
Abstract
Throughout our lives, epigenetic processes shape our development and enable us to adapt to a constantly changing environment. Identifying and understanding environmentally induced epigenetic change(s) that may lead to adverse outcomes is vital for protecting public health. This review, therefore, examines the present understanding of epigenetic mechanisms involved in the mammalian life cycle, evaluates the current evidence for environmentally induced epigenetic toxicity in human cohorts and rodent models and highlights the research considerations and implications of this emerging knowledge for public health and regulatory toxicology. Many hundreds of studies have investigated such toxicity, yet relatively few have demonstrated a mechanistic association among specific environmental exposures, epigenetic changes and adverse health outcomes in human epidemiological cohorts and/or rodent models. While this small body of evidence is largely composed of exploratory in vivo high-dose range studies, it does set a precedent for the existence of environmentally induced epigenetic toxicity. Consequently, there is worldwide recognition of this phenomenon, and discussion on how to both guide further scientific research towards a greater mechanistic understanding of environmentally induced epigenetic toxicity in humans, and translate relevant research outcomes into appropriate regulatory policies for effective public health protection.
Introduction
The belief that the sequence of our DNA (our genome) is the sole determinant of our life-long development and health has long been regarded as too simplistic. Darwin’s ‘On the Origin of the Species’ highlighted the role of the environment in the process of evolution, the natural selection of advantageous genetic mutations through environmental pressures (Darwin Citation1859). It has also been recognized that the pace of such change is sometimes too rapid to be accounted for by natural selection. First Lamarck, and then Waddington, suggested that alternative mechanisms, now termed epigenetics, must also contribute to phenotypic outcomes (Noble Citation2006). Epigenetics encompasses the mechanisms that regulate gene expression without altering the underlying DNA sequence, and the complete epigenetic status of a cell at any given time is termed the epigenome. Epigenetic programing is fundamental for normal mammalian development, and provides a more subtle mechanism by which the environment can rapidly alter gene expression within single or multiple generations. It is the complex interaction among our genome, epigenome and environment that shapes our development into unique individuals, and thus influences our health and potentially the health of our future offspring.
The induction of adverse DNA mutations by environmental factors that lead to disease, particularly cancer, is well established and recognized by regulatory bodies worldwide. As such, testing procedures and safety guidelines are in place to protect human health against the adverse effects of environmentally induced genetic mutations. There are, however, no current established regulatory procedures or guidelines in chemical safety programs for determining environmentally induced epigenetic toxicity. We, therefore, wanted to examine both the potential public health concerns and the resulting regulatory implications of such toxicity. For this purpose, we searched the current literature on epigenetic mechanisms involved in the mammalian life cycle and environmentally induced epigenetic toxicity in human epidemiological cohorts and rodent models in PubMed using the terms in Supplementary Table 1; defining environmentally induced epigenetic toxicity as environmental factors (chemicals, radiation and lifestyle factors (including alcohol, nutrition and smoking)) that induce adverse effects (growth retardation, fertility problems, hormonal changes, reproductive and/or other organ- and system-specific abnormalities (including immune disorders, obesity, diabetes and cancer)) and are associated with epigenetic mechanisms (histone modifications, DNA methylation, non-coding RNAs (ncRNAs) and other epigenetic machinery (DNA/RNA binding and modifying proteins)). Relevant papers were selected based on the inclusion criteria in Supplementary Table 1. These were then further evaluated and collated to provide a critical review that summarizes the main epigenetic mechanisms important during the mammalian life cycle, evaluates the current evidence for environmentally induced epigenetic toxicity in human cohorts and rodents models, and highlights the research considerations and implications of this emerging knowledge for public health and regulatory toxicology.
Environmentally induced epigenetic toxicity is an issue that has been highlighted as a current research focus within Public Health England’s (PHE's) Center for Radiation, Chemical and Environmental Hazards (CRCE), the wider PHE organization as part of the early life priority ‘ensuring every child has the best start in life’, and the international research and governmental communities. By identifying and contributing to the knowledge gaps PHE, and similar organizations worldwide, can ensure that they stay fully aware of the current situation and are in the best position to give advice and put forward any necessary recommendations and policies to protect public health against such toxicity.
Epigenetic mechanisms
Histones are a family of highly conserved, small, basic (positively charged) proteins around which (negatively charged) DNA winds to form nucleosomes, the main structural units of chromatin (the protein-DNA complex) that enable DNA to be packaged into the nucleus (Kornberg & Lorch Citation1999). Both the tails and globular domains of histones can undergo multiple posttranslational modifications, including acetylation, methylation, phosphorylation, ADP ribosylation, ubiquitination and sumoylation, giving rise to the so-called histone code (Strahl & Allis Citation2000; Turner Citation2000; Jenuwein & Allis Citation2001; Bhaumik et al. Citation2007; Kouzarides Citation2007; Suganuma & Workman Citation2011). The functional groups are added or removed by a range of enzymes: acetylases, methyltransferases, kinases, ADP ribosyltransferases, ubiquitin ligases, sumo ligases; and deacteylases, demethylases, phosphatases, ADP ribosyl hydrolases, deubiquitinases, sumo deconjugating enzymes, respectively (Wang & Dasso Citation2009; Khare et al. Citation2012). These modifications alter the interaction between DNA and DNA binding proteins (such as transcription factors and RNA polymerases) so that it is the overall combined effect of the histone code that determines an active or repressive chromatin structure and thus regulates gene expression. In general, histone acetylation and demethylation are associated with active chromatin (euchromatin), while histone deacteylation and methylation are associated with repressive chromatin (heterochromatin).
DNA methylation involves the addition of a methyl group, primarily to the C5 of cytosine within cytosine–phosphate–guanine dinucelotides (CpGs) to produce 5-methylcytosine (5mC). Due to the high mutagenic potential of 5mC to spontaneously deaminate to thymine, the mammalian genome is largely depleted of CpGs (Bird Citation1980; Bird & Taggart Citation1980), with the exception of distinct regions known as CpG islands (CGIs) (Bird et al. Citation1985). These CpG rich CGIs are non-randomly distributed throughout the mammalian genome, being predominantly associated with regulatory elements such as promoters (Deaton & Bird Citation2011). DNA methylation also alters the interaction between DNA and DNA binding proteins to regulate gene expression. Methylation at CGIs is generally associated with transcriptional repression, although the local CpG density within a specific promoter can contribute to the actual transcriptional effect (Hackett & Surani Citation2013; Messerschmidt et al. Citation2014). DNA is methylated by a family of DNA methyltransferases (DNMTs) (Bestor & Ingram Citation1983; Li et al. Citation1992, 1996; Okano et al. Citation1998, Citation1999; Aapola et al. Citation2000; Gowher et al. Citation2005). DNMT1, the maintenance DNMT, preferentially methylates hemimethylated DNA and thus maintains DNA methylation following DNA replication. De novo methylation is established by the de novo DNMTs 3A and 3B, which preferentially methylate unmethylated DNA. The third member of the DNMT3 family, DNMT3L, does not possess any DNMT activity, but can help to recruit, and stimulate the activity of, DNMT3A and 3B. Much less is known about the mechanisms of DNA demethylation. It has long been suggested that 5mC can be removed by both passive (through lack of maintenance during replication) and active (enzymatic) mechanisms. Yet, specific DNA demethylase enzyme(s) in mammals remained elusive until the discovery of the 10–11 translocation (TET) enzyme family. This family, which can oxidize 5mC to 5-hydroxymethylcytosine (5hmC) and further to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) (Tahiliani et al. Citation2009; Ito et al. Citation2011), has fueled research into indirect active DNA demethylation pathways. Consequently, a range of mechanisms for the demethylation of DNA have been proposed (described in relation to mammalian development in Dean Citation2014; Messerschmidt et al. Citation2014; Messerschmidt, Citation2016).
The ncRNA superfamily encompasses a number of families broadly classified according to their length: long non-coding RNAs (lncRNAs) (>200 nt) and short non coding RNAs (sncRNAs) (<200 nt), which include microRNAs (miRNAs, single stranded, ∼19–25 nt), piwi-interacting RNAs (piRNAs, single stranded, ∼24–30 nt) and endogenous short interfering RNAs (esiRNAs, double stranded, ∼21–22 nt). The vast majority of the mammalian genome is composed of so-called non-coding DNA (ncDNA), with only 1–5% coding for proteins. It was widely believed that these 20–30 000 protein-coding genes were the sole mediators and executors of all cellular functions. The remaining 95–99% of the genome was regarded as ‘junk’ DNA. However, a functional role for ncDNA was inferred from the strong correlation between increasing ncDNA abundance and increasing organism complexity (Mattick Citation2007). Prokaryote genomes contain only 10% ncDNA, more complex fungi and animals >50%, rising to >98% in complex mammals (including mice and humans) (Carey Citation2011). Indeed, over the last decade, the previous gene-centric dogma, central to molecular biology, has been shown to be incorrect. NcDNA is transcribed into ncRNAs, which play major roles in regulating gene expression. While lncRNAs do this in a variety of ways, including chromosome remodeling, and transcriptional or post-transcriptional regulation (Galupa & Heard Citation2015; Kanduri Citation2016; Taylor et al. Citation2015), sncRNAs predominantly mediate gene expression at the post-transcriptional level (Cook & Blelloch Citation2013; Hale et al. Citation2014). In general, miRNAs repress gene expression by binding to mRNAs in a sequence-specific manner and either inducing their degradation or inhibiting their translation (Ivey & Srivastava Citation2015), whereas piRNAs and esiRNAs bind to complementary transposable elements (TEs) and induce their degradation (Watanabe et al. Citation2006, Citation2008). Thus, miRNAs are involved in fine tuning gene expression, whilst piRNAs and esiRNAs play a primary role in maintaining genome stability.
All these mechanisms play critical roles throughout normal mammalian development, particularly during early embryo and germ cell development (Cook & Blelloch Citation2013; Beaujean Citation2014; Dean Citation2014; Hale et al. Citation2014; Luk et al. Citation2014; Messerschmidt et al. Citation2014; Mukherjee et al. Citation2014; O'Doherty & McGettigan Citation2014; Grote & Herrmann Citation2015; Hogg & Western Citation2015; Marcho et al. Citation2015). As with any rapidly developing field, there is a continuous generation of new information that must be incorporated as appropriate, such as novel histone or DNA modifications and ncRNAs families implicated in the epigenetic regulation of the mammalian life cycle (Dean Citation2014; Hale et al. Citation2014; Messerschmidt et al. Citation2014; Sun et al. Citation2014; Garcia-Lopez et al. Citation2015; Sharma et al. Citation2016); and new functions for existing epigenetic mechanisms within mammalian development (Gou et al. Citation2014; Kuramochi-Miyagawa et al. Citation2008; Watanabe et al. Citation2008, Citation2011; Li et al. Citation2015, Pantano et al. Citation2015; Williams et al. Citation2015; Zhang et al. Citation2015).
Role of epigenetics in the mammalian life cycle
Normal mammalian development is a cyclical series of events that enable the continued production of multiple generations (described in Gilbert Citation2014). A new generation begins with the fusion of the male (spermatozoa) and female (oocyte) gametes (fertilization) to produce a single cell (zygote) containing one haploid female pronucleus and one haploid male pronucleus. All the different cell types that comprise the new individual develop from this zygote and so contain the same unique genome. To enable de-differentiation of the highly specialized mature male and female gametes and subsequent generation of multiple specialized cell types capable of performing a wide range of diverse functions, these genomes require differential modifications so that functionally relevant genes can be switched on/off. This is achieved through epigenetic re-programing and programing. Thus, throughout development, epigenetic processes regulate the expression and silencing of specific genes to drive the mammalian life cycle. Two key global epigenetic re-programing and programing events occur, one after fertilization in the early embryo in all cells, and the second in germ cells only during primordial germ cell (PGC) development ().
Figure 1. Epigenetics and the mammalian life cycle. Following fertilization epigenetic marks are removed to generate a totipotent zygote, capable of producing all the different cell types required to form a new individual. Re-establishment of new epigenetic marks then redirects and drives development of the zygote through embryogenesis and ultimately into a mature adult. Primordial germ cells (PGCs) undergo a second epigenetic re-programing and programing to ensure correct imprinting and development of mature germ cells according to the gender of the embryo, and enable the successful production of subsequent generations in a continuous cycle. Such epigenetic processes can be perturbed by environmental factors, potentially leading to an adverse phenotypic outcome.
