
Abstract
Ras converting enzyme 1 (Rce1) is an integral membrane endoprotease localized to the endoplasmic reticulum that mediates the cleavage of the carboxyl-terminal three amino acids from CaaX proteins, whose members play important roles in cell signaling processes. Examples include the Ras family of small GTPases, the γ-subunit of heterotrimeric GTPases, nuclear lamins, and protein kinases and phosphatases. CaaX proteins, especially Ras, have been implicated in cancer, and understanding the post-translational modifications of CaaX proteins would provide insight into their biological function and regulation. Many proteolytic mechanisms have been proposed for Rce1, but sequence alignment, mutational studies, topology, and recent crystallographic data point to a novel mechanism involving a glutamate-activated water and an oxyanion hole. Studies using in vivo and in vitro reporters of Rce1 activity have revealed that the enzyme cleaves only prenylated substrates and the identity of the a2 amino residue in the Ca1a2X sequence is most critical for recognition, preferring Ile, Leu, or Val. Substrate mimetics can be somewhat effective inhibitors of Rce1 in vitro. Small-molecule inhibitor discovery is currently limited by the lack of structural information on a eukaryotic enzyme, but a set of 8-hydroxyquinoline derivatives has demonstrated an ability to mislocalize all three mammalian Ras isoforms, giving optimism that potent, selective inhibitors might be developed. Much remains to be discovered regarding cleavage specificity, the impact of chemical inhibition, and the potential of Rce1 as a therapeutic target, not only for cancer, but also for other diseases.
Introduction
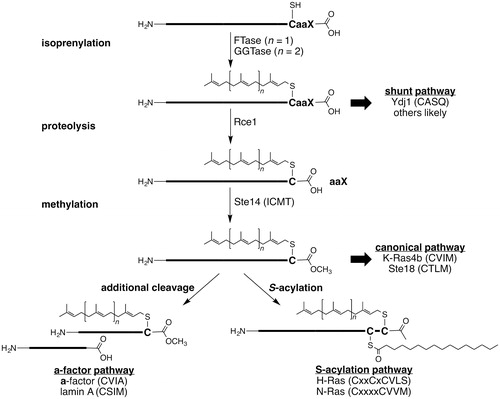
Many eukaryotic proteins are isoprenylated with either farnesyl (C15) or geranylgeranyl (C20) groups. The CaaX-type prenyl proteins are historically defined by a carboxyl-terminal CaaX motif where C is cysteine, a is an aliphatic amino acid, and X is one of several amino acid residues. Proteins containing this motif are often subject to an ordered series of post-translational modifications (PTMs): C15 or C20 isoprenylation of the cysteine, endoproteolysis by Rce1 to remove aaX, and carboxyl methylation (). Deviations from this standard pathway occur. Some proteins are isoprenylated, but not cleaved (i.e. shunt pathway), whereas other proteins are subject to additional PTMs after the canonical set of modifications (e.g. a-factor and S-acylation pathways). For the relatively few CaaX-type proteins examined in detail, disruption of the PTMs has profound effects on their function and localization. The CaaX-type proteins play an important role in cell signaling processes; their members comprise the Ras family of small GTPases, the γ-subunit of heterotrimeric GTPases, nuclear lamins, and protein kinases and phosphatases (Gao et al. Citation2009; Wang and Casey Citation2016). The CaaX proteins Ras, RhoA, RhoC, RheB, Rac proteins, CDC42, and PTP4A3 have been implicated in cancer (Winter-Vann and Casey Citation2005). Understanding the PTMs of CaaX proteins is therefore expected to provide insight into their regulation and biological function. This review focuses on the activity, mechanism, and inhibition of Ras converting enzyme (Rce1), which mediates the endoproteolytic cleavage of the carboxyl-terminal three amino acids from CaaX proteins.
Figure 1. Overview of post-translational modifications associated with CaaX proteins. Both farnesyl (C15) and geranylgeranyl (C20) can be added to CaaX proteins. There are multiple classes of isoprenylated CaaX proteins: those with motifs that resist cleavage (shunt), those with motifs that are cleaved (canonical), and those additionally modified by cleavage (a-factor) or S-acylation. Motifs shown are for indicated yeast (Ydj1p, Ste18p, a-factor) or human proteins (all others).
