Acetylation in epigenetic regulation: HATs, HDACs, BET proteins, and HDAC inhibitors
Epigenetic mechanisms are most commonly defined as stable changes in gene expression profile or cellular phenotype that are caused by modifications of chromosomes without alterations in the DNA sequence. Although the original definition of epigenetics covered only heritable changes in chromosome structure (Berger et al. Citation2009), it is now commonly extended to reversible modifications of chromatin that are dynamically induced by environmental factors. The cellular mechanisms responsible for epigenetic regulation include methylation of chromosomal DNA and a network of post-translational modifications of histone proteins. DNA methylation typically occurs at CpG dinucleotides and causes transcriptional repression either by recruitment of co-repressors or through disruption of transcription factor binding to DNA (Jones Citation2012). In contrast, post-translational histone modifications, among which acetylation, methylation, and phosphorylation are the best characterized, either suppress or enhance transcription by modulating the availability of gene promoters for transcription factors and RNA polymerase II (Gardner et al. Citation2011).
Histone proteins are acetylated on their N-terminal lysine residues by histone acetyltransferases (HATs). This process leads to opening of chromatin structure, increased accessibility of gene promoters, and recruitment of transcriptional regulators recognizing acetylated lysines, such as bromodomain-containing BET (bromodomain and extra-terminal) proteins, and is generally associated with enhanced transcription. Histone deacetylases (HDACs) counteract the activity of HATs and remove acetyl groups from histones, leading to termination of transcriptional processes (Verdin and Ott Citation2015). Four classes of HDACs are expressed in mammals: class I (HDACs 1–3 and 8), class II (HDACs 4–7, 9 and 10), and class IV (HDAC11) are zinc-dependent enzymes, whereas the activity class III HDACs (sirtuins Sirt1–7) is NAD-dependent (de Ruijter et al. Citation2003). Although required for transcriptional activation, histone acetylation alone is not sufficient for gene induction and thus global or promoter-specific increases in the acetylation status of histone tails are not always directly translated into enhanced transcription (Wang et al. Citation2009). This is exemplified by transcriptomic studies of HDAC inhibitors in cancer cell lines, in which comparable numbers of genes were up- and downregulated, despite global histone hyperacetylation (Glaser et al. Citation2003; Peart et al. Citation2005). Recent advances in mass spectrometric technologies have also revealed that protein acetylation is not restricted to histones; in fact, the prevalence and importance of acetylation is comparable to that of phosphorylation (Kim and Yang Citation2011). Approximately 1700 proteins, including transcription factors, signal transduction regulators, and structural proteins undergo reversible acetylation, which modulates their activity, subcellular localization, and stability (Choudhary et al. Citation2009). Posttranslational modification of proteins by reversible acetylation has therefore emerged as one of the critical processes in maintaining cellular homeostasis and shaping responses to environmental stimuli.
Given a central role of protein acetylation in regulating gene expression at multiple levels, it is not surprising that several proteins responsible for attachment (“writers”), recognition (“readers”), and removal (“erasers”) of acetyl groups are important modulators of immune responses. Alterations in HAT/HDAC activity and expression, as well as aberrant histone acetylation marks have been identified in a number of chronic immune-mediated inflammatory diseases, including asthma, chronic obstructive pulmonary disease, colitis, systemic lupus erythematosus, and rheumatoid arthritis (Grabiec et al. Citation2008; Zhang and Zhang Citation2015). These observations have generated a great interest in the therapeutic potential of targeting acetylation regulators and, indeed, inhibitors of HDACs have uniformly demonstrated potent anti-inflammatory properties in vitro in cell types relevant to pathology of inflammatory disorders and in animal models of these diseases (Grabiec et al. Citation2011; Shakespear et al. Citation2011). The involvement of sirtuins in immunity, in particular Sirt1, is more complex and both inhibitors and activators of sirtuin activity have shown immune modulatory effects depending on the model and cell type tested (Chen et al. Citation2015). More recently, anti-inflammatory effects of small-molecule acetylated histone mimetics, which target BET proteins, have also been reported (Prinjha et al. Citation2012; Klein et al. Citation2016).
Epigenetics of bacterial infections
Characterization of the essential contributions of DNA and histone modifications in shaping the immune response, and observations that epigenetic marks are dynamically regulated by environmental cues have stimulated interest in the potential roles of epigenetic mechanisms in microbial infections. Early studies of gastric cancer related to Helicobacter pylori infection identified alterations in DNA methylation at the promoter regions of tumor suppressor genes (Maekita et al. Citation2006). Although it remains controversial whether aberrant DNA methylation is directly caused by bacteria or is secondary to ongoing inflammation, these observations provided the first proof of principle that epigenetic changes can be directly linked to bacterial infections. Subsequent reports found that Escherichia coli, Campylobacter rectus and Mycobacterium leprae also regulate DNA methylation patterns and/or DNA methyltrasferase expression in host cells (Bobetsis et al. Citation2007; Tolg et al. Citation2011; Masaki et al. Citation2013). Intriguingly, M. leprae is capable of reprograming differentiated Schwann cells into stem cell-like cells. These changes are associated with alterations in DNA methylation and promote bacillary dissemination (Masaki et al. Citation2013). These observations raise the possibility that bacteria can dramatically alter host cell functions through epigenetic reprograming of cellular transcriptional programs (Pereira et al. Citation2016).
Several enzymatically active bacterial effector molecules have been shown to directly modulate epigenetic histone marks. The recently discovered examples include the SET (Suppressor of variegation, Enhancer of zeste and Trithorax) domain proteins BaSET and BtSET produced and delivered into host cells by Bacillus anthracis and Burkholderia thailandensis, respectively, which display histone methyltransferase activity (Li, Lu, et al. Citation2013; Mujtaba et al. Citation2013). Upon delivery to the nucleus, BaSET methylates histone H1, leading to inhibition of NF-κB, whereas BtSET localizes to the nucleolus and promotes transcription of rRNA genes through methylation of histone H3K4. Based on these findings, histone methylation by bacterial effectors has been proposed as a novel virulence strategy and, indeed, SET domain proteins have been identified in other bacterial pathogens, including Legionella spp. and Chlamydia spp. (Rolando et al. Citation2015).
Bacteria can also modulate epigenetic marks in host cells indirectly through their effects on mitogen-activated protein kinase (MAPK) signaling. MAPKs activate downstream kinases that phosphorylate histone H3S10, which is associated with H3 acetylation and linked to activation of transcription (Sawicka and Seiser Citation2012). B. anthracis and Mycobacterium tuberculosis have been shown to modulate MAPK-dependent changes in histone phosphorylation and acetylation, which cause significant changes in inflammatory activation of epithelial cells and macrophages (Pennini et al. Citation2006; Raymond et al. Citation2009). Pathogens can therefore induce epigenetic changes in host cells through manipulation of signaling pathways activated during infection, highlighting an intricate cross-talk between epigenetics and signaling events that has already been identified in other pathologies (Grabiec and Reedquist Citation2013; Singh and Ellenrieder Citation2013).
For more detailed discussion of advances in these areas of patho-epigenetics of microbial infection, we refer the readers to excellent recent reviews (Bierne et al. Citation2012; Rolando et al. Citation2015; Niller and Minarovits Citation2016). Here, we will focus on the interactions between bacteria and the protein acetylation system of the host. We will discuss the recently discovered strategies used by pathogenic microorganisms to evade the immune response through manipulation of histone acetylation marks, and the effects of compounds that modulate protein acetylation, in particular HDACi, on the outcome of bacterial infections.