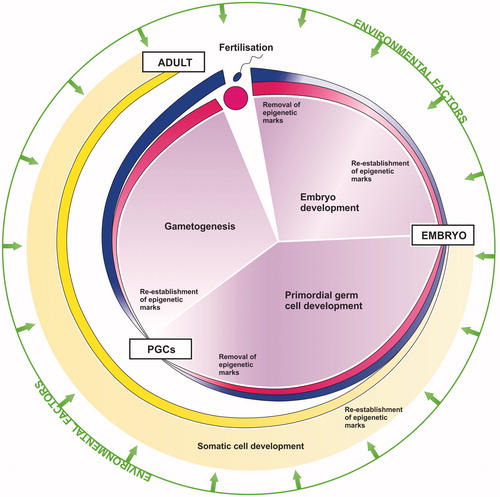
The first re-programing event immediately following fertilization de-differentiates the mature parental gametes so that the resulting zygote becomes totipotent, capable of producing all the different cell types required to form a new individual. This involves global DNA demethylation and changes in histone modifications and non-coding RNA expression to effectively ‘reset’ the zygote epigenome and thus genome. Epigenetic programing via DNA methylation, histone modifications and non-coding RNAs then redirects and drives development of the zygote through embryogenesis and ultimately into a mature adult. The dynamics of this first re-programing and programing event are particularly well highlighted by the functional switching of sncRNAs from a focus on guarding the genome to guiding development. The early zygote has low levels of miRNAs, but high expression of piRNAs and esiRNAs, with esiRNAs being the predominate species (Garcia-Lopez et al. Citation2015). As early development progresses and the embryonic genome is activated, miRNA expression increases and piRNA and esiRNA expression decreases, so that the pattern of sncRNA expression is reversed (Ohnishi et al. Citation2010; Suh & Blelloch Citation2011). Immediately following fertilization the quiescent embryo genome requires protection, hence higher expression of piRNAs and esiRNAs, both of which are involved in maintaining genome stability (Watanabe et al. Citation2006, Citation2008). Once transcription within the embryo genome is activated, however, there is rapid cell proliferation and differentiation, processes for which miRNA-mediated regulation is essential (Ivey & Srivastava Citation2015), hence increased expression of miRNAs.
It is worth noting that some genomic regions escape this first global ‘reset’, including imprinted control regions (ICRs) and the most active TEs. The continued transcriptional silencing of such regions is essential for early embryogenesis and genome stability, respectively. Normal embryo development requires a maternal and paternal contribution, generation of uniparental embryos by pronuclear transplantation results in developmental failure (Barton et al. Citation1984; McGrath & Solter Citation1984). This is because the male and female pronuclei are differentially imprinted; that is certain genes carry a parent-of-origin epigenetic mark so that they are only expressed from a single parental allele, ensuring the correct dosage of maternally and paternally derived gene products (reviewed in Bartolomei & Ferguson-Smith Citation2011). Imprinted genes are generally widely and highly expressed during prenatal development and predominantly down-regulated after birth. The placenta and brain, in particular, express many of the imprinted genes, in keeping with experimentally identified roles for imprinted genes in prenatal nutrient acquisition and growth regulation, neurodevelopment, and postnatal energy homeostasis and behavior. It is, therefore, vital for normal embryo development that these imprints are maintained during the first global epigenetic re-programing event. Similarly, the activation of TEs would be deleterious for the embryonic genome and so the continued silencing of such loci through early embryogenesis is essential for genome stability and subsequent normal development.
The second re-programing and programing event involves PGCs only. It is essential for the generation of subsequent germ cells (and thus the successful production of future generations) that parental imprints within PGCs are removed and new imprints are re-established according to the gender of the embryo. A female must reprogram the paternally imprinted genes on the chromosomes inherited from her father into maternally imprinted genes and vice versa. Thus, PGCs undergo a second round of global demethylation and changes in histone modification and non-coding RNA expression, this time including removal of the parent-specific imprints. Once again some of the most active TEs also escape this second phase of epigenetic re-programing, thereby maintaining genome stability. The mechanisms protecting specific genomic regions from epigenetic re-programing are not yet well-understood. However, some recent insights are presented and discussed in Hatanaka et al. (Citation2015) and Leseva et al. (Citation2016). Following sex determination, epigenetic programing via DNA methylation, histone modifications and non-coding RNAs redirect and drive differentiation of the PGCs along the distinct spermatozoal or oocyte lineage. Like the early zygote, mature gametes are transcriptionally quiescent. Thus, a similar sncRNA switch, this time back to increased genome protection makes functional sense. Indeed, PGCs, which represent an interim stage of the developing germ line that requires miRNA-mediated proliferation and differentiation with some pi/esiRNA-mediated genome protection, express greater levels of miRNAs and reduced levels of piRNAs and esiRNAs compared with the mature gametes and early zygote (Garcia-Lopez et al. Citation2015). Ultimately, the mature gametes come together to enable the cycle to begin again and so on.
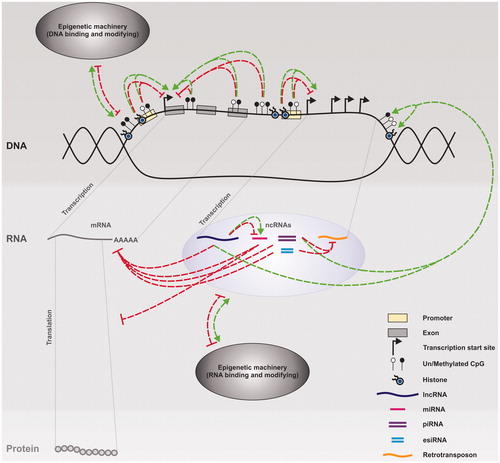
Finally, it is important to note that histone modifications, DNA methylation and ncRNAs are not separate processes; they are closely linked throughout mammalian development. The different epigenetic pathways converge and interact to regulate gene expression at multiple levels (). Thus, all the machinery involved in these pathways, including DNA and RNA binding and modifying proteins, are epigenetically important. This combination of interactive pathways helps to confer efficiency, redundancy, robustness and fidelity onto epigenetic re-programing/programing during development, and thus the successful and continued production of multiple generations.
Figure 2. Epigenetic regulation of gene expression. A range of epigenetic processes including DNA methylation, histone modifications and non-coding RNAs (ncRNAs), (such as long ncRNAs (lncRNAs), microRNAs (miRNAs), piwi-interacting RNAs (piRNAs) and endogenous short interfering RNAs (esiRNAs)) form an efficient and robust complex interactive network that regulates gene expression at the transcriptional and post-transcriptional levels. Inhibitory processes are shown in red and positive in green. Both histone modifications and DNA methylation changes can inhibit or promote transcription of mRNAs and ncRNAs. ncRNAs can then inhibit the production of protein from mRNAs either via mRNA degradation and/or reduced translation, and/or protect genome stability via the degradation of transposable elements (retrotransposons). Some ncRNAs can also promote changes in DNA methylation, histone modifications, and/or the expression of other ncRNAs. To complete the network, DNA/RNA binding and modifying proteins can both inhibit or promote gene expression whilst their own expression is under the control of the DNA modifications and ncRNAs they promote and/or interact with.
While such complex networks exist to maintain cellular homeostasis, they are open to environmental perturbation (). Indeed, numerous environmental factors have been shown to induce epigenetic changes via a variety of mechanisms. These include both direct and indirect processes that ultimately lead to alterations in DNA methylation, histone modification and/or ncRNA expression. For example, certain metals and environmental chemicals can directly bind and alter the activity of DNMTs, TETs and histone modifiers (Chervona & Costa Citation2012; Hou et al. Citation2012; Wang & Wang Citation2013; Ruiz-Hernandez et al. Citation2015). Both chemically induced DNA adducts and those generated as a consequence of reactive oxygen species damage resulting from chemical exposure can interfere with the ability of epigenetic machinery to interact with DNA (Baccarelli & Bollati Citation2009; Ruiz-Hernandez et al. Citation2015). Reactive oxygen species are also proposed to stimulate the production of alpha-ketoglutarate, a co-substrate for TETs and one of the two main classes of histone demethylases (Chervona & Costa Citation2012; Ruiz-Hernandez et al. Citation2015). Poor nutrition (reduced folate), detoxification of arsenic and consumption of glutathione during the detoxification of reactive oxygen species can all lead to reduced levels of the DNMT co-substrate S-adenosylmethionine (Hou et al. Citation2012; Ruiz-Hernandez et al. Citation2015). Similarly, histone acetylases require the co-substrate acetyl-CoA, and altered glycolytic production of acetyl-CoA can lead to changes in histone acetylation (Wellen et al. Citation2009; Moussaieff et al. Citation2015). Endocrine disrupting chemicals can bind to nuclear receptors, and either directly induce activity altering modifications to the epigenetic machinery (such as histone methyl transferases); or act as transcription factors, binding to hormone-responsive promotor elements and promoting expression of ncRNAs and/or epigenetic machinery (Kabir et al. Citation2015; Trevino et al. Citation2015). Additional epigenetic mechanisms are likely to be elucidated as research continues. It is when these environmentally induced perturbations exceed the capacity of the cell to maintain homeostasis that epigenetic toxicity occurs, potentially leading to altered phenotypes and adversely impacting public health.
Environmental factors and epigenetic perturbation
Epigenetic processes are susceptible to perturbation throughout an individual’s life (). Even in adulthood the same epigenetic processes drive cell proliferation, differentiation, function and adaptation. Environmentally induced epigenetic changes later in life can, therefore, also influence an individual’s health. Indeed, numerous adult onset diseases have been associated with abnormal epigenetic changes, including cancer, diabetes, and neurological, renal, cardiac and respiratory conditions (Hamm & Costa Citation2015); and epigenetic processes play a key role in initiating the onset of puberty, changes to which can also increase the risk of some of these adult onset diseases (Rzeczkowska et al. Citation2014). However, certain stages in development and cell types can be thought of as particularly sensitive to epigenetic change due to the resulting severity of the outcome for the individual or the potential for affecting multiple generations.
Early life
Early embryo and germ line development can be affected by in utero exposures. Environmentally induced epigenetic changes during the vital first developmental re-programing and programing event could result in incorrect erasure and/or re-establishment of the embryo epigenome. This could lead to activation of TEs, and abnormal imprinting and gene expression. Such changes could have far-reaching consequences on embryo viability and development (including early germ line formation), and thus subsequent future health and fertility. It is also important to consider, however, early ex utero exposures. Early childhood and adolescence are also periods of significant growth and development, so it is easy to envisage how environmentally induced epigenetic changes during these stages could have detrimental effects on future health and fertility.
Gametogenesis and pre-conception
Spermatogenesis occurs continuously in adult males and even though females are born with their full complement of oocytes, oocyte maturation occurs in a continuous cycle throughout the reproductive life of adult females. Thus, environmental exposures to those oocytes and spermatozoa that go onto produce an embryo (pre-conception exposures) are also an important consideration. This represents an adulthood exposure to the parent, but a very early life exposure to the offspring. Environmentally induced epigenetic changes in the sperm or egg prior to conception could, therefore, affect embryo viability and development and the subsequent future health and fertility of the next generation. Indeed, murine spermatozoal miRNAs and/or esiRNAs have been shown to function in fertilization and early embryo development (Liu et al. Citation2012, Citation2016; Yuan et al. Citation2016), and to influence phenotypic outcomes in subsequent progeny (Rassoulzadegan et al. Citation2006; Wagner et al. Citation2008; Grandjean et al. Citation2009). Moreover, subsequent studies have further demonstrated that early life trauma or parental stress can induce altered miRNA expression in sperm, which in turn can transmit behavioral and metabolic abnormalities through to the next murine generation (Rodgers et al. Citation2013; Gapp et al. Citation2014; Rodgers et al. Citation2015). It is also worth noting that some of these RNA-mediated phenotypes have been shown to be dependent upon the remaining member of the DNMT family, DNMT2 (Kiani et al. Citation2013). Although DNMT2 has demonstrated some weak DNMT activity, it is predominantly associated with RNA, particularly tRNA, methylation, potentially contributing to the regulation of RNA stability and translation (Motorin et al. Citation2010).