Clinical relevance of Rce1
CaaX proteins are abundant eukaryotic proteins with diverse biological functions. They are involved in cancer, development, aging, parasitic growth, and many other biological pathways (Wang and Casey Citation2016). Ras GTPases are some of the more prominently studied CaaX proteins because of their involvement in cancer. Because Ras function optimally requires post-translational modification, there is intense interest for developing therapeutic inhibitors for all modification steps (Silvius Citation2002; Winter-Vann and Casey Citation2005; Konstantinopoulos et al. Citation2007; Berndt et al. Citation2011; Cox et al. Citation2015; Wang and Casey Citation2016). Such agents include farnesyl and geranylgeranyl transferase inhibitors (FTIs and GGTIs), dual prenyl transferase inhibitors (DPIs), Rce1 protease inhibitors (RPIs), and carboxyl methyl transferase inhibitors (MTIs). FTIs are the most advanced of these agents and have reached clinical trials, but their utility as anti-cancer agents has not been substantiated (Gelb et al. Citation2006; Cox et al. Citation2015). The development and testing of RPIs and MTIs substantially lag that of prenyl transferase inhibitors. Rce1 is also being investigated as a potential biomarker in various types of cancer, but its utility as a disease predictor remains far from being understood (Huang et al. Citation2016; Li et al. Citation2017; Shi et al. Citation2017).
Studies investigating the importance of Rce1 in the cancer setting have largely relied on rce1−/− mouse embryonic fibroblasts (MEFs; the mouse homozygous knockout is inviable), tissue-specific mouse knockouts, and small molecule agents that interfere with Rce1 activity (Chen Citation1998, Citation1999; Bergo et al. Citation2002, Citation2004; Wahlstrom et al. Citation2007). Loss of Rce1 activity is associated with Ras mislocalization, diminished growth of Ras-transformed fibroblasts in culture and in nude mice, and hypersensitivity to FTIs. Chemical inhibition of Rce1 yields similar results. Collectively, these observations suggest that Rce1-deficiency correlates with a reduced ability to maintain the transformed state. It is unclear though whether outcomes observed with knockout approaches are directly due to defects in Ras signaling alone, or more likely, due to a broad impact of Rce1 inhibition on multiple CaaX proteins. The latter likely underlies the varied outcomes observed for tissue-specific knockout studies. Loss of Rce1 in hematopoietic tissue exacerbates K-Ras induced myeloproliferative disease (Wahlstrom et al. Citation2007). Loss of Rce1 in cardiac tissue results in high rates of lethal cardiomyopathy (Bergo et al. Citation2004). Loss of Rce1 in retina compromises photoreceptor function and is associated with retinal degeneration (Christiansen et al. Citation2011). Whether chemical-based inhibition of Rce1 will yield similar outcomes remains undetermined.
There is additional disease-related biology outside of cancer in which Rce1 is involved (Gelb et al. Citation2006). Rce1 is essential for growth of Plasmodium sp. and Trypanosoma brucei, the causative agents of malaria and African sleeping sickness, respectively (Gelb et al. Citation2003; Gillespie et al. Citation2007). Inhibitors that preferentially target parasite Rce1 over human could thus have potential clinical value in this setting (Mokry et al. Citation2009). In the case of certain bacterial infections, mammalian Rce1 regulates the modification and effectiveness of bacterial effector proteins injected into the eukaryotic host (Fueller et al. Citation2006; Price et al. Citation2010). Prokaryotic Rce1 itself appears to regulate the pathogenicity of bacterial species associated with high mortality rates, such as Staphylococcus aureus and Streptococcus pneumoniae (Frankel et al. Citation2010). Hence, the clinical importance of Rce1 may extend beyond cancer to several forms of infectious disease. Multiple developmental processes also appear to rely on Rce1. Loss of mouse Rce1 activity (i.e. rce1−/−) results in late embryonic lethality, and tissue specific knockouts result in various disease states (Kim et al. Citation1999; Bergo et al. Citation2004; Wahlstrom et al. Citation2007; Christiansen et al. Citation2011).