Bacterial regulation of the host acetylation system
In recent years, great progress has been made in our understanding of how bacteria hijack the acetylation system to manipulate transcriptional regulation in host cells in order to evade elimination by the immune system (Hamon and Cossart Citation2008; Bierne et al. Citation2012). A number of unique mechanisms utilized by pathogenic microorganisms targeting changes in histone and non-histone protein acetylation have been identified. They include direct and indirect effects on epigenetic histone marks, modulation of HAT and HDAC expression, and production of metabolites that regulate the activity of acetylation system components ().
Table 1. Changes in epigenetic histone acetylation marks and/or expression of acetylation regulators in host cells induced by pathogenic bacteria.
Modulation of histone acetylation
Several pathogens, including M. tuberculosis, H. pylori and Listeria monocytogenes have been shown to manipulate host cell antibacterial responses and evade the immune system by affecting histone acetylation status. Initial evidence that bacteria regulate host gene expression by altering acetylation of histones was provided by Wang et al., who demonstrated that M. tuberculosis inhibits interferon-γ-dependent HLA-DR gene expression by inducing histone hypoacetylation at the HLA-DRα promoter (Wang et al. Citation2005). While M. tuberculosis infection had no effect on HDAC1 and HDAC2 expression, it led to upregulation of the corepressor protein, mammalian Sin3A, which in turn promoted the formation of an HDAC-containing complex and histone deacetylation at the promoter region of the HLA-DRα gene (Wang et al. Citation2005). Decreased histone acetylation levels have also been reported upon infection with H. pylori (Ding et al. Citation2010). Chronic infection with H. pylori is the major cause of human gastric diseases, including peptic ulcers and malignancies, and alterations in chromatin structure have been shown to contribute to the pathogenic effects of H. pylori on cellular proliferation and survival (Xia et al. Citation2008; Fehri et al. Citation2009). H. pylori-induced global deacetylation of histone H3K23 in gastric epithelial cells that was accompanied by decreased histone H3S10 phosphorylation and correlated with differential regulation of c-Jun and HSP70 expression (Ding et al. Citation2010). The functional consequences of these changes in epigenetic histone marks, however, have not been characterized.
Thus far, the most comprehensive mechanistic evidence of bacterial regulation of histone acetylation in host cells has been provided for the intracellular pathogens L. monocytogenes and Legionella pneumophila. L. monocytogenes, which causes significant morbidity in pregnant women and newborns, has developed sophisticated strategies to facilitate its entry and survival within infected cells, among which the manipulation of the host transcriptional machinery plays an important role (Niller and Minarovits Citation2016). Infection of HeLa cells with L. monocytogenes caused rapid global deacetylation of histone H4 and specific deacetylation of histone H3K18 (Hamon et al. Citation2007; Eskandarian et al. Citation2013). Histone H4 deacetylation was associated with reduced H3S10 phosphorylation, and was dependent on the virulence factor listeriolysin-O (LLO) (Hamon et al. Citation2007). LLO-induced changes in histone modifications strongly correlated with reduced expression of a small cluster of host genes, including important regulators of the immune response (e.g. the chemokine CXCL2 and the phosphatase DUSP4). Notably, other bacterial toxins that belong to the same family, perfringolysin from Clostridium perfringens and pneumolysin from Streptococcus pneumoniae, had a similar effect on H3S10 phosphorylation (Hamon et al. Citation2007). Although their effects on histone H4 acetylation have not been tested in this study, it is likely they would be similar to those generated by LLO given the similarity between the mechanisms of action of these bacterial effector molecules.
In contrast, Eskandarian et al. demonstrated that histone H3K18 deacetylation is mediated by the L. monocytogenes protein InlB, which activates the receptor c-Met on host cells (Eskandarian et al. Citation2013). The signaling cascade triggered by InlB-activated c-Met converged on nuclear translocation and recruitment of the class III deacetylase Sirt2 to gene promoter regions. Histone H3K18 deacetylation by Sirt2 downregulated the expression of a subset of genes involved in transcriptional regulation and immune responses, thereby promoting L. monocytogenes invasion and survival. The role of Sirt2-dependent transcriptional reprograming during L. monocytogenes infection has been confirmed in vivo: bacterial titers in infected mice lacking Sirt2 expression were significantly reduced compared to WT mice (Eskandarian et al. Citation2013). Specific histone acetylation marks are also reduced in host cells by L. pneumophila, the etiological factor of severe pneumonia called Legionnaires’ disease. The L. pneumophila effector molecule RomA has been shown to cause trimethylation of histone H3K14, which prevents acetylation of this residue and represses innate immune genes, such as toll-like receptor 5 (TLR5) and interleukin-6 (IL6) (Rolando et al. Citation2013). Collectively, these studies have provided clear biochemical evidence that pathogenic bacteria utilize a broad range of strategies targeting epigenetic changes in histone acetylation to promote their survival within the infected host ().
Figure 1. Microbial pathogens developed multiple strategies to evade the immune response through manipulation of the host acetylation system. 2-Aminoacetophenone, a quorum sensing molecule released by Pseudomonas aeruginosa induces HDAC1 expression in host cells (Bandyopadhaya et al. Citation2016). Similarly, Anaplasma phagocytophilum, through its effector molecule ankyrin A upregulates HDAC1 and promotes HDAC1 recruitment to gene promoters (Garcia-Garcia et al. Citation2009; Rennoll-Bankert et al. Citation2015). The metalloproteinase NleC produced by enteropathogenic and enterohaemorrhagic Escherichia coli degrades the host HAT p300 (Shames et al. Citation2011), whereas listeriolysin-O secreted by Listeria monocytogenes promotes histone deacetylation in infected cells through an unknown mechanism (Hamon et al. Citation2007). RomA, the effector molecule of Legionella pneumophila, blocks acetylation by inducing histone H3 trimethylation (Rolando et al. Citation2013). All these processes lead to global or promoter-specific histone hypoacetylation, condensation of chromatin structure, and suppressed transcription of genes responsible for host defense against microbial infection. 2-AA: 2-aminoacetophenone; Ac: acetylated; 3-Me: tri-methylated; LLO: listeriolysin-O; TF: transcription factor.
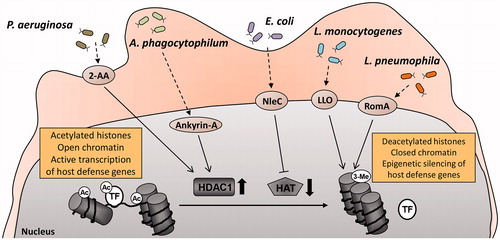
Manipulation of HDAC expression
An alternative strategy used by many pathogenic bacteria to subvert host cell responses utilizes indirect effects on histone acetylation marks by modulation of HAT and HDAC expression. In particular, HDAC1 appears to be the key acetylation system component targeted by pathogens to evade eradication by the immune system.