Somatic versus germ cell change
If an epigenetic change occurs in a somatic cell or in a germ cell that is correctly ‘reset’ during germ line development in the subsequent offspring, then only one generation will be affected. However, if an epigenetic change becomes permanently established in the germ line, it will be transmitted across future generations, even in the absence of the original stimulus. As mentioned earlier, there are a number of loci within PGCs that escape the two global DNA demethylation re-programing events. Recent data has extended this list to include specific regions not associated with TEs that are preferentially located within CGIs, enhancers, promoters and gene bodies (Seisenberger et al. Citation2012; Hackett et al. Citation2013; Tang et al. Citation2015). These both provide evidence that DNA methylation can be stably inherited, and highlight specific loci protected from global epigenetic erasure that could be involved in transmitting epigenetic information across generations. In addition, not all of the spermatozoal histones are replaced with protamines during spermiogenesis, up to 15% of histones within human spermatozoa and 1% in mouse spermatozoa are retained (Miller et al. Citation2010; Carrell Citation2012). These retained histones are not randomly distributed throughout the male genome, they are particularly enriched at the promoters of developmental genes. Thus, spermatozoal chromatin may also transmit unique epigenetic information that helps to activate and regulate early embryo development and contribute to epigenetic inheritance across multiple generations.
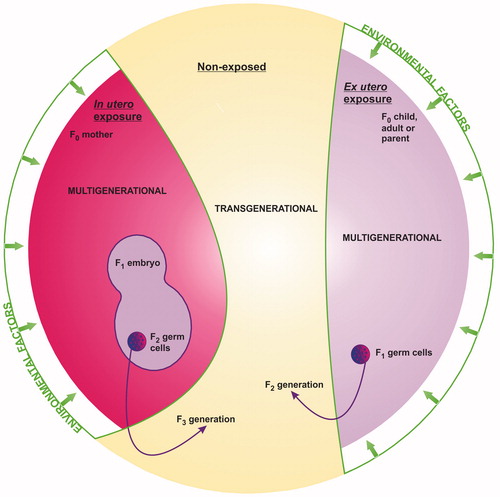
This so-called transgenerational epigenetic inheritance is a hotly debated topic. Much of the controversy is concerned with multigenerational versus transgenerational epigenetic toxicity (or epigenetic effect(s) versus epigenetic inheritance) (). If an environmental exposure is truly transgenerational, then: (1) following an in utero exposure the resulting change penetrates to at least the F3 generation (great-great grandchildren), where F0 is the exposed mother and F3 represents the first non-exposed generation; or (2) following an ex utero exposure in childhood/adulthood the resulting change penetrates to at least the F2 generation (the great-grandchildren), where F0 is the exposed generation and F2 represents the first non-exposed generation. A transgenerational change thus represents epigenetic inheritance. Conversely, a multigenerational change represents an epigenetic effect that affects multiple generations, all of which were exposed to the original stimulus, either directly as the exposed mother or child/adult or indirectly as an embryo or PGC within the exposed generation. With respect to public health, however, this is mere semantics; both could have far-reaching consequences for the present and future health of human populations. It is not necessarily important whether an environmental exposure induces adverse outcomes in multiple exposed or non-exposed generations. The fact that it may cause epigenetic toxicity in any generation makes it relevant to public health.
Figure 3. Multigenerational versus transgenerational epigenetic toxicity following in utero or ex utero environmental exposure. Penetration of adverse effect(s) to the first non-exposed generation (the F3 generation following in utero exposure, or the F2 generation following ex utero exposure) represents true epigenetic inheritance and thus transgenerational toxicity. Adverse effect(s) that affect multiple generations, all of which were exposed either directly or indirectly to the original factor represent epigenetic effect(s) and thus multigenerational toxicity.
Current evidence for putative environmentally induced epigenetic toxicity
Human cohorts
For many decades, there have been epidemiological studies linking the risks of adverse phenotypes to a wide range of environmental factors. For example, pre-conception, in utero or childhood undernutrition have been associated with an increased risk of developing various adult-onset diseases, including psychiatric disorders, type 2 diabetes and obesity, in numerous famine cohorts such as the Dutch Hunger Winter, the Chinese Great Leap Forward and the Överkalix cohorts (Lumey et al. Citation2011; Pembrey et al. Citation2014). Likewise, metabolic syndromes, hormonal perturbation, reduced sperm quality and increased cancer risks have been associated with chemical and radiation exposures in the survivors of human disasters, including the Seveso dioxin release, the Hiroshima and Nagasaki atomic bombs and the Chernobyl nuclear accident (Warner et al. Citation2011, Citation2013; Foley et al. Citation2015; Kamiya et al. Citation2015). In addition, large human cohorts that have recruited participants at birth for long-term follow up with comprehensive data and sample collection, such as the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort (Boyd et al. Citation2013), provide a wealth of information to investigate epidemiological links between specific environmental exposures and public health outcomes. There have been over 1200 publications using data from ALSPAC (ALSPAC 2016), many of which have linked pre-conception and in utero environmental factors (including chemicals, pharmaceuticals, nutrition, smoking, anxiety and depression) with adverse outcomes (such as asthma and other respiratory disorders, increased weight/BMI-related issues and behavioral problems) (Golding Citation2010; Boyd et al. Citation2013). In particular, studies examining the parental and grandparental smoking status have demonstrated an interesting gender-specific association between paternal pre-conception or grandmaternal in utero exposure to cigarette smoke and increased weight-related issues in the subsequent sons and grandsons, but not daughters or granddaughters (Pembrey et al. Citation2014). Gender-specific effects were also observed in a number of the famine cohorts, with an increased risk of obesity occurring in female descendants only (Lumey et al. Citation2011; Pembrey et al. Citation2014).
The association of later life health with pre-conception, in utero or childhood environmental factors has led to the “Developmental Origins of Health and Disease” (DOHaD) hypothesis (Wadhwa et al. Citation2009). The DOHaD hypothesis evolved from Barker’s “thrifty phenotype” hypothesis originally proposed over 25 years ago (reviewed in Barker Citation2007), and was formed “to recognise….the concept that the early life environment has widespread consequences for later health” (Wadhwa et al. Citation2009). This concept of phenotypic plasticity, particularly as a consequence of different environments in early life, implicates epigenetic mechanisms and has fueled research into environmentally induced epigenetic toxicity. We therefore sought to review the existing literature (up to 7 March 2016) to evaluate the current evidence for such toxicity.
We identified 76 human cohort studies within PubMed that associated adverse phenotype(s) (such as birth outcomes, organ-specific abnormalities/disorders, cancer or mortality) with epigenetic change(s) (mainly DNA methylation to-date, but also histone modification, epigenetic machinery and miRNAs) and exposure to environmental factor(s) (including air pollution, chemicals, fibers, radiation or lifestyle factors (alcohol, nutrition or smoking)) at various stages of development (pre-conception, in utero, neonatal, childhood, adolescence, adulthood and lifetime). These studies can be ranked into categories that demonstrate (1) associations between environmental exposure(s), global epigenetic change(s) and adverse phenotype(s); (2) associations between environmental exposure(s), specific epigenetic changes(s) and adverse phenotypes(s) (including further subcategories, such as size and nature of cohort, type of sample analyzed, use of case controls, dose- and/or time-dependent changes, single changes within relevant biological pathways, multiple changes within the same biological pathway, correlation of changes with gene expression at mRNA and/or protein level, use of additional mechanistically relevant controls, robust statistical analysis, validation in independent cohorts); or (3) associations between environmental exposure(s), specific epigenetic changes(s) and adverse phenotypes(s) that were confirmed in a relevant in vivo and/or in vitro system. Studies falling into category 3 provide more comprehensive mechanistic evidence for environmentally induced epigenetic toxicity and are shown in . Specifically, these studies provide mechanistic associations between bisphenol A (BPA), formaldehyde, arsenic, nickel, or cigarette smoke, epigenetic changes and adverse phenotypes, and potential mechanisms of epigenetic toxicity in humans. The smoking evidence is particularly compelling as it has been established by multiple independent groups investigating different epigenetic mechanisms. In addition, studies within categories 1 and 2 may also enable the identification of markers and/or mechanisms of environmentally induced epigenetic toxicity through a weight of evidence approach (e.g. the same association(s) reported in multiple independent cohorts or multiple association(s) that contribute to the same biological pathway), and are, therefore, included in Supplementary Table 2.
Table 1. Current evidence for putative environmentally induced epigenetic toxicity in human epidemiological cohorts.
Furthermore, while human epidemiological cohort studies can demonstrate association, they do not prove causality. They are also complicated by the fact that the biological material under study, often blood cells, is not from the target organ/tissue. Interpretation of the findings with respect to mechanistic connections between environmental factors, epigenetic changes and adverse effects in the target organ/tissue can therefore be challenging. However, as stated above, all 10 of the studies shown in further confirmed identified associations between environmental exposures, specific epigenetic changes and adverse phenotypes in a relevant in vivo and/or in vitro system; and seven of the 10 studies either directly sampled the target tissue or validated the same change in the blood and target tissue of an appropriate in vivo model. As such, these studies are of greater mechanistic relevance, and provide more comprehensive evidence for putative causal relationships.
Rodent models
The deleterious effects of exposures to environmental factors in human cohorts have stimulated investigations of environmentally induced toxicity in rodent models. In the late 1990s/early 2000s, these studies began to associate adverse phenotypes resulting from environmental exposures with epigenetic mechanisms (Issa et al. Citation1996; Tao et al. Citation1998, Citation1999; Choi et al. Citation1999; Bielawski et al. Citation2002; Govindarajan et al. Citation2002; Chen et al. Citation2004; Marwick et al. Citation2004; Pulling et al. Citation2004; Vuillemenot et al. Citation2004; Wu et al. Citation2004). Then in 2005, Anway et al. demonstrated that in utero exposure to the environmental chemical vinclozolin (a fungicide used in the wine industry) reduced sperm quantity, quality and thus male fertility with a >90% penetrance through multiple generations (to at least the F4 generation). The effect appeared to be transmitted through the male germ line, with altered DNA methylation patterns detected in the testes of the F1 generation and the sperm of the F2 and F3 generations. This was the first study to show phenotypic and epigenetic changes in generations not directly exposed to the original stimulus. As expected, this study generated some controversy, particularly when subsequent groups failed to reproduce the same results (Gray & Furr Citation2008; Schneider et al. Citation2008; Inawaka et al. Citation2009; Schneider et al. Citation2013). These disparities may have been due to the use of different routes of administration and variations in rat strains/stocks. Nevertheless, over the ensuing decade the hundreds of subsequent studies have provided more evidence for environmentally induced epigenetic toxicity.