Regulation of Rce1
Little is known about the regulation of Rce1 activity, and it is likely to have a general house-keeping function. Consistent with the widespread tissue expression of CaaX proteins, Rce1 is expressed in multiple tissues with some variation in expression levels (Otto et al. Citation1999; Young et al. Citation2001; Uhlen et al. Citation2015; Tissue Expression of RCE1, The Human Protein Atlas Citation2017). There is evidence, however, that Rce1 levels can be regulated by the USP17 deubiquitinating enzyme, whose activity leads to proteasomal degradation of Rce1 and hence reduced protein levels (Burrows et al. Citation2009; Jaworski et al. Citation2014). In wild-type MEF cells, the presence of USP17 results in down regulation of Rce1 activity, resulting in relocalization of GFP H-Ras from the plasma membrane to the cytosol and down regulation of Ras/MEK/ERK pathway signaling. Interestingly, these studies suggest that USP17 has differential impact on distinct splice variant isoforms of Rce1 in mammals (Rce1 Iso1 and Rce1 Iso2). Rce1 Iso2 protein levels appear to be regulated by USP17, whereas Rce1 Iso1 is unaffected. Nevertheless, both isoforms of Rce1 are required to reconstitute GFP H-Ras to the plasma membrane in rce1−/− MEF cells, and USP17 on its own could not block cell proliferation in the absence of Rce1, suggesting a strong and direct link between the Rce1 and USP17 proteins. Further studies are required to understand the roles of different isoforms of Rce1 and their effect on the oncogenic Ras signaling pathway, as this would impact drug discovery efforts.
Identification and homologs of Rce1
The Ras converting enzyme (Rce1) is alternatively known as FACE-2 and Type II CAAX prenyl endopeptidase. Prior to its genetic identification, attempts to purify Rce1 using traditional biochemical approaches did not lead to its identification (Young et al. Citation2001). In retrospect, the main difficulties hindering the purification of Rce1 were its membrane-associated nature and the contaminating presence of Ste24p (FACE-1/Type I CAAX prenyl endopeptidase/Afc1), an evolutionarily unrelated protease with partially overlapping CaaX protease activity (Tam et al. Citation1998; Trueblood et al. Citation2000). The inability to biochemically purify or significantly enrich Rce1 is a complicating issue that continues to this date.
The genes encoding Rce1 and Ste24 were eventually both identified in Saccharomyces cerevisiae through an elegant genetic screen involving the a-factor mating pheromone as a reporter (Boyartchuk et al. Citation1997). Functional orthologs have subsequently been identified in other eukaryotic organisms (humans, mice, plant, parasite, worm, pathogenic fungi, etc.) (Kim et al. Citation1999; Otto et al. Citation1999; Cadiñanos, Schmidt, et al. 2003; Cadiñanos, Varela, et al. 2003; Mokry et al. Citation2009; Esher et al. Citation2016). Prokaryotic orthologs also exist, and while not expected to possess farnesyl-dependent specificity, at least one can cleave a synthetic farnesylated substrate in vitro (Pei and Grishin Citation2001; Manolaridis et al. Citation2013).
Collectively, Rce1 orthologs vary in their primary sequence identity (9–63%) and size (30–37 kDa) (Pei and Grishin Citation2001; Dore and Schmidt Citation2013). They all are predicted to be multi-span membrane proteins. Sequence divergence for transmembrane segments among homologs of any type is a common occurrence, and this may in part explain the low degree of sequence homology between Rce1 orthologs. Despite their low homology, Rce1 orthologs all have several conserved amino acids that are proposed to serve as catalytic residues: two adjacent glutamates and two separate histidines (Pei and Grishin Citation2001; Manolaridis et al. Citation2013). Other highly conserved residues include an arginine often positioned four residues away from the pair of glutamates, an aromatic residue (Phe or Tyr) often positioned four residues from the first histidine, and a asparagine/aspartate positioned near the second histidine. A cysteine near the second histidine is conserved in eukaryotic orthologs, but not in prokaryotic ones. The importance of conserved residues for the activity of eukaryotic Rce1 has been investigated for multiple eukaryotic orthologs, which are all expected to be involved in the cleavage of CaaX-type proteins (Dolence et al. Citation2000a; Plummer et al. Citation2006; Mokry et al. Citation2009). With exception of the proposed catalytic residues, the roles of other conserved residues are not yet fully understood and complicated by different observations. For example, the cysteine conserved in eukaryotic Rce1 orthologs has been reported to be essential for activity in one study but completely dispensable for activity in other studies (Dolence et al. Citation2000a; Plummer et al. Citation2006; Hildebrandt et al. Citation2013). Whether conserved residues are important for the activities of the prokaryotic orthologs remains an open question. Where examined, prokaryotic orthologs also appear to be associated with cleavage activities, including protection against antimicrobial peptides (Ellermeier and Losick Citation2006; Kjos et al. Citation2010). Rce1 has distinct, but somewhat similar sequence motifs to the PrsW family of proteases, the domain of unknown function protein DUF2324 from the Pfam family, and the 7-transmembrane-spanning APH-1 subunit of γ-secretase (Pei et al. Citation2011).