Anaplasma phagocytophilum is an intracellular pathogen that survives and propagates within neutrophils and causes human granulocytic anaplasmosis. Because infection with A. phagocytophilum dramatically alters major neutrophil functions, involvement of a global epigenetic mechanism affecting a broad range of host defense genes was hypothesized. Indeed, A. phagocytophilum upregulated HDAC1 in host cells, causing a global increase of HDAC activity (Garcia-Garcia et al. Citation2009). The A. phagocytophilum effector molecule ankyrin A promoted recruitment of HDAC1 to the promoters of host defense genes which led to a specific reduction of histone H3 acetylation at these regions and epigenetic suppression of target gene expression (Garcia-Garcia et al. Citation2009; Rennoll-Bankert et al. Citation2015). Silencing of HDAC1 expression or inhibition of enzyme activity restored defense gene expression in infected cells and prevented intracellular propagation of bacteria (Garcia-Garcia et al. Citation2009). Importantly, these effects were specific for HDAC1, providing evidence that specific manipulation of a single HDAC family member by pathogenic microorganisms is sufficient for epigenetic reprograming of a plethora of host genes required for protection against infection.
A similar strategy is used by Pseudomonas aeruginosa, an opportunistic pathogen that typically infects and colonizes inflamed airways (e.g. in cystic fibrosis) and burn wounds. The quorum sensing signal released by P. aeruginosa, 2-aminoacetophenone, induced expression of HDAC1 in human THP-1 monocytes, which led to global histone H3K18 hypoacetylation (Bandyopadhaya et al. Citation2016). After subsequent challenge with bacterial molecules, changes in epigenetic acetylation marks resulted in dampened induction of inflammatory cytokines and chemokines, including TNF, IL-1β and MCP-1, thus significantly impairing host cell responses to infection (Bandyopadhaya et al. Citation2016). Similar to A. phagocytophilum infection, these processes were fully reversible by the HDAC1 knockdown or inhibition of class I HDACs, confirming the central role of HDAC1 induction in promoting tolerance to P. aeruginosa through regulation of the host epigenome.
There is preliminary evidence that the periodontal pathogen Porphyromonas gingivalis also modulates expression of HDAC1. Global profiling of HDAC expression revealed that mRNA and protein levels of HDAC1 are elevated in gingival tissues from patients with chronic periodontitis compared to healthy controls and increased expression of HDAC1 colocalizes with TNF-expressing cells (Cantley et al. Citation2016). However, this is likely to be cell type-specific as infection of gingival epithelial cells with oral pathogens in vitro downregulated HDAC1 and HDAC2 (Yin and Chung Citation2011), and increased levels of acetylated histone H3 were detected in epithelial cells in the gingival tissue in murine periodontitis (Martins et al. Citation2016). Additional studies are therefore needed to understand the complex manner in which P. gingivalis affects HDAC expression in different types of gingival cells, and whether the differences reported in patients with periodontitis are caused by oral pathogenic bacteria or are secondary to the ongoing chronic inflammation. In this context, it is important to note that HDAC1 is also upregulated by cytokines in the absence of infection, and correlates with expression of inflammatory mediators in synovial tissue from rheumatoid arthritis patients (Kawabata et al. Citation2010; Angiolilli et al. Citation2016).
The sirtuin family member Sirt1 is also targeted by bacteria to subvert the host acetylation system. The intracellular pathogen Salmonella enterica serovar Typhimurium has been shown to target Sirt1 complexed with regulators of the mTOR pathway to lysosomal degradation, which resulted in impairment of autophagy (Ganesan et al. Citation2017). Sirt1 was also downregulated in M. tuberculosis-infected macrophages, promoting intracellular growth of bacteria and persistent inflammatory response. Consistently, Sirt1 activators reduced lung pathology in mice infected with M. tuberculosis, whereas deletion of myeloid Sirt1 increased susceptibility to infection (Cheng et al. Citation2017). This effect was specific for Sirt1 as myeloid Sirt2 deletion had no effect on chronic M. tuberculosis infection (Cardoso et al. Citation2015). In contrast, mice lacking Sirt1 in myeloid cells had similar mortality upon infection with Streptococcus pneumoniae as wild-type mice (Crotty Alexander et al. Citation2013), indicating a unique role Sirt1 in host responses to certain intracellular pathogens. Notably, downregulation of Sirt1 by pathogenic bacteria modulated host cell responses through changes in the acetylation status of transcription factors and signaling molecules (Cheng et al. Citation2017; Ganesan et al. Citation2017). These observations confirmed that subversion of the host acetylation system by bacteria dysregulates anti-microbial host responses also through non-epigenetic mechanisms.
Pathogenic bacteria also manipulate epigenetic regulatory mechanisms of the host through proteolytic degradation of a HAT family member. Enteropathogenic and enterohaemorrhagic E. coli effector protein NleC is a zinc-dependent metalloproteinase that is delivered to host cells and targets intracellular signaling to suppress the inflammatory response. NleC has been shown to specifically bind and degrade the host HAT p300, which dampens IL-8 production by infected cells (Shames et al. Citation2011). Overexpression of p300 antagonized NleC-dependent IL-8 suppression by enteropathogenic E. coli (Shames et al. Citation2011), confirming that microbial pathogens can target both arms of the host histone acetylation system – HATs and HDACs – to manipulate the inflammatory response through epigenetic reprograming ().
HDAC inhibition by short chain fatty acids
Pathogenic as well as commensal bacteria can also modulate the host cell acetylation system through their metabolic products. Short chain fatty acids (SCFAs), which are the main fermentation products of anaerobic bacteria, are potent inhibitors of class I/II HDACs and regulate multiple aspects of the immune response (Corrêa-Oliveira et al. Citation2016). Recent studies have revealed central roles of SCFAs, such as butyric acid or propionic acid, in mediating communication between commensal bacteria and the host immune system. Commensal microbes promote generation of regulatory T cells (Tregs) in mice by releasing SCFAs which prevent exaggerated inflammatory responses in the gut and thus maintain intestinal immune homeostasis (Arpaia et al. Citation2013; Furusawa et al. Citation2013). Butyrate treatment enhanced histone H3 acetylation at the promoter region of the transcription factor FoxP3 (Furusawa et al. Citation2013), which is critical for Treg differentiation, suggesting involvement of an epigenetic mechanism. The suppressive role of multiple HDAC family members in FoxP3 induction has been well-documented (Beier et al. Citation2012) and, in line with this model, only SCFAs capable of inhibiting HDAC activity potentiated the generation of Tregs (Arpaia et al. Citation2013). Modulation of the host acetylation system by bacterial metabolites can therefore be seen as a novel mechanism of host-microbe interactions, which balances pro- and anti-inflammatory signals in the mucosal tissue of the gut. Although a functional link between metabolite production by commensal microbiota and epigenetic regulation in host cells has only been provided for SCFAs, several other microbial metabolites can potentially modulate host epigenetic mechanisms. These include vitamins, isothiocyanates, polyphenols, and choline (Krautkramer et al. Citation2017). Commensal bacteria have also been shown to induce epigenetic reprograming in mucosal immune cells through changes in DNA and histone methylation, though the underlying mechanisms remain to be characterized (Woo and Alenghat Citation2017).