We identified 147 rodent studies within PubMed (up to 7 March 2016) demonstrating both adverse phenotype(s) (such as reduced fertility, reproductive, developmental or other organ-specific abnormalities, or adult onset diseases (cancer, diabetes, obesity or immune disorders)) and epigenetic change(s) (histone modification, DNA methylation, epigenetic machinery or miRNAs) following exposure to environmental factor(s) (including chemicals, radiation or lifestyle factors (alcohol, nutrition, or smoking)) at various stages of development (pre-conception, in utero, neonatal, lactation, childhood, adolescence and adulthood). These studies can be ranked into categories that demonstrate (1) environmentally induced adverse phenotype(s) and global epigenetic change(s); (2) environmentally induced adverse phenotype(s) and specific epigenetic change(s) (including further subcategories, such as dose- and/or time-dependent changes, single changes within relevant biological pathways, multiple changes within the same biological pathway, correlation of changes with gene expression at mRNA and/or protein level, use of additional mechanistically relevant controls, robust statistical analysis); (3) environmentally induced adverse phenotype(s) and specific epigenetic change(s) that were reversed by an inhibitor/treatment, absent in a knock out/down model, and/or mechanistically linked in a relevant in vitro system; or (4) the same environmentally induced adverse phenotype(s) and specific epigenetic change(s) in both rodent model(s) and human cohort(s). Studies falling into categories 3 and/or 4 provide more comprehensive mechanistic evidence for environmentally induced epigenetic toxicity and are shown in . In total, 18 of the 40 studies shown in validated loss of both epigenetic change(s) and adverse effect(s) following appropriate inhibition, treatment or knock out/down; thereby demonstrating a definitive causal link between exposure (largely high dose) to BPA, formaldehyde, cadmium, methylmercury, 2,3,7,8-tetra-chloro-dibenzo-p-dioxin (TCDD), urethane, vinyl carbamate, alcohol, caffeine, overnutrition, cigarette smoke, nicotine or 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK), epigenetic changes and adverse phenotypes. While the remaining studies do not necessarily demonstrate causality, they do provide additional mechanistic associations between exposure (again largely high dose) to BPA, formaldehyde, nickel, phthalates, TCDD, alcohol, undernutrition, 2-amino-1-methyl-6-phenyl-imidazo[4,5-b]pyridine (PhIP), peanut, cigarette smoke, nicotine or NKK, epigenetic changes and adverse phenotypes. The BPA, TCDD, alcohol and smoking-related evidence is particularly compelling as it has been established by multiple independent groups investigating different epigenetic mechanisms. In addition, studies within categories 1 and 2 may also enable identification of markers and/or mechanisms of environmentally induced epigenetic toxicity through a weight of evidence approach (e.g. the same epigenetic change(s) reported in multiple independent studies or multiple epigenetic changes that contribute to the same biological pathway), and, therefore, are included in Supplementary Table 3. While much of the work to date has focused on rodent models, there is also some evidence for environmentally induced epigenetic toxicity in other mammalian species, including sheep and monkeys (Wu et al. Citation2008; Begum et al. Citation2013; Lie et al. Citation2014a, Citation2014b).
Table 2. Current evidence for putative environmentally induced epigenetic toxicity in rodent models.
Other exposures of environmental origin that have been extensively investigated in both human cohorts and rodent models include stress, recreational drugs, maternal immune activation/infection and parental age. While this large body of work also provides some comprehensive evidence for, and specific mechanisms of, epigenetic toxicity, it was considered outside the scope of this review. Likewise, pharmaceutical drug-induced epigenetic toxicity in human cohorts and rodent models has also been reported. However, such drugs are not environmental factors and so were not considered any further. Moreover, there were additional studies that have investigated environmentally induced epigenetic changes without simultaneously assessing any phenotypic/toxicological endpoints and/or focused on acute exposures (less than 24 h). While such studies are more difficult to interpret with respect to epigenetic toxicity (and so were not included in this review), they may still provide useful mechanistic data.
It is important to note that not all environmentally induced adverse phenotypes have been associated with epigenetic changes. Indeed, 18 human cohort and 20 rodent studies within PubMed reported a lack of epigenetically related toxicity (Supplementary Table 4). These studies do not necessarily rule out an epigenetic mechanism of toxicity per se, but they do exclude a link between the specific epigenetic, exposure and/or adverse endpoints/markers investigated.
The literature also contains many in vitro studies investigating the role of specific epigenetic mechanisms in adverse outcomes induced by environmental exposures. In addition, concurrent in vivo and in vitro studies have improved our knowledge of the mechanisms involved in the mammalian life cycle, including recent insights into early human development (Laird Citation2013; Slieker et al. Citation2015; Surani Citation2015; Williams et al. Citation2015), and epigenetic inheritance in simpler model systems such as Caenorhabditis elegans, Drosophila melanogaster and Danio rerio (zebrafish) (Padilla et al. Citation2014; Somer & Thummel Citation2014; Williams et al. Citation2014). Thus, although comparison and integration of these different human cohort, animal, and in vitro studies is complicated by the different doses, routes of administration, timings and lengths of exposures, the various strains, species, cell types or statistical methods used, and/or the specific adverse outcome(s) or marker(s) selected, the current evidence demonstrates a mechanistic association between specific environmental exposures, epigenetic changes and adverse health outcomes in human epidemiological cohorts and model systems. More studies that incorporate some form of inhibition/treatment/cessation into the experimental design are vital for establishing whether or not an exposure truly represents an epigenetic toxicant. Although potentially complex, the use of a systems biology approach, incorporating genome-wide analysis at multiple levels (histone code, methylome, non-coding RNA expression, transcriptome, proteome and metabolome) would provide comprehensive analysis of epigenetic changes and associated phenotypic outcomes. Subsequent identification of human relevant mechanisms of epigenetic toxicity can then be further validated as appropriate in smaller more focused studies. The challenge now is to fully identify and investigate the specific functional epigenetic mechanisms and biological pathways relevant to humans so that the risk(s) of environmentally induced epigenetic toxicity to public health can be properly assessed and addressed.
Research considerations for public health
There is no doubt that humans are exposed to a range of environmental factors during everyday life. However, to determine whether, or not, environmentally induced epigenetic toxicity is a real concern for public health there are a number of important considerations.
Mechanisms and model systems
The nature of the research means that studies in humans are mainly restricted to epidemiological cohorts. These cohorts often have limited availability of appropriate biosample at appropriate collection windows (Bakulski & Fallin Citation2014). Ex vivo or in vitro experiments on human embryonic tissue and cells are quite rightly restricted by ethics, and can only model a small part of the in vivo situation. Thus, the majority of whole system studies have been performed in animal models, predominantly rodents. While the rodent and human life cycles share many commonalities they do exhibit subtle differences, both with each other and other mammalian species (Kristensen et al. Citation2013; Laird Citation2013; Leitch et al. Citation2013; Surani Citation2015). Such differences are of critical importance when considering mechanistic relevance to humans.
Nevertheless, such work provides valuable data. The key is integrating and interpreting it correctly, in terms of differences/similarities at both the system (in vitro versus in vivo) and species (human versus non-human) levels. It is important to continue supplementing non-human models with human-derived data, incorporating new technologies to maximize data collection, and updating and re-evaluating current conclusions as novel information becomes available.
Priority questions: What are the detailed mechanism(s) that link a particular exposure to a specific adverse effect? Do these mechanism(s) function in humans? What are the potential human disease outcome(s)?
Dose, route, metabolism, timing and mixtures of exposure
Whether, or not, an environmental exposure elicits a response depends on a range of interacting variables, including dose, route of administration, metabolism and additional/pre-existing exposures (mixtures). Much of the animal studies to date on environmental chemicals have involved administration of high doses, many orders of magnitude above likely exposures in real human populations. Similarly, the first animal study to demonstrate that an in utero exposure could induce adverse phenotypes in subsequent generations administered the environmental chemical vinclozolin via the intraperitoneal route (Anway et al. Citation2005). Not only is it unlikely that a human would receive this, or any, environmental exposure via the same route of administration, but also the way in which an exposure is administered determines its overall effect. Indeed, oral administration of vinclozolin to pregnant females (the likely route of administration for humans given that vinclozolin is a fungicide used in the wine industry) failed to induce any adverse phenotypes in subsequent generations (Schneider et al. Citation2008). Thus, while it is important to fully characterize the hazard, further studies at doses and routes of administration similar to those found in humans are necessary for determining relevance to public health. It is worth noting, however, that establishing the actual level of human exposure to a particular factor and mimicking it in experimental models is not always easy.
Metabolism is another major factor that can determine the overall effect of an environmental exposure. Distinct species and model systems metabolize xenobiotics differently depending on their specific enzymatic profiles. Thus, metabolism of the parent environmental compound can vary greatly between models, resulting in the production of different types and levels of metabolites. It is, therefore, important to assess if the model system being used accurately replicates the in vivo human metabolic environment, and if not to conduct further studies using any additional appropriate metabolites.
The timing of an environmental exposure can also be a major contributing factor to the significance of the overall outcome. As discussed earlier, some life stages may be more susceptible than others to a particular exposure. There was the assumption that in utero exposure represents the most vulnerable period in an individual’s life with respect to their future development. However, it is now becoming clear that pre-conception and postnatal exposures can also be detrimental to future health across multiple generations.
Finally, humans are continuously exposed to a mixture of different environmental factors. Multiple exposures at multiple life stages could be additive and/or cumulative. While a particular low-level exposure may have no adverse effects, multiple low-level exposures over multiple stages of life may induce adverse phenotypes. Thus, once individual exposures have been characterized, it may also be relevant to assess the effect of (1) mixtures of environmental exposures, (2) multiple lifetime exposures of single environmental exposures, and (3) multiple lifetime exposures of mixtures of environmental exposures.
Priority questions: What dose(s) via what route(s) of administration are human populations actually exposed to, and what metabolite(s) are formed? Do low-level exposures induce epigenetic toxicity? Is any part of the life cycle particularly more sensitive to an exposure than others? Are multiple exposures additive and/or cumulative?
Human variability
It is known that different strains of laboratory animals can respond very differently to the same environmental exposure. Such differences are largely driven by diverse genetic backgrounds. Individual humans demonstrate substantial genetic differences and so an individual’s underlying genetics could have a huge effect on his/her susceptibility to a particular environmental exposure, depending on his/her metabolism, adaptability, gender, age, previous exposures, etc. Thus, not all humans will respond to an environmental exposure in the same way. Indeed, there are many examples in the literature of how single polymorphisms (SNPs) can affect the response of individuals and populations to different environmental exposures. These include not only mutations that alter the function of gene products but also those that lead to changes in DNA methylation or miRNA binding sites.
Priority question: How can we identify particularly susceptible individuals or populations?
Adaptive versus adverse epigenetic change
Epigenetic change offers a much more versatile, reversible and rapid way for adapting to changes in our environment compared with genetic mutation. This view of epigenetically mediated phenotypic plasticity falls into the realms of Lamarckian inheritance, the idea that an organism can pass on acquired characteristics to future offspring. The benefits of such inheritance are obvious and even Darwin himself proposed a hypothetical mechanism for the transmission of somatic tissue characteristics via the germ cells to subsequent generations, which he termed pangenesis (Darwin Citation1868). Indeed, not all environmentally induced epigenetic changes have proved detrimental. Hughes et al. (Citation2009) reported that calorie restriction during adolescence and young adulthood as a result of the Dutch Hunger Winter was associated with altered DNA methylation and reduced risk of colorectal cancer in later life. Tyagi et al. (Citation2015) described a protective effect of early (in utero through lactation) exposure to dietary omega-3 fatty acids on neuroplasticity in rats that involved specific changes in DNA methylation. Such benefits are not just limited to nutrition; advantageous chemically induced epigenetic changes have also been reported. Zeybel et al. (Citation2012) demonstrated a beneficial heritable histone modification-mediated reprograming of hepatic wound healing in male rats that reduced fibrotic scarring in the subsequent offspring of rats treated with the hepatotoxin carbon tetrachloride. In addition, it is possible to have adverse outcomes that are reversible. Indeed, Nishihara et al. (Citation2013) provided epidemiological data to support the benefits of smoking cessation, with DNA methylation-related colon cancer risk reverting back to that of non-smokers within 10 years of cessation. Thus, the reversibility of environmentally induced changes is also critically important when determining the true potential adversity of an environmental exposure. Such findings, both positive and negative, have been incorporated into UK governmental advice and recommendations (Boyd et al. Citation2013).
Furthermore, epigenetic changes may themselves have no consequences on gene expression (as demonstrated in a recent in vitro study by Ramos et al. (Citation2015). Thus, environmentally induced epigenetic changes may not always be mechanistically involved in adverse effects; instead, they may act as markers of past exposures (Bakulski & Fallin Citation2014), or represent adaptations involved in maintaining cellular homeostasis. Such changes may still prove useful for regulatory purposes and also do not rule out the possibility of later onset effects. For example, robust epigenetic changes induced by environmental exposures in the absence of a phenotypic consequence essentially become the ‘new normal’ epigenome, which could have greater susceptibility to disease development and/or future exposures (as demonstrated in two in vivo rat models (Greathouse et al. Citation2012; Wong et al. Citation2015)). Characterizing normal variation within the epigenome between non-disease states would provide valuable data to begin establishing limits of epignetic change above which cellular homeostasis fails and epigenetic toxicity is induced. The considerable inter- and intra-human/mammalian variation combined with the vast number of potential epigenetic changes make this a very challenging task. However, defining the limits of normal variation for specific epigenetic changes of relevance would be a sensible and pragmatic starting point.