Topology and structure of Rce1
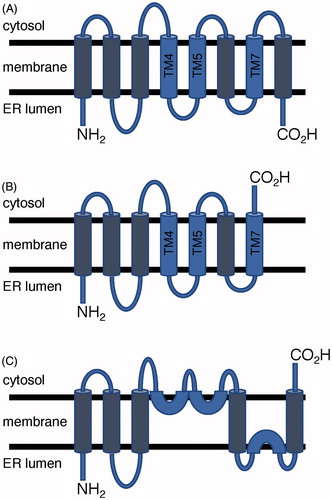
Rce1 is an integral membrane protein that is localized to the endoplasmic reticulum (ER) in eukaryotes (Schmidt et al. Citation1998; Bracha-Drori et al. Citation2008). Prokaryotic orthologs are envisaged to be located to the plasma membrane (Xiang et al. Citation2017). Hydropathy and multiple-sequence alignment across Rce1 orthologs from Homo sapiens (HsRce1), S. cerevisiae Rce1 (ScRce1), and several other species predicted seven or eight trans-membrane segments depending on species () (Hildebrandt et al. Citation2013). Topology studies carried out on ScRce1 and based on maleimide labeling of cysteines in ScRce1 and in an HA tag placed at either the N- or C-terminal end of the enzyme suggested that the N-terminus projected into the ER lumen, whereas the C-terminus was located on the cytosolic side. This orientation was further supported by an N-linked glycosylation assay. Such an orientation would require an odd number of transmembrane spans (). Maleimide labeling of natural and introduced cysteines indicated that ScRce1 might contain several reentrant helices, which do not span the bilayer, on the cytosolic face of the membrane (). The topology study relied heavily on mutants that introduced non-natural cysteines into ScRce1, which could introduce subtle changes that disrupt α-helices and helix hairpins leading to alternative topologies, misfolded protein, or both (Monne et al. Citation1999; Norholm et al. Citation2011). The maleimide reagent could also react with cysteines in large pockets that extend deeply into the membrane, which is a structural feature of Rce1 from an archaeon (vide infra).
Figure 2. Postulated topology models of Rce1. (A) Topology with 8 transmembrane helices found in MmRce1 crystal structure and predicted for HsRce1; (B) 7 transmembrane helices predicted for ScRce1; and (C) Proposed alternative topology for ScRce1 based on substituted cysteine accessibility experiments. Conserved catalytic glutamate and histidine residues are located on blue-colored helices. (see color version of this figure at www.tandfonline.com/ibmg).
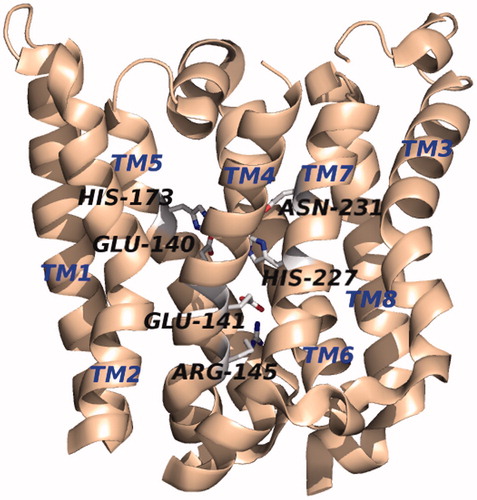
The crystal structure of an Rce1 homolog from the archaea Methanococcus maripaludis (MmRce1) was elucidated at 2.5-Å resolution in complex with a monoclonal antibody Fab fragment (PDB accession number 4CAD) () (Manolaridis et al. Citation2013). Its structure, measuring approximately 35 × 26 × 46 Å, is made up of 8 transmembrane α-helices linked by short loops plus two peripheral membrane α-helices (topology shown in ). The C- and N-termini point into the ER lumen, which is in agreement with the hydropathy predictions, but contradicts the topology data reported for ScRce1 (Hildebrandt et al. Citation2013). At its core is a conical cavity with a large volume of 1400 Å3 that encompasses the catalytic site and opens to the cytosol. Inside the cavity, the conserved residues E140, E141, H173, H227, and N231 reside within the span of the membrane, and each project their side chains into the cavity, except E141, suggesting that it might not be directly involved in catalysis. The highly-conserved residue R145 sits at the base of the cone, separating the active site from the ER lumen. A water molecule is located in the cavity approximately 10 Å from the cytosolic surface of the membrane and is bridged by E140 and H173, which are positioned opposite one another. Additionally, one side of the cavity is open to the membrane via a gap between two of the transmembrane helices (TM2 and TM4).