Whereas SCFAs are clearly beneficial for maintaining the homeostatic balance between the commensal microbiota of the intestine and the host, high concentrations of these bacterial metabolites may contribute to pathology during bacterial infections through their effects on neutrophils and structural cells of the infected tissue. For example, SCFAs impaired the effector functions of neutrophils infected with Aggregatibacter actinomycetemcomitans (Corrêa et al. Citation2017), an opportunistic facultative anaerobic pathogen involved in aggressive periodontitis. SCFA treatment suppressed neutrophil cytokine production, bacterial phagocytosis and killing of A. actinomycetemcomitans both in vitro and in vivo. These effects were associated with histone H3K9 hyperacetylation and most likely can be attributed to HDAC inhibition by SCFAs. Consistently, the HDAC inhibitors SAHA and MS-275 disabled neutrophil functions to a similar degree (Corrêa et al. Citation2017). SCFAs produced by oral pathogens could also affect gingival tissue cells to perpetuate inflammation and alveolar bone resorption in periodontal disease. Large quantities of butyric acid and propionic acid are present in gingival crevices of patients with severe periodontitis, and SCFA concentrations correlate with clinical parameters of disease activity and inflammation (Niederman et al. Citation1996, Citation1997). In vitro, butyric acid has been shown to inhibit proliferation and/or induce apoptosis of gingival epithelial cells and fibroblasts (Tsuda et al. Citation2010; Chang et al. Citation2013). Although the contribution of HDAC inhibitory activity of SCFAs to the reported anti-proliferative effects has not been evaluated in these studies, the ability of HDACi to upregulate cell cycle inhibitors and induce cell cycle arrest under inflammatory conditions is well documented (Nishida et al. Citation2004; Jüngel et al. Citation2006).
SCFAs produced by oral pathogens may also contribute to other pathologies associated with periodontal disease through their ability to reactivate latent viruses and induce lytic replication, thus contributing to systemic dissemination of viruses. SCFAs from P. gingivalis enhanced replication of Epstein-Barr virus (EBV), Kaposi’s Sarcoma-associated herpesvirus (KPHV), and human immunodeficiency virus (HIV), and these effects were dependent on epigenetic changes in histone acetylation induced by SFCAs (Imai et al. Citation2009, Citation2012; Yu et al. Citation2014). In that regard, it is noteworthy that the ability of HDAC inhibitors to reactivate latent viruses can also be used to the patients’ benefit: HDAC inhibitors are currently tested as a pharmacological strategy to purge HIV from its latent reservoir in memory CD4 T cells (Matalon et al. Citation2011). The presence of this replication-competent provirus reservoir is considered the main obstacle to HIV eradication and the HDAC inhibitor SAHA has been demonstrated to disrupt HIV latency in vivo in patients on antiretroviral therapy (Archin et al. Citation2012).
HDAC inhibitors and bacterial infections
HDAC inhibitor effects on immune cell responses
HDAC inhibitors have been thoroughly tested in multiple models of chronic inflammatory diseases, showing protective effects in both prophylactic and therapeutic protocols (Dinarello et al. Citation2011; Grabiec et al. Citation2011). Anti-inflammatory effects of HDAC inhibitors have also been reported in models of lethal septic shock induced by systemic administration of LPS or Pam3CSK4 (Ciarlo et al. Citation2013). Comparatively little is known about the effects of these compounds in microbial infections. In light of the well-established ability of HDAC inhibitors to suppress cytokine production in response to inflammatory agonists, including TLR ligands, questions regarding the influence of these compounds on the quality of immune responses to pathogens have arisen. In a pioneering study addressing this problem, Roger et al. demonstrated that the HDAC inhibitor valproate increases mortality of mice after non-severe infection with Klebsiella pneumoniae or Candida albicans (Roger et al. Citation2011). In vitro, HDAC inhibitors impaired a broad range of innate immune responses of mouse bone marrow-derived macrophages (BMDMs) and dendritic cells induced by TLR ligands and other bacterial products (Roger et al. Citation2011). Phagocytosis and killing of E. coli and Staphylococcus aureus by BMDMs was also reduced by HDAC inhibition and associated with decreased production of reactive oxygen and nitrogen species (Mombelli et al. Citation2011). These observations suggested that therapeutic application of HDAC inhibitors might significantly compromise the immune system and render patients more susceptible to bacterial infections.
However, subsequent studies revealed that the effects of HDAC inhibitors on antibacterial responses in vitro and in vivo strongly depend on compound selectivity and the timing of treatment. Whereas priming with SAHA or trichostatin A (TSA) impaired phagocytosis of E. coli by human macrophages, HDAC inhibitor co-treatment had no effect on this process. Instead, simultaneous treatment with HDAC inhibitors promoted clearance of intracellular bacteria by enhancing generation of mitochondrial reactive oxygen species. Interestingly, only the HDAC6-specific inhibitor tubastatin A, but not MS-275, which targets only class I HDACs, improved bacterial killing by macrophages (Ariffin et al. Citation2015), indicating a unique and non-redundant role for HDAC6 in this process. The importance of HDAC6 in anti-bacterial responses by innate immune cells is supported by observations in the cecal ligation and puncture (CLP)-induced lethal sepsis model. Consistent with in vitro observations, tubastatin A improved the survival of animals after CLP, which was associated with improved bacterial clearance from the blood, attenuated acute liver injury and reduced systemic cytokine levels, whereas class I HDAC-selective MS-275 had no effect (Li et al. Citation2015). HDAC6 inhibition also restored innate immune cell populations in the bone marrow, including macrophages and neutrophils, after CLP-induced sepsis (Zhao et al. Citation2016). A similar specificity has been reported in B. anthracis infections: the HDAC8-specific inhibitor PCI-34051, but not other isoform-selective HDAC inhibitors, reversed the inhibitory effect of the Anthrax lethal toxin (LeTx) on IL-1β production and overcame the resistance of mouse macrophages to LeTx-induced pyroptosis (Ha et al. Citation2014, Citation2016). These observations suggested that HDAC8 inhibition could partly restore immune cell functions during B. anthracis infection. Taken together, these results indicate that whereas global inhibition of HDAC activity might impair innate immune responses, selective targeting of individual HDACs could facilitate elimination of certain microbial pathogens ().
Figure 2. HDAC inhibitors regulate activation of several cell types involved in antimicrobial responses. Activation of inflammatory potential of macrophages is suppressed by inhibitors of class I/II HDACs, but specific inhibition of HDAC6 and HDAC8 facilitates elimination of certain pathogens. SCFAs produced by bacteria and class II HDAC inhibitors stimulate regulatory T cells, which protect the commensal microbiota from elimination by the immune system but could result in blunted immune response to pathogenic microorganisms. Similarly, SCFAs and HDAC inhibitors impair antimicrobial responses of neutrophils. Different classes of HDAC inhibitors and SCFAs stimulate epithelial cell production of CAMPs which contribute to pathogen elimination and stimulate the host immune response. HDAC inhibitors and BET protein inhibitors also prevent infection-related bone resorption by suppressing osteoclast function. CAMPs: cationic antimicrobial peptides; HDACi: HDAC inhibitors; ROS: reactive oxygen species; SCFAs: short chain fatty acids; Tregs: regulatory T cells.