Priority questions: What is the ‘normal’ epigenome? What are the real short- and/or long-term consequence(s) of environmentally induced epigenetic change(s)?
Implications for regulatory toxicology and intervention
All the previous considerations are vital for effective regulatory toxicology and potential intervention. A robust, dose-dependent, causal relationship among a specific environmental exposure, an epigenetic change and an adverse public health outcome is required to classify a chemical as an epigenetic toxicant. While a similar robust, dose-dependent relationship is also crucial to facilitate the development of testing methods for hazard assessment, relevant environmental regulation and/or appropriate medical intervention, establishing causality is not necessarily essential. Epigenetic changes that act as markers of exposure and/or predictors of future toxicity, but do not in themselves directly induce an adverse effect, may also be useful in risk assessment and intervention. As yet there is no precedent for epigenetically mediated toxicity in chemical regulation. However, environmental exposures during critical windows of human development and subsequent non-genotoxic/epigenetic changes that may lead to later life adverse health outcomes (such as obesity, diabetes and cancer) across multiple generations continues to be of high concern for all the Organization for Economic Co-operation and Development (OECD) member countries, the International Agency on Research for Cancer (IARC) and the European Commission (EFSA Citation2013; Greally & Jacobs Citation2013; Herceg et al. Citation2013; OECD Citation2014). Epigenetics thus offers an opportunity for improved chemical regulation and public health protection. For example, epigenetic changes have the potential to provide (1) more sensitive/earlier end points of toxicity within existing regulatory test guidelines (TGs), (2) novel markers of exposure/predictors of future toxicity for the development of in vitro chemical screening assays and (3) novel end points of toxicities for which we are currently rarely able to conduct definitive regulatory testing due to animal welfare, cost and time considerations (such as non-genotoxic carcinogens, multigenerational toxicity and chronic low-dose exposures that impact health in later life). As such, epigenetics is high on the agenda of the OECD expert working group on an integrated approach to testing and assessment (IATA) for non-genotoxic carcinogens (NGTxC) and the OECD endocrine disrupters testing and assessment advisory group (EDTA AG) (Greally & Jacobs Citation2013; Jacobs et al. Citation2015). The interest of OECD member countries in epigenetics within the former expert group is particularly strong, where epigenetic mechanisms are playing a pivotal role in the development of tools for improved chemical safety assessment (Jacobs et al. Citation2015). Thus, the incorporation of epigenetics within chemical regulation could ultimately reduce animal usage and the time and cost of chemical research and development, while simultaneously improving the protection of public health worldwide.
The current body of evidence for environmentally induced epigenetic toxicity is predominantly a collection of human epidemiological data and exploratory in vivo high (often single) dose range studies, performed, not for regulatory purposes, but to investigate the theoretical potential and putative mechanisms of epigenetic toxicity in biological systems. Nevertheless, such studies have helped to identify putative markers and assays that may be useful for biomonitoring such exposures and toxicities, and testing the epigenetic toxicity potential of environmental exposures, respectively. Those that may be of particular interest for development into regulatory strategies/procedures are presented in and . A number of reporter mouse models, including the Agouti Avy, CabpIAP and AuxinFu, have also been used to investigate environmentally induced epigenetic changes in in vivo exploratory research (Dolinoy et al. Citation2007; Waterland et al. Citation2006; Rosenfeld Citation2010). The Avy model in particular has been put forward as a potential epigenetic toxicological tool (Dolinoy Citation2008; Rosenfeld Citation2010; Faulk et al. Citation2013). However, the suitability of the Avy mouse as a true reporter model has since been questioned following both lack of reproducibility (Rosenfeld et al. Citation2013), and the suggestion that it may possess a “thrifty” genotype, naturally predisposing offspring to a metabolic syndrome when food is plentiful, thus confounding results following environmental exposures (Greally & Jacobs Citation2013). The AxinFu mouse may, therefore, represent a more promising reporter model in the development of regulatory toxicological assays for potential environmentally induced epigenetic toxicity.
In addition, current OECD human health-related TGs have the potential for adaptation to incorporate adverse epigenetic endpoints (Greally & Jacobs Citation2013). Inclusion of such endpoints as non-compulsory measurements would (1) encourage better use of existing studies by adding a valuable non-genotoxic/epigenetic component that is not currently included, (2) provide an extensive sample resource and (3) initiate the collection of epigenetic data in a regulatory context. This would begin to address regulatory concerns and help to develop formal epigenetically relevant toxicity TGs. Such an approach would also respect the 3Rs. An overview of the relevant TGs that could be explored for adaptation (updated from Greally & Jacobs Citation2013), is provided in . These include not only the in vitro endocrine TGs and in vivo reproductive and developmental TGs (Greally & Jacobs Citation2013) but also a newly revised genotoxicity TG (TG 483) where spermatogonal samples could be further utilized for RNA analyzes (Linschooten et al. Citation2009; Marczylo et al. Citation2012; Metzler-Guillemain et al. Citation2015). The potential for such adaptations is already acknowledged at the OECD, but more applied development of key markers and subsequent validation work is needed to formally incorporate epigenetic endpoints into TGs. Examples of such proposed adaptations are provided within the OECD Endocrine Disruptor Conceptual Framework (Supplementary Table 5; OECD Citation2012). Similar discussions on the incorporation of epigenetic toxicity into the regulatory framework are also a current focus of industry supported workshops (van Ravenzwaay et al. Citation2014; ECETOC Citation2016; Miousse et al. Citation2015).
Table 3. Current OECD human health related TGs that have the potential for adaptation to include epigenetic endpoints.
When considering temporal aspects to reproductive, developmental and endocrine disrupting effects, the TGs currently in use may not incorporate either the most relevant life stage(s), the most sensitive endpoints for epigenetic perturbation and/or sufficient duration of observation to detect later-onset effects. Thus, it is likely that the full spectrum of potential effects will not be identified. While some tests in the OECD Conceptual Framework do cover exposure during critical periods of development in utero, to date, they may not address effects that might be induced by exposure during neonatal or pubertal development, and/or emerge during later life stages.
For regulatory purposes, there are major limitations to simply extending lifetime rodent studies beyond the standard 2 years to examine later-onset epigenetic adverse outcomes, including financial, ethical and scientific considerations. In addition to the increased costs and animal welfare issues associated with housing and monitoring up to the natural death of the animals, there is also the confounding factor of increased background pathology with age. Infectious pathologies and increased incidences of certain tumor types, such as pituitary tumors or lymphomas/leukaemias, are known to occur with increasing age, and the high spontaneous incidences of leukemias/lymphomas and mammary gland tumors in laboratory animals from inbred colonies are known to vary considerably between studies (Innes et al. Citation1967; Greaves Citation2000). Thus, an overview of non-neoplastic, pre-neoplastic, neoplastic and benign pathologies within each model system is essential for the accurate interpretation of life-long rodent studies. Such overviews are, however, rarely included in the relevant literature associated with extended lifetime studies. Consequently, these studies are rarely considered acceptable to many regulatory authorities and are not specifically requested for ethical reasons, the numbers of animals needed to ensure appropriate statistical power would be both unworkable, and unethical. There are other more time and financially economical ways in which lifetime, multigenerational and/or transgenerational toxicity could be assessed, for example using zebrafish models, both respecting the 3Rs and satisfying sufficient statistical power. Already widely used in the pharmaceutical industry for developmental toxicity screening, zebrafish models appear to be a promising alternative to mice. The main epigenetic pathways, including histone modifications, DNA methylation and sncRNAs, are also conserved in zebrafish (Mishima Citation2012; Kamstra et al. Citation2015; Williams et al. Citation2014).
Furthermore, it is also important to consider the broader repercussions of regulatory toxicology. Of course, it is vital that any environmental toxicant is carefully regulated or even banned from use. However, the situation is often not as straightforward as this. For example, chemicals can have far-reaching benefits to society, from agriculture to medicine and food/product safety to the development of new technologies. Regulations are frequently a fine balance between maximizing the ‘modern life’ benefits of an environmental chemical while minimizing any potential adverse effects. This is where dose/level of exposure and relevance to humans is crucial, but considerations of alternative replacements are also important. Do we run the risk of banning a chemical that does not demonstrate any epigenetic toxicity at the levels found within the environment or human populations only to replace it with an alternative whose characteristics and potential effects we know even less about? Surely, it is better for both society and public health to fully assess the potential epigenetic effects of environmental exposures and introduce regulations and TGs based on relevant data and proven mechanisms of toxicity.
Conclusions
Despite the fact that the seminal work on environmentally induced epigenetic toxicity was published over a decade ago, this area of research largely remains in its infancy, striving to elucidate putative mechanisms of epigenetic toxicity and identify mechanistic associations in human cohorts. This might well be expected since the fundamental idea that environmental factors can induce phenotypic changes across multiple generations challenges the central dogma of genetic inheritance, moving into what was a controversial area of genetics, but is now better accepted and established, Lamarckism. While many hundreds of studies have investigated environmentally induced epigenetic toxicity, relatively few (47 to-date) have demonstrated a mechanistic association among specific environmental exposures, epigenetic changes and adverse phenotypes. Even fewer (18 of the 47 to-date) further established causality, all of which were studies in rodent models. Moreover, the majority of these 47 studies represent exploratory in vivo high (often single) dose range experiments, many orders of magnitude above likely human exposures. Nevertheless, they do set a precedent for the existence of environmentally induced epigenetic toxicity, and provide tools for linking environmental exposures to adverse health outcomes. Thus, although the current literature remains incomplete regarding specific mechanisms of epigenetically mediated environmentally induced toxicity in humans at doses relevant to human exposures, there is sufficient information to perform retrospective epigenetic analysis of existing regulatory studies and identify future research needs. For example, epigenetic mechanisms and markers identified within the 47 studies highlighted in this review could be further examined and potentially validated in existing regulatory studies. Improved human exposure data could be collected and used to facilitate the use of more relevant doses, routes of administration, metabolites and mixtures. Guidelines could be developed for improved molecular (in vivo, in vitro and epidemiological) and bioinformatic study designs that incorporate suitable new technologies (such as omics platforms) and appropriate inhibitors/treatments/cessations (for exposure/change/effect validation) to definitively identify detailed epigenetic mechanisms that link, or act as markers of, environmental exposures and adverse health outcomes. Collaboration between scientists from academia, industry, and governmental and regulatory bodies will promote further research within a regulatory context, and drive the development and implementation of epigenetically relevant integrated testing strategies or policies for the continued protection of public health.
Supplementary_Material_CRT_Revisions.docx
Download MS Word (456.5 KB)Acknowledgements
The authors gratefully acknowledge the comments offered by three reviewers selected by the Editor and anonymous to the authors. Their comments were very helpful in revising the paper.
Declaration of interest
The authors’ affiliation is as shown on the cover page. This work was funded by the National Institute for Health Research Health Protection Research Unit (NIHR HPRU) in Health Impact of Environmental Hazards at King’s College London, in partnership with PHE. The authors have sole responsibility for the writing and content of the paper, and the views expressed are those of the authors and do not necessarily reflect those of the NIHR, PHE, the NHS or the Department of Health.
Related Research Data
References
- Aapola U, Kawasaki K, Scott HS, Ollila J, Vihinen M, Heino M, Shintani A, Kawasaki K, Minoshima S, Krohn K, et al. 2000. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics. 65:293–298.
- ALSPAC Peer-Reviewed Publications. Available from: http://www.bristol.ac.uk/alspac/researchers/research/papers/.
- Anway MD, Cupp AS, Uzumcu M, Skinner MK. 2005. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 308:1466–1469.
- Baccarelli A, Bollati V. 2009. Epigenetics and environmental chemicals. Curr Opin Pediatr. 21:243–251.
- Bakulski KM, Fallin MD. 2014. Epigenetic epidemiology: promises for public health research. Environ Mol Mutagen. 55:171–183.
- Barker DJ. 2007. The origins of the developmental origins theory. J Intern Med. 261:412–417.
- Bartolomei MS, Ferguson-Smith AC. 2011. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 3:a002592.