Figure 3. Crystal structure of MmRce1 (PDB: 4CAD). The protein structure is oriented with the cytosolic face at the top and that of the ER lumen on the bottom of the image. (see color version of this figure at www.tandfonline.com/ibmg).
Proteolytic mechanism
Early studies implicated Rce1 to be from the family of cysteine proteases based on the fact that mechanism-based cysteine protease inhibitors abolished activity and mutational studies showing that catalytic activity resulted when the only conserved cysteine among eukaryotic orthologs was changed to alanine (Ma et al. Citation1993; Chen et al. Citation1996; Dolence et al. Citation2000a). More recently, however, a study on ScRce1 found that the only conserved cysteine among eukaryotic Rce1 orthologs was not necessary for enzyme function, and a cysteine-free Rce1 mutant was fully functional in vivo (Plummer et al. Citation2006; Hildebrandt et al. Citation2013). Rce1 was also putatively assigned as a membrane-bound metalloprotease because of a conserved set of glutamates and histidines found across eukaryotic and prokaryotic orthologs (Pei and Grishin Citation2001). A mutational analysis study revealed the importance of these residues and the requirement of glutamate and histidine by ScRce1 in catalysis, suggesting either a zinc metalloenzyme or a novel proteolytic mechanism (Plummer et al. Citation2006). Indeed, several pieces of data implicate a novel proteolytic mechanism, rather than the four established canonical mechanisms of proteolysis: aspartate, cysteine, serine/threonine, and metal-based. Rce1 has little sequence similarity with other proteases and no conserved cysteine across eukaryotes and prokaryotes (Pei and Grishin Citation2001). EDTA does not affect the activities of MmRce1 or ScRce1, although excess Zn2+ inactivates ScRce1, but not MmRce1 (Plummer et al. Citation2006; Manolaridis et al. Citation2013). Phenanthroline inactivation of MmRce1 is the result of protein unfolding, and no Zn2+ was detected in MmRce1 in proton-induced X-ray emission (PIXE) spectroscopy, total reflection X-ray fluorescence (TXRF) spectroscopy, or X-ray crystallography experiments.
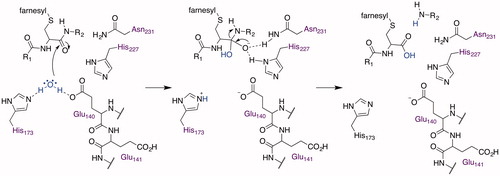
The crystal structure of MmRce1 suggested proteolysis through a novel mechanism, corroborating an earlier proposal (Plummer et al. Citation2006), involving an active site glutamate, two histidines, and an asparagine that are essential for catalytic activity () (Manolaridis et al. Citation2013). Computational docking studies, using a decapeptide containing the farnesylated canonical CaaX motif of RhoA (CLVS), suggested that the substrate adopts a β-hairpin structure to insert itself into the cavity. The farnesyl group fills the space between TM2 and TM4, which are decorated with non-polar amino acids, positioning the peptide bond between the cysteine and the first aliphatic amino acid near the water bridging H173 and E140 (). The model does not provide insight into the position of the C-terminal carboxylate and any interactions that it might have with MmRce1. The water nucleophile attacks the carbonyl of the amide of the prenylated cysteine (). H227 and N231, which form a structural element called an oxyanion hole, stabilize the resulting tetrahedral oxyanion intermediate that collapses to the carboxylate, jettisoning the amine, which is facilitated by proton transfer from H173 or E140, and completing the proteolysis. Although the proteolysis mechanism of Rce1 is novel, elements of the proposed mechanism, such as a glutamate-activated water and an oxyanion hole, are found in other membrane-associated proteases (Manolaridis et al. Citation2013): S2P (Feng et al. Citation2007), Ste24 (Pryor et al. Citation2013; Quigley et al. Citation2013), SGP (Fujinaga et al. Citation2004), and GlpG rhomboid protease (Baker et al. Citation2007; Ben-Shem et al. Citation2007; Wang and Ha Citation2007; Vinothkumar et al. Citation2010). The crystal structure model provides a foundation to gain coarse-level insight into the structure and mechanism of the eukaryotic Rce1; nevertheless, caution should be exercised with the mechanistic hypothesis because of species specific differences in the primary structure and topology of Rce1. For example, the primary structure of MmRce1 is only 13% identical to that of HsRce1 and 12% identical to ScRce1 based on Clustal Omega analysis (UnitProtKB), and the N- and C-termini of MmRce1 point into the ER lumen if adopting an MmRce1 topology (Manolaridis et al. Citation2013), or are located on opposite sides of the ER membrane if oriented like ScRce1 (Hildebrandt et al. Citation2013). Solved crystal structures of human and yeast Rce1 would contribute significantly to a better understanding of the catalytic mechanism of proteolysis.