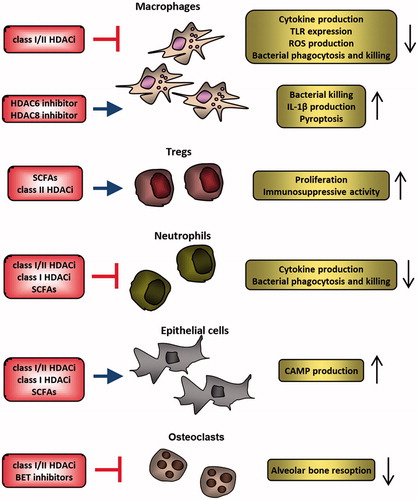
Modulators of sirtuin activity have also been tested in animal models of septic shock. In line with immune regulatory role of sirtuins, Sirt1 activation with resveratrol attenuated sepsis-induced acute kidney and lung injury (Li, Zhang, et al. Citation2013; Xu et al. Citation2016), whereas inhibition of Sirt1 during the hypoinflammatory phase of sepsis reversed endotoxin tolerance and enhanced immune responses (Vachharajani et al. Citation2014). It is important to note, however, that anti-inflammatory effects of sirtuin inhibition in sepsis have also been demonstrated. Administration of sirtinol and cambinol, which inhibit Sirt1 and Sirt2, prior to infection improved survival of mice during septic shock induced by K. pneumoniae challenge, which was associated with suppressed inflammatory activation of immune cells (Lugrin et al. Citation2013). The balance between pro- and anti-inflammatory functions of sirtuins therefore likely depends on the experimental model, timing of treatment, and/or compound specificity.
HDAC inhibitors in infection-related bone loss
In vivo studies provided evidence that HDAC inhibitors not only modulate inflammatory responses but may also prevent other pathological consequences of microbial infection, namely alveolar bone loss in periodontitis. Enhancement of osteoclast activity is an important feature of chronic inflammation in patients with periodontal disease that contributes to alveolar bone resorption and eventual tooth loss. The HDAC inhibitor 1179.4b which targets both class I and class II enzymes, protected mice from alveolar bone destruction in the P. gingivalis-induced model of periodontitis (Cantley et al. Citation2011). The observed effects can be attributed to class II HDAC family members as MS-275, which selectively targets class I HDACs, failed to protect mice from bone loss in this model. Surprisingly, alleviated alveolar bone resorption in 1179.4b-treated animals was not accompanied by reduced infiltration of immune cells to the periodontal tissue (Cantley et al. Citation2011), indicating that direct suppression of osteoclast formation rather than global suppression of the inflammatory response or bacterial outgrowth is responsible for the protective effects of HDAC inhibition. Indeed, HDAC inhibitors block osteoclast differentiation in vitro and suppress osteoclastogenesis in vivo (Rahman et al. Citation2003; Nakamura et al. Citation2005).
Interestingly, targeting different components of the acetylation system with the BET protein inhibitor JQ1 not only ameliorated alveolar bone resorption and reduced osteoclast formation in experimental P. gingivalis-induced periodontitis but also suppressed expression of inflammatory cytokines in the periodontal tissue (Meng et al. Citation2014). These observations suggest that the blockade of “readers”, rather than “erasers” of protein acetylation, might be more beneficial in simultaneous targeting of both inflammation and bone resorption in periodontal disease. However, contributions of individual HDAC isoforms to these processes, as well as the influence of acetylation modulators on bacterial clearance and plaque formation in periodontitis models remain to be determined.
Regulation of antimicrobial peptide production by HDAC inhibitors
While previous reports predominantly focused on the influence of blocking HDAC activity on immune cell function during infection, studies analyzing epithelial cell responses to bacterial challenges have identified HDAC inhibitors as potent regulators of antimicrobial peptide production (Yedery and Jerse Citation2015) (). Cationic antimicrobial peptides (CAMPs) are small (typically <10 kDa) effector molecules of the immune system that display microbicidal activity against a broad range of invading pathogens and modulate both innate and adaptive immune responses. Large quantities of CAMPs are produced by epithelial cells of mucosal tissues, which are the first line of interaction with pathogenic microorganisms (Doss et al. Citation2010).
Expression of the two major classes of mammalian CAMPs, defensins, and cathelicidins, is potently induced by HDAC inhibitors in colonic and airway epithelial cells (Schauber et al. Citation2003; Steinmann et al. Citation2009; Liu et al. Citation2013; Fischer et al. Citation2016; Miraglia et al. Citation2016). Although CAMP induction by HDAC inhibitors belonging to different chemical classes, including TSA, butyrate, phenylbutyrate, and MS-275, has been consistently observed across different types of epithelial cells (), several mechanisms underlying these effects have been proposed. Fischer er al. demonstrated that TSA upregulates human β-defensin 2 (hBD2) in the colonic epithelial cell line Caco-2 through a mechanism that involves phosphorylation of the IKKα/β complex, acetylation of the NF-κB p65 subunit and preferential phosphorylation of histone H3S10 at the hBD2 promoter (Fischer et al. Citation2016). In contrast, induction of the cathelicidin LL-37 by SCFAs has been shown to be dependent on MAP kinase signaling: both ERK and JNK inhibition prevented LL-37 upregulation by phenylbutyrate in bronchial epithelial cells (Steinmann et al. Citation2009), and ERK inhibition suppressed induction of LL-37 by butyrate and TSA in colonic epithelial cells (Schauber et al. Citation2003). Finally, a recent study utilized a luciferase-based reporter system to identify the involvement of the transcription factors STAT3 and HIF-1α in LL-37 upregulation by MS-275 in the colonic epithelial cell line HT-29 (Miraglia et al. Citation2016). It is currently unknown whether changes in the acetylation status of STAT3 and HIF-1α are responsible for LL-37 induction, but it is likely to be the case given that reversible acetylation affects transcription factors in multiple ways, including their stability, nuclear retention and gene target specificity (Glozak et al. Citation2005). While these studies support the mechanistic model in which HDAC inhibitors promote CAMP production though their direct effects on signaling pathways and transcription factors, it remains to be verified whether the observed distinct mechanisms of CAMP induction are specific for individual CAMP genes, or rather represent a cell type-specific phenomenon.
Table 2. Studies of histone deacetylase inhibitors (HDACi) in microbial infections.
The functional importance of CAMP induction by SCFAs has been validated in vivo in a model of bacillary dysentery caused by Shigella flexneri. Oral administration of butyrate reduced the severity of colon inflammation and bacterial load in the stool of infected rabbits (Raqib et al. Citation2006). Improvement of shigellosis clinical symptoms in butyrate-treated animals was associated with increased expression of cathelicidin CAP-18 (homologue of LL-37 in rabbits) in the intestinal epithelium and accumulation of the active form of CAP-18 in the stool (Raqib et al. Citation2006). Based on these findings, a clinical trial analyzing the effects of butyrate as an adjunct to antibiotics in patients with shigellosis has been launched in Bangladesh (ClinicalTrials.gov: NCT00800930). Compared to placebo, butyrate treatment led to early improvement of rectal inflammation, reduction of IL-8 and IL-1β levels in the stool, and elevated expression of LL-37 in rectal epithelial cells (Raqib et al. Citation2012). Although the contribution of butyrate HDAC inhibitory activity to the observed effects has not been tested in this setting, this is the first proof-of-concept study showing that induction of CAMPs by SCFAs might be clinically beneficial in the treatment of infections caused by a pathogen that is frequently found to be antibiotic resistant (Mandomando et al. Citation2009).