- Barton SC, Surani MA, Norris ML. 1984. Role of paternal and maternal genomes in mouse development. Nature. 311:374–376.
- Baselga-Escudero L, Pascual-Serrano A, Ribas-Latre A, Casanova E, Salvado MJ, Arola L, Arola-Arnal A, Blade C. 2015. Long-term supplementation with a low dose of proanthocyanidins normalized liver miR-33a and miR-122 levels in high-fat diet-induced obese rats. Nutr Res (New York, NY). 35:337–345.
- Beaujean N. 2014. Histone post-translational modifications in preimplantation mouse embryos and their role in nuclear architecture. Mol Reprod Dev. 81:100–112.
- Begum G, Davies A, Stevens A, Oliver M, Jaquiery A, Challis J, Harding J, Bloomfield F, White A. 2013. Maternal undernutrition programs tissue-specific epigenetic changes in the glucocorticoid receptor in adult offspring. Endocrinology. 154:4560–4569.
- Bestor TH, Ingram VM. 1983. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci USA. 80:5559–5563.
- Bhaumik SR, Smith E, Shilatifard A. 2007. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 14:1008–1016.
- Bielawski DM, Zaher FM, Svinarich DM, Abel EL. 2002. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcohol Clin Exp Res. 26:347–351.
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 40:91–99.
- Bird AP. 1980. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8:1499–1504.
- Bird AP, Taggart MH. 1980. Variable patterns of total DNA and rDNA methylation in animals. Nucleic Acids Res. 8:1485–1497.
- Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, Molloy L, Ness A, Ring S, Davey Smith G. 2013. Cohort profile: the 'children of the 90s' – the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 42:111–127.
- Buscariollo DL, Fang X, Greenwood V, Xue H, Rivkees SA, Wendler CC. 2014. Embryonic caffeine exposure acts via A1 adenosine receptors to alter adult cardiac function and DNA methylation in mice. PLoS One. 9:e87547.
- Carey N. 2011. The epigenetics revolution: how modern biology is rewriting our understanding of genetics, disease and inheritance. London: Icon Books Ltd.
- Carlin J, George R, Reyes TM. 2013. Methyl donor supplementation blocks the adverse effects of maternal high fat diet on offspring physiology. PLoS One. 8:e63549. Epub 2013/05/10.
- Carrell DT. 2012. Epigenetics of the male gamete. Fertil Steril. 97:267–274.
- Chao HH, Zhang XF, Chen B, Pan B, Zhang LJ, Li L, Sun XF, Shi QH, Shen W. 2012. Bisphenol A exposure modifies methylation of imprinted genes in mouse oocytes via the estrogen receptor signaling pathway. Histochem Cell Biol. 137:249–259.
- Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. 2004. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis. 25:1779–1786.
- Chervona Y, Costa M. 2012. The control of histone methylation and gene expression by oxidative stress, hypoxia, and metals. Free Radic Biol Med. 53:1041–1047.
- Chiou YH, Liou SH, Wong RH, Chen CY, Lee H. 2015. Nickel may contribute to EGFR mutation and synergistically promotes tumor invasion in EGFR-mutated lung cancer via nickel-induced microRNA-21 expression. Toxicol Lett. 237:46–54.
- Choi SW, Stickel F, Baik HW, Kim YI, Seitz HK, Mason JB. 1999. Chronic alcohol consumption induces genomic but not p53-specific DNA hypomethylation in rat colon. J Nutr. 129:1945–1950.
- Cook MS, Blelloch R. 2013. Small RNAs in germline development. Curr Top Dev Biol. 102:159–205.
- Darwin CR. 1859. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. 1st ed. London: John Murray.
- Darwin CR. 1868. The variation of animals and plants under domestication. 1st ed. London: John Murray.
- Dean W. 2014. DNA methylation and demethylation: a pathway to gametogenesis and development. Mol Reprod Dev. 81:113–125.
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev. 25:1010–1022.
- Dolinoy DC. 2008. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev. 66:S7–S11.
- Dolinoy DC, Huang D, Jirtle RL. 2007. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci USA. 104:13056–13061.
- ECETOC. 2015. The role of epigenetics in reproductive toxicology. Available from: http://www.ecetoc.org/eventsmanager/62/258/The-Role-of-Epigenetics-in-Reproductive-Toxicity/.
- ECETOC. 2016. Noncoding RNAs and risk assessment science.
- EFSA. 2013. Scientific opinion on the hazard assessment of endocrine disruptors: scientific criteria for identification of endocrine disruptors and appropriateness of existing test methods for assessing effects mediated by these substances on human health and the environment. EFSA J. 11:3132–3216.
- Faulk C, Barks A, Dolinoy DC. 2013. Phylogenetic and DNA methylation analysis reveal novel regions of variable methylation in the mouse IAP class of transposons. BMC Genomics. 14:48.
- Foley TP Jr, Limanova Z, Potlukova E. 2015. Medical consequences of Chernobyl with focus on the endocrine system: Part 1. Casopis Lekaru Ceskych. 154:227–231.
- Galupa R, Heard E. 2015. X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev. 31:57–66.
- Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, Farinelli L, Miska E, Mansuy IM. 2014. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci. 17:667–669.
- Garcia-Lopez J, Alonso L, Cardenas DB, Artaza-Alvarez H, Hourcade Jde D, Martinez S, Brieno-Enriquez MA, Del Mazo J. 2015. Diversity and functional convergence of small noncoding RNAs in male germ cell differentiation and fertilization. RNA. 21:946–962.
- Gilbert SF. 2014. Developmental biology. 10th ed. Sunderland, MA, USA: Sinauer Associates, Inc.
- Golding J. 2010. Republished paper: determinants of child health and development: the contribution of ALSPAC – a personal view of the birth cohort study. Postgrad Med J. 86:387–390.
- Gou LT, Dai P, Yang JH, Xue Y, Hu YP, Zhou Y, Kang JY, Wang X, Li H, Hua MM, et al. 2014. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res. 24:680–700.
- Govindarajan B, Klafter R, Miller MS, Mansur C, Mizesko M, Bai X, LaMontagne K Jr, Arbiser JL. 2002. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol Med (Cambridge, Mass). 8:1–8.
- Gowher H, Liebert K, Hermann A, Xu G, Jeltsch A. 2005. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem. 280:13341–13348.
- Grandjean V, Gounon P, Wagner N, Martin L, Wagner KD, Bernex F, Cuzin F, Rassoulzadegan M. 2009. The miR-124-Sox9 paramutation: RNA-mediated epigenetic control of embryonic and adult growth. Development. 136:3647–3655.
- Gray LE Jr, Furr J. 2008. Vinclozolin treatment induces reproductive malformations and infertility in male rats when administered during sexual but not gonadal differentiation; however, the effects are not transmitted to the subsequent generations. Biol Reprod. 78:227.
- Greally JM, Jacobs MN. 2013. In vitro and in vivo testing methods of epigenomic endpoints for evaluating endocrine disruptors. Altex. 30:445–471.
- Greathouse KL, Bredfeldt T, Everitt JI, Lin K, Berry T, Kannan K, Mittelstadt ML, Ho SM, Walker CL. 2012. Environmental estrogens differentially engage the histone methyltransferase EZH2 to increase risk of uterine tumorigenesis. Mol Cancer Res: MCR. 10:546–557.
- Greaves P. 2000. Histopathology of preclinical toxicity studies: interpretation and relevance in drug safety evaluation. 2nd ed. New York: Elsevier Science.
- Grote P, Herrmann BG. 2015. Long noncoding RNAs in organogenesis: making the difference. Trends Genet. 31:329–335.
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA. 2013. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 339:448–452.
- Hackett JA, Surani MA. 2013. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond. B, Biol Sci. 368:20110328.
- Halappanavar S, Nikota J, Wu D, Williams A, Yauk CL, Stampfli M. 2013. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J Immunol. 190:3679–3686.
- Hale BJ, Yang CX, Ross JW. 2014. Small RNA regulation of reproductive function. Mol Reprod Dev. 81:148–159.
- Hamm CA, Costa FF. 2015. Epigenomes as therapeutic targets. Pharmacol Ther. 151:72–86.
- Hatanaka Y, Inoue K, Oikawa M, Kamimura S, Ogonuki N, Kodama EN, Ohkawa Y, Tsukada Y, Ogura A. 2015. Histone chaperone CAF-1 mediates repressive histone modifications to protect preimplantation mouse embryos from endogenous retrotransposons. Proc Natl Acad Sci USA. 112:14641–14646.
- Herceg Z, Lambert MP, van Veldhoven K, Demetriou C, Vineis P, Smith MT, Straif K, Wild CP. 2013. Towards incorporating epigenetic mechanisms into carcinogen identification and evaluation. Carcinogenesis. 34:1955–1967.
- Hogg K, Western PS. 2015. Refurbishing the germline epigenome: out with the old, in with the new. Semin Cell Dev Biol. 45:104–113.
- Hou L, Zhang X, Wang D, Baccarelli A. 2012. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 41:79–105.
- Hughes LA, van den Brandt PA, de Bruine AP, Wouters KA, Hulsmans S, Spiertz A, Goldbohm RA, de Goeij AF, Herman JG, Weijenberg MP, et al. 2009. Early life exposure to famine and colorectal cancer risk: a role for epigenetic mechanisms. PLoS One. 4:e7951.
- Ignacio C, Mooney SM, Middleton FA. 2014. Effects of acute prenatal exposure to ethanol on microRNA expression are ameliorated by social enrichment. Front Pediatr. 2:00103. DOI: 10.3389/fped.2014.00103.
- Inawaka K, Kawabe M, Takahashi S, Doi Y, Tomigahara Y, Tarui H, Abe J, Kawamura S, Shirai T. 2009. Maternal exposure to anti-androgenic compounds, vinclozolin, flutamide and procymidone, has no effects on spermatogenesis and DNA methylation in male rats of subsequent generations. Toxicol Appl Pharmacol. 237:178–187.
- Innes JRM, Garner FM, Stokey JL. 1967. Respiratory disease in rats. In: Cotchin E, Roe FJC, editors. Pathology of laboratory rats and mice. 2nd ed. Oxford and Edinburgh: Blackwell Scientific. p. 229–257.
- Issa JP, Baylin SB, Belinsky SA. 1996. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res. 56:3655–3658.
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 333:1300–1303.
- Ivey KN, Srivastava D. 2015. microRNAs as developmental regulators. Cold Spring Harb Perspect Biol. 7:a008144.
- Izzotti A, Balansky R, D'Agostini F, Longobardi M, Cartiglia C, Micale RT, La Maestra S, Camoirano A, Ganchev G, Iltcheva M, et al. 2014. Modulation by metformin of molecular and histopathological alterations in the lung of cigarette smoke-exposed mice. Cancer Med. 3:719–730.
- Jacobs M, Louekari K, Colacci A, Luijten M, Hakkert B, Paparella M, Vasseur P. 2015. The international regulatory need for tests and information to develop an Integrated Approach to Testing and Assessment (IATA) of non-genotoxic carcinogens. Toxicol Lett. 238:S123.
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science. 293:1074–1080.
- Jiang Y, Xia W, Yang J, Zhu Y, Chang H, Liu J, Huo W, Xu B, Chen X, Li Y, et al. 2015. BPA-induced DNA hypermethylation of the master mitochondrial genePGC-1α contributes to cardiomyopathy in male rats. Toxicology. 329:21–31.
- Jin H, Chen JX, Wang H, Lu G, Liu A, Li G, Tu S, Lin Y, Yang CS. 2015. NNK-induced DNA methyltransferase 1 in lung tumorigenesis in A/J mice and inhibitory effects of (-)-epigallocatechin-3-gallate. Nutr Cancer. 67:167–176.
- Kabir ER, Rahman MS, Rahman I. 2015. A review on endocrine disruptors and their possible impacts on human health. Environ Toxicol Pharmacol. 40:241–258.
- Kamiya K, Ozasa K, Akiba S, Niwa O, Kodama K, Takamura N, Zaharieva EK, Kimura Y, Wakeford R. 2015. Long-term effects of radiation exposure on health. Lancet (London, England). 386:469–478.