Figure 4. Crystal structure of the MmRce1 catalytic site (PDB: 4CAD), showing the locations of the critical residues E140, E141, R145, H173, H227, and N231. Selected N–O distances between residues are given in Ångströms and marked by black dashed lines. The hydrogen bond between H227 and N231 reported by Manolaridis et al. is shown as a yellow line. (see color version of this figure at www.tandfonline.com/ibmg).
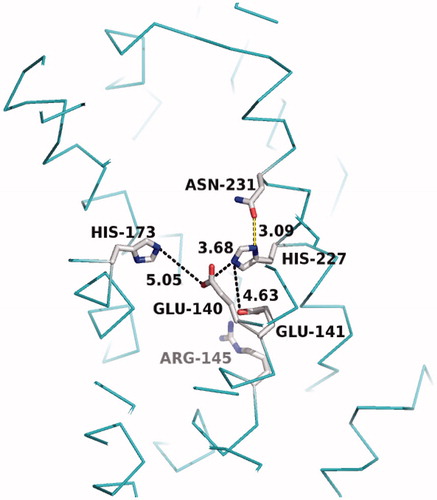
Figure 5. Proposed basic catalytic mechanism of MmRce1 proteolysis.
Reporter substrates for measuring Rce1 activity
Various reporter substrates have been developed to monitor Rce1 activity both in vivo and in vitro (). These reporters invariably take the format of an isoprenylated polypeptide with an aaX extension.
Table 1. Reporter substrates used to monitor Rce1 activity in vivo and in vitro.
In vivo reporters
Many in vivo studies of Rce1 activity have relied on the yeast a-factor mating pheromone (36-mer polypeptide) and the genetic tractability that the yeast system provides. This includes the study that ultimately uncovered the identity of the RCE1 gene (Boyartchuk et al. Citation1997). The a-factor reporter has since been used in various studies to systematically explore Rce1 and Ste24 specificity (Trueblood et al. Citation2000; Cadiñanos, Schmidt, et al. 2003; Cadiñanos, Varela, et al. 2003; Plummer et al. Citation2006; Bracha-Drori et al. Citation2008; Mokry et al. Citation2009). Through use of this reporter, there has been extensive but not exhaustive evaluation of CaaX protease specificity; collectively only ∼75 CaaX combinations have been evaluated out of the 8000 possible CaaX sequence combinations (Boyartchuk and Rine Citation1998; Trueblood et al. Citation2000; Cadiñanos et al. Citation2003b; Young et al. Citation2005; Plummer et al. Citation2006; Krishnankutty et al. Citation2009; Mokry et al. Citation2009; Hildebrandt et al. Citation2016b). The utility of the a-factor reporter is tempered by the high sensitivity of the biological assays used to detect the reporter that could potentially lead to false-positives for cleavable CaaX motifs, an inability to distinguish between defective farnesylation and defective proteolysis for certain CaaX motifs, and the evaluation of CaaX motifs outside of their natural context. As an example, the CASQ motif has been widely regarded as an example of a Ste24-specific motif, but the amount of a-factor produced in the context of CASQ is only 1–2% relative to wild-type a-factor (CVIA), and the motif is not cleaved in its natural context as part of the yeast Hsp40 chaperone Ydj1 (Hildebrandt et al. Citation2016b). Despite these limitations, the a-factor reporter system has been one of the more useful genetic systems for exploring Rce1 activity and specificity.
By comparison to a-factor, Ras-based genetic methods are not widely used to study Rce1 activity due to their qualitative nature and reliance on rather complicated genetic set ups. Nevertheless, such methods have demonstrated that the absence of Rce1 modulates the thermosensitivity of yeast (Boyartchuk et al. Citation1997). More commonly, GFP-tagging of Ras and other Ras-related GTPases is used to assess the role of Rce1 in regulating small GTPase subcellular localization (Boyartchuk et al. Citation1997; Choy et al. Citation1999; Michaelson et al. Citation2005; Manandhar et al. Citation2007; Bracha-Drori et al. Citation2008; Roberts et al. Citation2008; Mokry et al. Citation2009; Hanker et al. Citation2010; Manandhar et al. Citation2010; Gentry et al. Citation2015; Mohammed et al. Citation2016). In the yeast system, GFP-Ras2 is cytosolic in the absence of farnesylation and has diffuse membranous patterns of intracellular distribution in the absence of either Rce1p or the ICMT (Ste14p) (Boyartchuk et al. Citation1997; Manandhar et al. Citation2010); the absence of Ste24p does not impact Ras2p localization. An altered response to heat shock is observed for the rce1 deletion relative to wild-type, suggesting that mislocalization of Ras disrupts the Ras signaling circuit (Boyartchuk et al. Citation1997).