Conclusions and future perspectives
Clear evidence has been provided in the last decade that protein acetylation in host cells is dynamically regulated during infection and plays a crucial role in mounting an effective immune response against pathogens. Pathogenic microorganisms developed sophisticated strategies of immune evasion and the host acetylation system has emerged as an important target of bacterial virulence factors (, ). Thus far, however, experimental efforts have predominantly focused on changes in epigenetic histone acetylation marks induced by bacteria and their transcriptional consequences. Future studies should therefore be extended to non-histone targets of HATs and HDACs to generate a more comprehensive map of changes in the host acetylome during infection. In that regard, it is noteworthy that HATs and HDACs also regulate other types of histone lysine acylations, such as crotonylation and butyrylation (Sabari et al. Citation2017), but the role of these modifications in host–microbe interactions remains to be investigated. Another pressing question that remains to be answered is whether changes in histone acetylation (or other epigenetic mechanisms) can be maintained in host cells after pathogen clearance and resolution of inflammation. Although long-lasting effects of pathogen-induced epigenetic changes remain largely understudied, the concept of epigenetic memory of bacterial infection has been supported by some recent examples (Pereira et al. Citation2016), as well as the discovery of macrophage “innate immune memory” that is mediated through epigenetic reprograming (Saeed et al. Citation2014).
In light of our increasing knowledge regarding the many roles of protein acetylation in immunity, the introduction of HDAC inhibitors into clinical practice in oncology has raised questions about potential risks of increased sensitivity to infections in HDAC inhibitor-treated patients. Although infections were commonly reported in patients with cutaneous T-cell lymphoma treated with HDAC inhibitors (Piekarz et al. Citation2009), most of these adverse events could be attributed to disease-related impairment of cellular immunity rather than direct effects of these compounds on the immune system. Similarly, the risk for infections was not increased in patients with graft-versus-host disease after hemopoietic stem-cell transplantation treated with SAHA (vorinostat) (Choi et al. Citation2014), suggesting that HDAC inhibition has no detrimental effects on the quality of immune responses to infection. Long-term effects of epigenetic therapies on the immune system, however, remain to be determined.
Finally, it is important to note that in most cases manipulation of the protein acetylation system in host cells by specific pathogens has not been studied in the context of global effects of acetylation modulators (HDAC inhibitors, BET inhibitors) on the quality of immune responses to a broad range of different pathogens (). Future studies need to be rationally designed to simultaneously analyze both aspects of acetylation biology of infections. Only integration of these two lines of experimental evidence will allow prediction whether pharmacological modulators of the acetylation system can be used as a therapeutic strategy to promote the pathogen elimination or shape the immune response to prevent excessive collateral damage of host tissues.
Acknowledgements
We would like to thank Dr. Anu Goenka (Manchester Collaborative Centre for Inflammation Research, The University of Manchester) for critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Angiolilli C, Grabiec AM, Ferguson BS, Ospelt C, Malvar Fernandez B, van Es IE, van Baarsen LGM, Gay S, McKinsey TA, Tak PP, et al. 2016. Inflammatory cytokines epigenetically regulate rheumatoid arthritis fibroblast-like synoviocyte activation by suppressing HDAC5 expression. Ann Rheum Dis. 75:430–438.
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, et al. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 487:482–485.
- Ariffin JK, das Gupta K, Kapetanovic R, Iyer A, Reid RC, Fairlie DP, Sweet MJ. 2015. Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages. Antimicrob Agents Chemother. 60:1521–1529.
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. 2013. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 504:451–455.
- Bandyopadhaya A, Tsurumi A, Maura D, Jeffrey KL, Rahme LG. 2016. A quorum-sensing signal promotes host tolerance training through HDAC1-mediated epigenetic reprogramming. Nat Microbiol. 1:16174.
- Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. 2012. Histone deacetylases 6 and 9 and sirtuin-1 control foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 5:ra45.
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. 2009. An operational definition of epigenetics. Genes Dev. 23:781–783.
- Bierne H, Hamon M, Cossart P. 2012. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med. 2:a010272.
- Bobetsis YA, Barros SP, Lin DM, Weidman JR, Dolinoy DC, Jirtle RL, Boggess KA, Beck JD, Offenbacher S. 2007. Bacterial infection promotes DNA hypermethylation. J Dent Res. 86:169–174.
- Cantley MD, Bartold PM, Marino V, Fairlie DP, Le GT, Lucke AJ, Haynes DR. 2011. Histone deacetylase inhibitors and periodontal bone loss. J Periodont Res. 46:697–703.
- Cantley MD, Dharmapatni AA, Algate K, Crotti TN, Bartold PM, Haynes DR. 2016. Class I and II histone deacetylase expression in human chronic periodontitis gingival tissue. J Periodont Res. 51:143–151.
- Cardoso F, Castro F, Moreira-Teixeira L, Sousa J, Torrado E, Silvestre R, Castro AG, Saraiva M, Pais TF, Cardona P-J. 2015. Myeloid sirtuin 2 expression does not impact long-term Mycobacterium tuberculosis control. PLoS One. 10:e0131904.
- Chang M-C, Tsai Y-L, Chen Y-W, Chan C-P, Huang C-F, Lan W-C, Lin C-C, Lan W-H, Jeng J-H. 2013. Butyrate induces reactive oxygen species production and affects cell cycle progression in human gingival fibroblasts. J Periodont Res. 48:66–73.
- Chen X, Lu Y, Zhang Z, Wang J, Yang H, Liu G. 2015. Intercellular interplay between Sirt1 signalling and cell metabolism in immune cell biology. Immunology. 145:455–467.
- Cheng CY, Gutierrez NM, Marzuki MB, Lu X, Foreman TW, Paleja B, Lee B, Balachander A, Chen J, Tsenova L, et al. 2017. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol. 2:eaaj1789.
- Choi SW, Braun T, Chang L, Ferrara JLM, Pawarode A, Magenau JM, Hou G, Beumer JH, Levine JE, Goldstein S, et al. 2014. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 15:87–95.
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 325:834–840.
- Ciarlo E, Savva A, Roger T. 2013. Epigenetics in sepsis: targeting histone deacetylases. Int J Antimicrob Agents. 42(Suppl): S8–S12.
- Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MAR. 2016. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology. 5:e73.
- Corrêa RO, Vieira A, Sernaglia EM, Lancellotti M, Vieira AT, Avila-Campos MJ, Rodrigues HG, Vinolo MAR. 2017. Bacterial short-chain fatty acid metabolites modulate the inflammatory response against infectious bacteria. Cell Microbiol. 19:e12720.
- Crotty Alexander LE, Marsh BJ, Timmer AM, Lin AE, Zainabadi K, Czopik A, Guarente L, Nizet V. 2013. Myeloid cell sirtuin-1 expression does not alter host immune responses to Gram-negative endotoxemia or Gram-positive bacterial infection. PLoS One. 8:e84481.
- Dinarello CA, Fossati G, Mascagni P. 2011. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med. 17:333–352.
- Ding S-Z, Fischer W, Kaparakis-Liaskos M, Liechti G, Merrell DS, Grant PA, Ferrero RL, Crowe SE, Haas R, Hatakeyama M, Goldberg JB. 2010. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS One. 5:e9875.
- Doss M, White MR, Tecle T, Hartshorn KL. 2010. Human defensins and LL-37 in mucosal immunity. J Leukoc Biol. 87:79–92.
- Eskandarian HA, Impens F, Nahori M-A, Soubigou G, Coppée J-Y, Cossart P, Hamon MA. 2013. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science. 341:1238858.