- Kamstra JH, Alestrom P, Kooter JM, Legler J. 2015. Zebrafish as a model to study the role of DNA methylation in environmental toxicology. Environ Sci Pollut Res. 22:16262–16276.
- Kanduri C. 2016. Long noncoding RNAs: lessons from genomic imprinting. Biochim Biophys Acta. 1859:102–111.
- Kassie F, Kalscheuer S, Matise I, Ma L, Melkamu T, Upadhyaya P, Hecht SS. 2010. Inhibition of vinyl carbamate-induced pulmonary adenocarcinoma by indole-3-carbinol and myo-inositol in A/J mice. Carcinogenesis. 31:239–245.
- Khare SP, Habib F, Sharma R, Gadewal N, Gupta S, Galande S. 2012. HIstome-a relational knowledgebase of human histone proteins and histone modifying enzymes. Nucleic Acids Res. 40:D337–D342.
- Kiani J, Grandjean V, Liebers R, Tuorto F, Ghanbarian H, Lyko F, Cuzin F, Rassoulzadegan M. 2013. RNA-mediated epigenetic heredity requires the cytosine methyltransferase Dnmt2. PLoS Genet. 9:e1003498.
- Kilcoyne KR, Smith LB, Atanassova N, Macpherson S, McKinnell C, van den Driesche S, Jobling MS, Chambers TJ, De Gendt K, Verhoeven G, et al. 2014. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci USA. 111:E1924–E1932.
- Kornberg RD, Lorch Y. 1999. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 98:285–294.
- Kouzarides T. 2007. Chromatin modifications and their function. Cell. 128:693–705.
- Kristensen DG, Skakkebaek NE, Rajpert-De Meyts E, Almstrup K. 2013. Epigenetic features of testicular germ cell tumours in relation to epigenetic characteristics of foetal germ cells. Int J Dev Biol. 57:309–317.
- Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Perera FP, Champagne FA. 2015. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci USA. 112:6807–6813.
- Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW, et al. 2008. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 22:908–917.
- Laird DJ. 2013. Humans put their eggs in more than one basket. Nat Cell Biol. 15:13–15.
- Lei H, Oh SP, Okano M, Juttermann R, Goss KA, Jaenisch R, Li E. 1996. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 122:3195–3205.
- Leitch HG, Tang WW, Surani MA. 2013. Primordial germ-cell development and epigenetic reprogramming in mammals. Curr Top Dev Biol. 104:149–187.
- Leseva M, Knowles BB, Messerschmidt DM, Solter D. 2016. Erase-maintain-establish: natural reprogramming of the mammalian epigenome. Cold Spring Harb Symp Quant Biol. [Epub ahead of print]. DOI: 10.1101/sqb.2015.80.027441.
- Leu YW, Chu PY, Chen CM, Yeh KT, Liu YM, Lee YH, Kuo ST, Hsiao SH. 2014. Early life ethanol exposure causes long-lasting disturbances in rat mesenchymal stem cells via epigenetic modifications. Biochem Biophys Res Commun. 453:338–344.
- Li E, Bestor TH, Jaenisch R. 1992. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 69:915–926.
- Li F, Xiao Y, Huang F, Deng W, Zhao H, Shi X, Wang S, Yu X, Zhang L, Han Z, et al. 2015. Spatiotemporal-specific lncRNAs in the brain, colon, liver and lung of macaque during development. Mol Biosyst. 11:3253–3263.
- Li G, Wang H, Liu AB, Cheung C, Reuhl KR, Bosland MC, Yang CS. 2012a. Dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-induced prostate carcinogenesis in CYP1A-humanized mice. Cancer Prev Res (Philadelphia, Pa). 5:963–972.
- Li Y, Xiao D, Dasgupta C, Xiong F, Tong W, Yang S, Zhang L. 2012b. Perinatal nicotine exposure increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: role of angiotensin II receptors. Stroke. 43:2483–2490.
- Li Y, Xiao D, Yang S, Zhang L. 2013. Promoter methylation represses AT2R gene and increases brain hypoxic-ischemic injury in neonatal rats. Neurobiol Dis. 60:32–38.
- Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE, Zhang S, Mac Laughlin SM, Kleemann DO, Walker SK, et al. 2014a. Impact of embryo number and maternal undernutrition around the time of conception on insulin signaling and gluconeogenic factors and microRNAs in the liver of fetal sheep. Am J Physiol Endocrinol Metab. 306:E1013–E1024.
- Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE, Zhang S, Maclaughlin SM, Kleemann DO, Walker SK, et al. 2014b. Periconceptional undernutrition programs changes in insulin-signaling molecules and microRNAs in skeletal muscle in singleton and twin fetal sheep. Biol Reprod. 90:5. DOI: 10.1095/biolreprod.113.109751.
- Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF, Wang YC. 2010. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 120:521–532.
- Linschooten JO, Van Schooten FJ, Baumgartner A, Cemeli E, Van Delft J, Anderson D, Godschalk RW. 2009. Use of spermatozoal mRNA profiles to study gene-environment interactions in human germ cells. Mutat Res. 667:m76.
- Liu W, Niu Z, Li Q, Pang RT, Chiu PC, Yeung WS. 2016. MicroRNA and embryo implantation. Am J Reprod Immunol. 75:263–271.
- Liu WM, Pang RT, Chiu PC, Wong BP, Lao K, Lee KF, Yeung WS. 2012. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci U.S.A. 109:490–494.
- Luk AC, Chan WY, Rennert OM, Lee TL. 2014. Long noncoding RNAs in spermatogenesis: insights from recent high-throughput transcriptome studies. Reproduction. 147:R131–R141.
- Lumey LH, Stein AD, Susser E. 2011. Prenatal famine and adult health. Annu Rev Public Health. 32:237–262.
- Marcho C, Cui W, Mager J. 2015. Epigenetic dynamics during preimplantation development. Reproduction. 150:R109–R120.
- Marczylo EL, Amoako AA, Konje JC, Gant TW, Marczylo TH. 2012. Smoking induces differential miRNA expression in human spermatozoa: a potential transgenerational epigenetic concern? Epigenetics. 7:432–439.
- Martinez-Arguelles DB, Culty M, Zirkin BR, Papadopoulos V. 2009. In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology. 150:5575–5585.
- Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. 2004. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 31:633–642.
- Mattick JS. 2007. A new paradigm for developmental biology. J Exp Biol. 210:1526–1547.
- McGrath J, Solter D. 1984. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 37:179–183.
- Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. 2010. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 31:252–258.
- Messerschmidt DM. 2016. A twist in zygotic reprogramming. Nat Cell Biol. 18:139–140.
- Messerschmidt DM, Knowles BB, Solter D. 2014. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 28:812–828.
- Metzler-Guillemain C, Victorero G, Lepoivre C, Bergon A, Yammine M, Perrin J, Sari-Minodier I, Boulanger N, Rihet P, Nguyen C. 2015. Sperm mRNAs and microRNAs as candidate markers for the impact of toxicants on human spermatogenesis: an application to tobacco smoking. Syst Biol Reprod Med. 61:139–149.
- Middleton FA, Varlinskaya EI, Mooney SM. 2012. Molecular substrates of social avoidance seen following prenatal ethanol exposure and its reversal by social enrichment. Dev Neurosci. 34:115–128.
- Miller D, Brinkworth M, Iles D. 2010. Paternal DNA packaging in spermatozoa: more than the sum of its parts? DNA, histones, protamines and epigenetics. Reproduction. 139:287–301.
- Miousse IR, Currie R, Datta K, Ellinger-Ziegelbauer H, French JE, Harrill AH, Koturbash I, Lawton M, Mann D, Meehan RR, et al. 2015. Importance of investigating epigenetic alterations for industry and regulators: an appraisal of current efforts by the Health and Environmental Sciences Institute. Toxicology. 335:11–19.
- Mishima Y. 2012. Widespread roles of microRNAs during zebrafish development and beyond. Dev Growth Differ. 54:55–65.
- Motorin Y, Lyko F, Helm M. 2010. 5-Methylcytosine in RNA: detection, enzymatic formation and biological functions. Nucleic Acids Res. 38:1415–1430.
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, et al. 2015. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21:392–402.
- Mukherjee A, Koli S, Reddy KV. 2014. Regulatory non-coding transcripts in spermatogenesis: shedding light on 'dark matter'. Andrology. 2:360–369.
- Nishihara R, Morikawa T, Kuchiba A, Lochhead P, Yamauchi M, Liao X, Imamura Y, Nosho K, Shima K, Kawachi I, et al. 2013. A prospective study of duration of smoking cessation and colorectal cancer risk by epigenetics-related tumor classification. Am J Epidemiol. 178:84–100.
- Noble D. 2006. The music of life biology beyond genes. New York: Oxford University Press.
- O'Doherty AM, McGettigan PA. 2014. Epigenetic processes in the male germline. Reprod Fertil Dev. 27:725–738.
- OECD. 2012. Guidance document on standardised test guidelines for evaluating chemicals for endocrine disruption. France: OECD Publishing.
- OECD. 2014. Detailed review paper on the state of the science on novel in vitro and in vivo screening and testing methods and endpoints for evaluating endocrine disruptors. France: OECD Publishing.
- Ohnishi Y, Totoki Y, Toyoda A, Watanabe T, Yamamoto Y, Tokunaga K, Sakaki Y, Sasaki H, Hohjoh H. 2010. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res. 38:5141–5151.
- Okano M, Bell DW, Haber DA, Li E. 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 99:247–257.
- Okano M, Xie S, Li E. 1998. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 19:219–220.
- Onishchenko N, Karpova N, Sabri F, Castren E, Ceccatelli S. 2008. Long-lasting depression-like behavior and epigenetic changes of BDNF gene expression induced by perinatal exposure to methylmercury. J Neurochem. 106:1378–1387.
- Ostrow KL, Michailidi C, Guerrero-Preston R, Hoque MO, Greenberg A, Rom W, Sidransky D. 2013. Cigarette smoke induces methylation of the tumor suppressor gene NISCH. Epigenetics. 8:383–388.
- Padilla PA, Garcia AM, Ladage ML, Toni LS. 2014. Caenorhabditis elegans: an old genetic model can learn new epigenetic tricks. Integr Comp Biol. 54:52–60.
- Pandey M, Sahay S, Tiwari P, Upadhyay DS, Sultana S, Gupta KP. 2014a. Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: potential targets for cancer control. Toxicol Appl Pharmacol. 280:296–304.
- Pandey M, Sultana S, Gupta KP. 2014b. Involvement of epigenetics and microRNA-29b in the urethane induced inception and establishment of mouse lung tumors. Exp Mol Pathol. 96:61–70.
- Pantano L, Jodar M, Bak M, Ballesca JL, Tommerup N, Oliva R, Vavouri T. 2015. The small RNA content of human sperm reveals pseudogene-derived piRNAs complementary to protein-coding genes. RNA. 21:1085–1095.
- Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF. 2015. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: preventive effects of resveratrol. Mol Carcinog. 54:261–269.
- Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C. 2011. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun. 25:S80–S91.
- Paul S, Banerjee N, Chatterjee A, Sau TJ, Das JK, Mishra PK, Chakrabarti P, Bandyopadhyay A, Giri AK. 2014. Arsenic-induced promoter hypomethylation and over-expression of ERCC2 reduces DNA repair capacity in humans by non-disjunction of the ERCC2-Cdk7 complex. Metallom: Integrated Biometal Sci. 6:864–873.
- Pembrey M, Saffery R, Bygren LO. 2014. Human transgenerational responses to early-life experience: potential impact on development, health and biomedical research. J Med Genet. 51:563–572.
- Peng C, Zhang W, Zhao W, Zhu J, Huang X, Tian J. 2015. Alcohol-induced histone H3K9 hyperacetylation and cardiac hypertrophy are reversed by a histone acetylases inhibitor anacardic acid in developing murine hearts. Biochimie. 113:1–9.
- Pulling LC, Vuillemenot BR, Hutt JA, Devereux TR, Belinsky SA. 2004. Aberrant promoter hypermethylation of the death-associated protein kinase gene is early and frequent in murine lung tumors induced by cigarette smoke and tobacco carcinogens. Cancer Res. 64:3844–3848.