In mammalian cells, Rce1-dependent localization is similarly observed for the H-, N-, and K-Ras isoforms (Choy et al. Citation1999; Kim et al. Citation1999; Bergo et al. Citation2002; Michaelson et al. Citation2005; Mohammed et al. Citation2016). The Rheb family of small GTPases also rely on Rce1 for their proper localization to the ER and Golgi (Hanker et al. Citation2010), as do the oncogenic Ras-like small GTPases RalA and RalB to the plasma membrane (Gentry et al. Citation2015). A reliance on Rce1 for proper localization of small GTPases is, however, not generalizable. It is often unclear whether perturbing Rce1 activity also impacts other associated post-translational modifications that occur to CaaX proteins (e.g. palmitoylation of H- and N-Ras (Hancock et al. Citation1990, Citation1991)). Rho-family proteins (i.e. Cdc42, Rac1, Rac2, and RhoA) are cytosolic, and this localization is independent of Rce1 activity (Michaelson et al. Citation2001, Citation2005). The localization of non-GTPase CaaX proteins can also vary in their dependency on Rce1 activity (Christiansen et al. Citation2011). Recently, GFP-Ras reporters have been useful for examining the impact of RPIs on Ras localization (Manandhar et al. Citation2010; Mohammed et al. Citation2016). Where examined, these inhibitors pharmacologically phenocopy effects observed with Rce1 knockouts and siRNA knockdowns. A major limitation of all localization studies, whether using knockouts or inhibitor-based approaches, is the possibility that altering the cleavage state of the entire prenylome at once may result in pleiotropic effects that confound interpretation of results. Moreover, delocalization effects can be difficult to quantify, especially when the signal is distributed across multiple locations. Despite these issues, GFP-based reporters continue to have great utility for studies of Rce1 and are potentially useful for future high-content HTS in association with inhibitor identification.
Recently, the yeast Hsp40 chaperone Ydj1p has been developed as yet another reporter, in this case one that is useful for both genetic and localization studies (Hildebrandt et al. et al. 2016b). Whereas the endogenous Ydj1p CaaX motif (i.e. CASQ) is subject only to isoprenylation and avoids subsequent PTMs, a process referred to as shunting, it can be made susceptible to Rce1 cleavage by mutation of the CaaX motif to those that are cleaved in the context of other reporters (e.g. CVIA and CTLM). Ydj1p thus has the potential to report on both cleavable and cleavage resistant CaaX motifs. Determining the specificity of Rce1 through use of various in vivo reporters is likely to help resolve how many CaaX proteins are shunted and how many are regulated by Rce1-mediated cleavage.
In vitro reporters
Rce1 in vitro activity can be monitored using a variety of farnesylated substrates, ranging from synthetic peptides modeled on CaaX proteins (e.g. Ras and a-factor) to recombinant proteins (e.g. GST-fusions) that have to be farnesylated in vitro prior to use. Early assays using synthetic peptides incorporated a radiolabeled amino acid within the aaX portion of the CaaX motif, enabling direct tracking of the liberated aaX tripeptide through HPLC and thin-layer chromatography methods [extensively reviewed in Young et al. Citation2001)]. Indirect tracking of Rce1-mediated activity has involved use of synthetic peptide substrates or recombinant protein substrates, and coupled proteolysis-methylation assays. In these types of assays, ICMT-dependent methylation of the proteolyzed product is used to mark cleaved substrates. Addition of a radioactive methyl group requires enrichment and quantification of the marked products, whereas addition of a non-radioactive methyl group to an a-factor-based reporter can be monitored directly using a biological assay (Tam et al. Citation2001).