- Fehri LF, Rechner C, Janssen S, Mak TN, Holland C, Bartfeld S, Brüggemann H, Meyer TF. 2009. Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation. Epigenetics. 4:577–586.
- Fischer N, Sechet E, Friedman R, Amiot A, Sobhani I, Nigro G, Sansonetti PJ, Sperandio B. 2016. Histone deacetylase inhibition enhances antimicrobial peptide but not inflammatory cytokine expression upon bacterial challenge. Proc Natl Acad Sci USA. 113:E2993–E3001.
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 504:446–450.
- Ganesan R, Hos NJ, Gutierrez S, Fischer J, Stepek JM, Daglidu E, Krönke M, Robinson N. 2017. Salmonella Typhimurium disrupts Sirt1/AMPK checkpoint control of mTOR to impair autophagy. PLoS Pathog. 13:e1006227.
- Garcia-Garcia JC, Barat NC, Trembley SJ, Dumler JS. 2009. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog. 5:e1000488.
- Gardner KE, Allis CD, Strahl BD. 2011. Operating on chromatin, a colorful language where context matters. J Mol Biol. 409:36–46.
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. 2016. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (London, England). 388:1545–1602.
- Glaser KB, Staver MJ, Waring JF, Stender J, Ulrich RG, Davidsen SK. 2003. Gene expression profiling of multiple histone deacetylase (HDAC) inhibitors: defining a common gene set produced by HDAC inhibition in T24 and MDA carcinoma cell lines. Mol Cancer Ther. 2:151–163.
- Glozak MA, Sengupta N, Zhang X, Seto E. 2005. Acetylation and deacetylation of non-histone proteins. Gene. 363:15–23.
- Grabiec AM, Reedquist KA. 2013. The ascent of acetylation in the epigenetics of rheumatoid arthritis. Nat Rev Rheumatol. 9:311–318.
- Grabiec AM, Tak PP, Reedquist KA. 2008. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther. 10:226.
- Grabiec AM, Tak PP, Reedquist KA. 2011. Function of histone deacetylase inhibitors in inflammation. Crit Rev Immunol. 31:233–263.
- Ha S-D, Han CY, Reid C, Kim SO. 2014. HDAC8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J Immunol. 193:1333–1343.
- Ha S-D, Reid C, Meshkibaf S, Kim SO. 2016. Inhibition of Interleukin 1β (IL-1β) expression by anthrax lethal toxin (LeTx) is reversed by histone deacetylase 8 (HDAC8) inhibition in murine macrophages. J Biol Chem. 291:8745–8755.
- Hamon MA, Batsché E, Régnault B, Tham TN, Seveau S, Muchardt C, Cossart P. 2007. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci USA. 104:13467–13472.
- Hamon MA, Cossart P. 2008. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe. 4:100–109.
- Imai K, Inoue H, Tamura M, Cueno ME, Inoue H, Takeichi O, Kusama K, Saito I, Ochiai K. 2012. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie. 94:839–846.
- Imai K, Ochiai K, Okamoto T. 2009. Reactivation of latent HIV-1 infection by the periodontopathic bacterium Porphyromonas gingivalis involves histone modification. J Immunol. 182:3688–3695.
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13:484–492.
- Jüngel A, Baresova V, Ospelt C, Simmen BR, Michel BA, Gay RE, Gay S, Seemayer CA, Neidhart M. 2006. Trichostatin A sensitises rheumatoid arthritis synovial fibroblasts for TRAIL-induced apoptosis. Ann Rheum Dis. 65:910–912.
- Kawabata T, Nishida K, Takasugi K, Ogawa H, Sada K, Kadota Y, Inagaki J, Hirohata S, Ninomiya Y, Makino H. 2010. Increased activity and expression of histone deacetylase 1 in relation to tumor necrosis factor-alpha in synovial tissue of rheumatoid arthritis. Arthritis Res Ther. 12:R133.
- Kim G-W, Yang X-J. 2011. Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem Sci. 36:211–220.
- Klein K, Kabala PA, Grabiec AM, Gay RE, Kolling C, Lin L-L, Gay S, Tak PP, Prinjha RK, Ospelt C, Reedquist KA. 2016. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis. 75:422–429.
- Krautkramer KA, Rey FE, Denu JM. 2017. Chemical signaling between gut microbiota and host chromatin: what is your gut really saying? J Biol Chem. 292:8582–8593.
- Li T, Lu Q, Wang G, Xu H, Huang H, Cai T, Kan B, Ge J, Shao F. 2013. SET-domain bacterial effectors target heterochromatin protein 1 to activate host rDNA transcription. EMBO Rep. 14:733–740.
- Li T, Zhang J, Feng J, Li Q, Wu L, Ye Q, Sun J, Lin Y, Zhang M, Huang R, et al. 2013. Resveratrol reduces acute lung injury in a LPS-induced sepsis mouse model via activation of Sirt1. Mol Med Rep. 7:1889–1895.
- Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X, Alam HB. 2015. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg. 78:378–385.
- Liu Q, Liu J, Roschmann KI, van Egmond D, Golebski K, Fokkens W, Wang D, van Drunen C. 2013. Histone deacetylase inhibitors up-regulate LL-37 expression independent of toll-like receptor mediated signalling in airway epithelial cells. J Inflamm. 10:15.
- Lugrin J, Ciarlo E, Santos A, Grandmaison G, dos Santos I, Le Roy D, Roger T. 2013. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim Biophys Acta. 1833:1498–1510.
- Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. 2006. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 12:989–995.
- Mandomando I, Jaintilal D, Pons MJ, Valles X, Espasa M, Mensa L, Sigauque B, Sanz S, Sacarlal J, Macete E, et al. 2009. Antimicrobial susceptibility and mechanisms of resistance in Shigella and Salmonella isolates from children under five years of age with diarrhea in rural Mozambique. Antimicrob Agents Chemother. 53:2450–2454.
- Martins MD, Jiao Y, Larsson L, Almeida LO, Garaicoa-Pazmino C, Le JM, Squarize CH, Inohara N, Giannobile WV, Castilho RM. 2016. Epigenetic modifications of histones in periodontal disease. J Dent Res. 95:215–222.
- Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A. 2013. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell. 152:51–67.
- Matalon S, Rasmussen TA, Dinarello CA. 2011. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol Med. 17:466–472.
- Meng S, Zhang L, Tang Y, Tu Q, Zheng L, Yu L, Murray D, Cheng J, Kim SH, Zhou X, Chen J. 2014. BET inhibitor JQ1 blocks inflammation and bone destruction. J Dent Res. 93:657–662.
- Miraglia E, Nylén F, Johansson K, Arnér E, Cebula M, Farmand S, Ottosson H, Strömberg R, Gudmundsson GH, Agerberth B, Bergman P. 2016. Entinostat up-regulates the CAMP gene encoding LL-37 via activation of STAT3 and HIF-1α transcription factors. Sci Rep. 6:33274.
- Mombelli M, Lugrin J, Rubino I, Chanson A-L, Giddey M, Calandra T, Roger T. 2011. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J Infect Dis. 204:1367–1374.
- Mujtaba S, Winer BY, Jaganathan A, Patel J, Sgobba M, Schuch R, Gupta YK, Haider S, Wang R, Fischetti VA. 2013. Anthrax SET protein: a potential virulence determinant that epigenetically represses NF-κB activation in infected macrophages. J Biol Chem. 288:23458–23472.