- Ramos MP, Wijetunga NA, McLellan AS, Suzuki M, Greally JM. 2015. DNA demethylation by 5-aza-2'-deoxycytidine is imprinted, targeted to euchromatin, and has limited transcriptional consequences. Epigenet Chromat. 8:11. DOI: 10.1186/s13072-015-0004-x.
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. 2006. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 441:469–474.
- Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. 2013. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 33:9003–9012.
- Rodgers AB, Morgan CP, Leu NA, Bale TL. 2015. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc Natl Acad Sci USA. 112:13699–13704.
- Rosenfeld CS. 2010. Animal models to study environmental epigenetics. Biol Reprod. 82:473–488.
- Rosenfeld CS, Sieli PT, Warzak DA, Ellersieck MR, Pennington KA, Roberts RM. 2013. Maternal exposure to bisphenol A and genistein has minimal effect on A(vy)/a offspring coat color but favors birth of agouti over nonagouti mice. Proc Natl Acad Sci USA. 110:537–542.
- Ruiz-Hernandez A, Kuo CC, Rentero-Garrido P, Tang WY, Redon J, Ordovas JM, Navas-Acien A, Tellez-Plaza M. 2015. Environmental chemicals and DNA methylation in adults: a systematic review of the epidemiologic evidence. Clin Epigenet. 7:55. DOI: 10.1186/s13148-015-0055-7.
- Rzeczkowska PA, Hou H, Wilson MD, Palmert MR. 2014. Epigenetics: a new player in the regulation of mammalian puberty. Neuroendocrinology. 99:139–155.
- Schneider S, Kaufmann W, Buesen R, van Ravenzwaay B. 2008. Vinclozolin-the lack of a transgenerational effect after oral maternal exposure during organogenesis. Reprod Toxicol. 25:352–360.
- Schneider S, Marxfeld H, Groters S, Buesen R, van Ravenzwaay B. 2013. Vinclozolin–no transgenerational inheritance of anti-androgenic effects after maternal exposure during organogenesis via the intraperitoneal route. Reprod Toxicol. 37:6–14.
- Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. 2012. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 48:849–862.
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. 2016. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 351:391–396.
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. 2013. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 22:843–851.
- Slieker RC, Roost MS, van Iperen L, Suchiman HE, Tobi EW, Carlotti F, de Koning EJ, Slagboom PE, Heijmans BT, Chuva de Sousa Lopes SM. 2015. DNA methylation landscapes of human fetal development. PLoS Genet. 11:e1005583.
- Somer RA, Thummel CS. 2014. Epigenetic inheritance of metabolic state. Curr Opin Genet Dev. 27:43–47.
- Somineni HK, Zhang X, Biagini Myers JM, Kovacic MB, Ulm A, Jurcak N, Ryan PH, Khurana Hershey GK, Ji H. 2016. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. J Allergy Clin Immunol. 137:797–805.e795.
- Song Y, Liu C, Hui Y, Srivastava K, Zhou Z, Chen J, Miller RL, Finkelman FD, Li XM. 2014. Maternal allergy increases susceptibility to offspring allergy in association with TH2-biased epigenetic alterations in a mouse model of peanut allergy. J Allergy Clin Immunol. 134:1339–1345.e7.
- Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature. 403:41–45.
- Suganuma T, Workman JL. 2011. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 80:473–499.
- Suh N, Blelloch R. 2011. Small RNAs in early mammalian development: from gametes to gastrulation. Development. 138:1653–1661.
- Sun W, Guan M, Li X. 2014. 5-Hydroxymethylcytosine-mediated DNA demethylation in stem cells and development. Stem Cells Dev. 23:923–930.
- Surani MA. 2015. Human germline: a new research frontier. Stem Cell Rep. 4:955–960.
- Susiarjo M, Xin F, Bansal A, Stefaniak M, Li C, Simmons RA, Bartolomei MS. 2015. Bisphenol a exposure disrupts metabolic health across multiple generations in the mouse. Endocrinology. 156:2049–2058.
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. 2009. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 324:930–935.
- Takeda T, Fujii M, Taura J, Ishii Y, Yamada H. 2012. Dioxin silences gonadotropin expression in perinatal pups by inducing histone deacetylases: a new insight into the mechanism for the imprinting of sexual immaturity by dioxin. J Biol Chem. 287:18440–18450.
- Tang WW, Dietmann S, Irie N, Leitch HG, Floros VI, Bradshaw CR, Hackett JA, Chinnery PF, Surani MA. 2015. A unique gene regulatory network resets the human germline epigenome for development. Cell. 161:1453–1467.
- Tao L, Ge R, Xie M, Kramer PM, Pereira MA. 1999. Effect of trichloroethylene on DNA methylation and expression of early-intermediate protooncogenes in the liver of B6C3F1 mice. J Biochem Mol Toxicol. 13:231–237.
- Tao L, Kramer PM, Ge R, Pereira MA. 1998. Effect of dichloroacetic acid and trichloroacetic acid on DNA methylation in liver and tumors of female B6C3F1 mice. Toxicol Sci. 43:139–144.
- Taylor DH, Chu ET, Spektor R, Soloway PD. 2015. Long non-coding RNA regulation of reproduction and development. Mol Reprod Dev. 82:932–956.
- Tong Z, Han C, Luo W, Li H, Luo H, Qiang M, Su T, Wu B, Liu Y, Yang X, et al. 2013. Aging-associated excess formaldehyde leads to spatial memory deficits. Sci Rep. 3:1807.
- Tong Z, Han C, Qiang M, Wang W, Lv J, Zhang S, Luo W, Li H, Luo H, Zhou J, et al. 2015. Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer's disease. Neurobiol Aging. 36:100–110.
- Trevino LS, Wang Q, Walker CL. 2015. Phosphorylation of epigenetic “‘readers, writers and erasers': implications for developmental reprogramming and the epigenetic basis for health and disease”. Prog Biophys Mol Biol. 118:8–13.
- Tryndyak VP, Marrone AK, Latendresse JR, Muskhelishvili L, Beland FA, Pogribny IP. 2016. MicroRNA changes, activation of progenitor cells and severity of liver injury in mice induced by choline and folate deficiency. J Nutr Biochem. 28:83–90.
- Turner BM. 2000. Histone acetylation and an epigenetic code. Bioessays. 22:836–845.
- Tyagi E, Zhuang Y, Agrawal R, Ying Z, Gomez-Pinilla F. 2015. Interactive actions of Bdnf methylation and cell metabolism for building neural resilience under the influence of diet. Neurobiol Dis. 73:307–318.
- van Ravenzwaay B, Kleinjans J, Vrijhof H. 2014. Editorial-epigenetics and chemical safety. Mutat Res Genet Toxicol Environ Mutagen. 764–765:1–2.
- Vucetic Z, Kimmel J, Totoki K, Hollenbeck E, Reyes TM. 2010. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology. 151:4756–4764.
- Vuillemenot BR, Pulling LC, Palmisano WA, Hutt JA, Belinsky SA. 2004. Carcinogen exposure differentially modulates RAR-beta promoter hypermethylation, an early and frequent event in mouse lung carcinogenesis. Carcinogenesis. 25:623–629.
- Wadhwa PD, Buss C, Entringer S, Swanson JM. 2009. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 27:358–368.
- Wagner KD, Wagner N, Ghanbarian H, Grandjean V, Gounon P, Cuzin F, Rassoulzadegan M. 2008. RNA induction and inheritance of epigenetic cardiac hypertrophy in the mouse. Dev Cell. 14:962–969.
- Wang B, Li Y, Tan Y, Miao X, Liu XD, Shao C, Yang XH, Turdi S, Ma LJ, Ren J, et al. 2012. Low-dose Cd induces hepatic gene hypermethylation, along with the persistent reduction of cell death and increase of cell proliferation in rats and mice. PLoS One. 7:e33853.
- Wang X, Wang H. 2013. Epigenotoxicity of environmental pollutants evaluated by a combination of DNA methylation inhibition and capillary electrophoresis-laser-induced fluorescence immunoassay. Anal Bioanal Chem. 405:2435–2442.
- Wang Y, Dasso M. 2009. SUMOylation and deSUMOylation at a glance. J Cell Sci. 122:4249–4252.
- Warner M, Mocarelli P, Brambilla P, Wesselink A, Samuels S, Signorini S, Eskenazi B. 2013. Diabetes, metabolic syndrome, and obesity in relation to serum dioxin concentrations: the Seveso women's health study. Environ Health Perspect. 121:906–911.
- Warner M, Mocarelli P, Samuels S, Needham L, Brambilla P, Eskenazi B. 2011. Dioxin exposure and cancer risk in the Seveso Women's Health Study. Environ Health Perspect. 119:1700–1705.
- Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. 2006. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 20:1732–1743.
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, Iida N, Hoki Y, Murphy PJ, Toyoda A, et al. 2011. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 332:848–852.
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, Chiba H, Kohara Y, Kono T, Nakano T, et al. 2008. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 453:539–543.
- Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG. 2006. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis. 44:401–406.
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 324:1076–1080.
- Williams TD, Mirbahai L, Chipman JK. 2014. The toxicological application of transcriptomics and epigenomics in zebrafish and other teleosts. Brief Funct Genomics. 13:157–171.
- Williams Z, Morozov P, Mihailovic A, Lin C, Puvvula PK, Juranek S, Rosenwaks Z, Tuschl T. 2015. Discovery and characterization of piRNAs in the human fetal ovary. Cell Rep. 13:854–863.
- Wong RL, Wang Q, Trevino LS, Bosland MC, Chen J, Medvedovic M, Prins GS, Kannan K, Ho SM, Walker CL. 2015. Identification of secretaglobin Scgb2a1 as a target for developmental reprogramming by BPA in the rat prostate. Epigenetics. 10:127–134.
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, et al. 2008. Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 28:3–9.
- Wu Q, Ohsako S, Ishimura R, Suzuki JS, Tohyama C. 2004. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biol Reprod. 70:1790–1797.
- Xie L, Wu M, Lin H, Liu C, Yang H, Zhan J, Sun S. 2014. An increased ratio of serum miR-21 to miR-181a levels is associated with the early pathogenic process of chronic obstructive pulmonary disease in asymptomatic heavy smokers. Mol Biosyst. 10:1072–1081.
- Yan YE, Liu L, Wang JF, Liu F, Li XH, Qin HQ, Wang H. 2014. Prenatal nicotinic exposure suppresses fetal adrenal steroidogenesis via steroidogenic factor 1 (SF-1) deacetylation. Toxicol Appl Pharmacol. 277:231–241.
- Yang P, Ma J, Zhang B, Duan H, He Z, Zeng J, Zeng X, Li D, Wang Q, Xiao Y, et al. 2012. CpG site-specific hypermethylation of p16INK4alpha in peripheral blood lymphocytes of PAH-exposed workers. Cancer Epidemiol Biomarkers Prev. 21:182–190.
- Yang SR, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, Caito S, Adenuga D, Henry R, Fromm G, et al. 2008. IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol. 38:689–698.
- Yoshioka W, Higashiyama W, Tohyama C. 2011. Involvement of microRNAs in dioxin-induced liver damage in the mouse. Toxicol Sci. 122:457–465.
- Yuan S, Schuster A, Tang C, Yu T, Ortogero N, Bao J, Zheng H, Yan W. 2016. Sperm-borne miRNAs and endo-siRNAs are important for fertilization and preimplantation embryonic development. Development. 143:635–647.
- Zeybel M, Hardy T, Wong YK, Mathers JC, Fox CR, Gackowska A, Oakley F, Burt AD, Wilson CL, Anstee QM, et al. 2012. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 18:1369–1377.
- Zhang J, Zhou Y, Ma L, Huang S, Wang R, Gao R, Wu Y, Shi H, Zhang J. 2013. The alteration of miR-222 and its target genes in nickel-induced tumor. Biol Trace Elem Res. 152:267–274.
- Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, Dai P, Huang DW, Chen R, Fu XD, et al. 2015. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res. 25:193–207.
- Zhou R, Chen F, Chang F, Bai Y, Chen L. 2013. Persistent overexpression of DNA methyltransferase 1 attenuating GABAergic inhibition in basolateral amygdala accounts for anxiety in rat offspring exposed perinatally to low-dose bisphenol A. J Psychiatr Res. 47:1535–1544.