- Nakamura T, Kukita T, Shobuike T, Nagata K, Wu Z, Ogawa K, Hotokebuchi T, Kohashi O, Kukita A. 2005. Inhibition of histone deacetylase suppresses osteoclastogenesis and bone destruction by inducing IFN-beta production. J Immunol. 175:5809–5816.
- Niederman R, Buyle-Bodin Y, Lu BY, Naleway C, Robinson P, Kent R. 1996. The relationship of gingival crevicular fluid short chain carboxylic acid concentration to gingival inflammation. J Clin Periodontol. 23:743–749.
- Niederman R, Buyle-Bodin Y, Lu BY, Robinson P, Naleway C. 1997. Short-chain carboxylic acid concentration in human gingival crevicular fluid. J Dent Res. 76:575–579.
- Niller HH, Minarovits J. 2016. Patho-epigenetics of infectious diseases caused by intracellular bacteria. Adv Exp Med Biol. 879:107–130.
- Nishida K, Komiyama T, Miyazawa S-I, Shen Z-N, Furumatsu T, Doi H, Yoshida A, Yamana J, Yamamura M, Ninomiya Y. 2004. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheum. 50:3365–3376.
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. 2005. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 102:3697–3702.
- Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. 2006. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol. 176:4323–4330.
- Pereira JM, Hamon MA, Cossart P. 2016. A lasting impression: epigenetic memory of bacterial infections? Cell Host Microbe. 19:579–582.
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, et al. 2009. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 27:5410–5417.
- Prinjha RK, Witherington J, Lee K. 2012. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 33:146–153.
- Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Kohashi O. 2003. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood. 101:3451–3459.
- Raqib R, Sarker P, Bergman P, Ara G, Lindh M, Sack DA, Nasirul Islam KM, Gudmundsson GH, Andersson J, Agerberth B. 2006. Improved outcome in shigellosis associated with butyrate induction of an endogenous peptide antibiotic. Proc Natl Acad Sci USA. 103:9178–9183.
- Raqib R, Sarker P, Mily A, Alam NH, Arifuzzaman ASM, Rekha RS, Andersson J, Gudmundsson GH, Cravioto A, Agerberth B. 2012. Efficacy of sodium butyrate adjunct therapy in shigellosis: a randomized, double-blind, placebo-controlled clinical trial. BMC Infect Dis. 12:111
- Raymond B, Batsche E, Boutillon F, Wu Y-Z, Leduc D, Balloy V, Raoust E, Muchardt C, Goossens PL, Touqui L. 2009. Anthrax lethal toxin impairs IL-8 expression in epithelial cells through inhibition of histone H3 modification. PLoS Pathog. 5:e1000359.
- Rennoll-Bankert KE, Garcia-Garcia JC, Sinclair SH, Dumler JS. 2015. Chromatin-bound bacterial effector ankyrin A recruits histone deacetylase 1 and modifies host gene expression. Cell Microbiol. 17:1640–1652.
- Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson A-L, Reymond MK, Miconnet I, et al. 2011. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 117:1205–1217.
- Rolando M, Gomez-Valero L, Buchrieser C. 2015. Bacterial remodelling of the host epigenome: functional role and evolution of effectors methylating host histones. Cell Microbiol. 17:1098–1107.
- Rolando M, Sanulli S, Rusniok C, Gomez-Valero L, Bertholet C, Sahr T, Margueron R, Buchrieser C. 2013. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe. 13:395–405.
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. 2003. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 370:737–749.
- Sabari BR, Zhang D, Allis CD, Zhao Y. 2017. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 18:90–101.
- Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, Cheng S-C, Ratter J, Berentsen K, van der Ent MA, et al. 2014. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 345:1251086.
- Sawicka A, Seiser C. 2012. Histone H3 phosphorylation – a versatile chromatin modification for different occasions. Biochimie. 94:2193–2201.
- Schauber J, Svanholm C, Termén S, Iffland K, Menzel T, Scheppach W, Melcher R, Agerberth B, Lührs H, Gudmundsson GH. 2003. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut. 52:735–741.
- Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. 2011. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 32:335–343.
- Shames SR, Bhavsar AP, Croxen MA, Law RJ, Mak SHC, Deng W, Li Y, Bidshari R, de Hoog CL, Foster LJ, Finlay BB. 2011. The pathogenic Escherichia coli type III secreted protease NleC degrades the host acetyltransferase p300. Cell Microbiol. 13:1542–1557.
- Singh SK, Ellenrieder V. 2013. Senescence in pancreatic carcinogenesis: from signalling to chromatin remodelling and epigenetics. Gut. 62:1364–1372.
- Steinmann J, Halldórsson S, Agerberth B, Gudmundsson GH. 2009. Phenylbutyrate induces antimicrobial peptide expression. Antimicrob Agents Chemother. 53:5127–5133.
- Tolg C, Sabha N, Cortese R, Panchal T, Ahsan A, Soliman A, Aitken KJ, Petronis A, Bägli DJ. 2011. Uropathogenic E. coli infection provokes epigenetic downregulation of CDKN2A (p16INK4A) in uroepithelial cells. Lab Investig. 91:825–836.
- Tsuda H, Ochiai K, Suzuki N, Otsuka K. 2010. Butyrate, a bacterial metabolite, induces apoptosis and autophagic cell death in gingival epithelial cells. J Periodontal Res. 45:626–634.
- Vachharajani VT, Liu T, Brown CM, Wang X, Buechler NL, Wells JD, Yoza BK, McCall CE. 2014. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol. 96:785–796.
- Verdin E, Ott M. 2015. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 16:258–264.
- Wang Y, Curry HM, Zwilling BS, Lafuse WP. 2005. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J Immunol. 174:5687–5694.
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. 2009. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 138:1019–1031.
- Woo V, Alenghat T. 2017. Host-microbiota interactions: epigenomic regulation. Curr Opin Immunol. 44:52–60.
- Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, Naumann M, Lendeckel U. 2008. Helicobacter pylori regulates p21(WAF1) by histone H4 acetylation. Biochem Biophys Res Commun. 369:526–531.
- Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai X, Zeng Z, Zhao K-S. 2016. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxid Med Cell Longev. 2016:7296092.
- Yedery R, Jerse A. 2015. Augmentation of cationic antimicrobial peptide production with histone deacetylase inhibitors as a novel epigenetic therapy for bacterial infections. Antibiotics (Basel). 4:44–61.
- Yin L, Chung WO. 2011. Epigenetic regulation of human β-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 4:409–419.
- Yu X, Shahir A-M, Sha J, Feng Z, Eapen B, Nithianantham S, Das B, Karn J, Weinberg A, Bissada NF, Ye F. 2014. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, ezh2, and suv39h1 to promote Kaposi's sarcoma-associated herpesvirus replication. J Virol. 88:4466–4479.
- Zhang Z, Zhang R. 2015. Epigenetics in autoimmune diseases: pathogenesis and prospects for therapy. Autoimmun Rev. 14:854–863.
- Zhao T, Li Y, Liu B, Pan B, Cheng X, Georgoff P, Alam HB. 2016. Inhibition of histone deacetylase 6 restores innate immune cells in the bone marrow in a lethal septic model. J Trauma Acute Care Surg. 80:34–40.