A more utilitarian version of the direct assay monitors cleavage of an internally quenched fluorogenic (IQF) nonapeptide modeled on the farnesylated COOH-terminus of K-Ras4b (Hollander et al. Citation2000). In this assay, cleavage directly correlates with increased fluorescence, which is monitored in real time using a standard microplate reader having fluorescence detection capability. This assay approach is convenient for kinetic studies, HTS studies, and benefits from being non-radioactive. The assay confirms many of the expected properties of Rce1, ranging from isoprenoid dependence to tolerance for certain substitutions within the CaaX motif (Hollander et al. & Mallon 2003; Hildebrandt et al. Citation2016a). A major limitation of the IQF approach, however, is that either a fluorophore or quencher must be accommodated within the aaX portion of the CaaX motif, and such placement impacts Rce1 specificity and/or activity (Porter et al. Citation2007). For example, yeast Rce1 can tolerate lysine ε-dinitrophenyl (a quencher) at a1, and to a much lesser extent at X, but not at a2 in the context of a farnesylated nonapeptide modeled on K-Ras4b; a similar observation has been made for human Rce1 (Porter and Schmidt, unpublished). The requirement of a non-natural amino acid within the aaX portion of the motif also limits the utility of this approach for Rce1 specificity studies since the entire combinatorial landscape of the CaaX motif cannot be explored. Nonetheless, the mix-and-measure nature of this assay continues to be useful and is a major reason that HTS-based small molecule RPI identification has been able to proceed (Manandhar et al. Citation2007).
Specificity of Rce1
Through use of the above in vivo and in vitro approaches to examine Rce1 activity, a snapshot of Rce1 sequence specificity is beginning to emerge. In terms of isoprenylation, Rce1 cleaves substrates modified with either C15 and C20, but not unprenylated substrates (Otto et al. Citation1999; Hildebrandt et al. Citation2016b). To the best of our knowledge, the specificity of Rce1 toward substrates with shorter and longer isoprenoids (e.g. C5 and C25) or alternative lipids (e.g. C14 and C16 acyl lipids) has not been examined. Earlier studies, where this topic might have been explored, would have also likely used membranes containing both Rce1 and Ste24 making specificity interpretations difficult. In terms of peptide length, a farnesylated tetrapeptide CaaX motif is the minimally effective length for Rce1 recognition, although Rce1 can also cleave certain isoprenylated tripeptides in vitro with kinetics that rival that of traditional farnesylated tetrapeptides (Hollander et al. Citation2003). The biological relevance of shorter motifs has not been explored in vivo.
In terms of amino acid specificity, the a2 position appears to be the most critical residue for ScRce1 recognition as inferred from extensive substitution analysis of the a-factor CVIA motif (Trueblood et al. Citation2000). In this context, the most favored amino acids are Ile, Leu, and Val (i.e. branched chain, aliphatic amino acids). This is followed by Cys and Met, which are weakly tolerated. The next most critical residue appears to be a1 for which there is more tolerance, with the best tolerances observed with Ala, Cys, Ile, Leu, and Val and the small polar residues Ser and Thr; some additional polar residues are weakly tolerated. These results suggest that aliphatic is an oversimplification of allowable residues at the a1 position. The traditional X position can be occupied by about half the amino acids without significant impact on Rce1p activity: Ala, Val, Ser, Thr, Asn, Gln, His, Phe, Tyr, Cys, and Met. Besides its high sensitivity, the a-factor-based assay used in the Trueblood study is subject to false-positive readouts. Another limitation is its coupled nature. Any sequence combination that reports negatively or weakly in the assay could result from incomplete farnesylation rather than incomplete proteolysis. Thus, Rce1 may tolerate other amino acids within the CaaX motif, but such substrates were not biologically available if incompletely farnesylated leading to a weak signal in the genetic-based assay.
Additional specificity information is likely to come from other assay approaches. These certainly could involve more comprehensive genetic screens involving a-factor to identify more cleavable CaaX combinations or Ydj1 to identify both cleavable and non-cleavable sequences, but these approaches will still be biased toward sequences that are efficiently farnesylated, or possibly even geranylgeranylated in some cases. Other reporters such as Ras could also be utilized. Intriguingly, the reported specificity preferences of ScRce1 observed with the a-factor reporter are similar to those inferred from a Ras-based yeast genetic screen intended to interrogate farnesyl transferase specificity (Stein et al. Citation2015). The Ras-based screen evaluated the farnesylation potential of all 8,000 possible CaaX variations. Analysis of the top performing sequences reveals that a2 is enriched for Ile, Val, Leu, Met, and Cys (). Thus, both Ras and a-factor reporters identify similar enrichment of amino acids at a2; considerable variation was observed at a1 and X. It is tempting to speculate that the top performing sequences in the Ras-based study are cleaved, but this was not directly assessed. Moreover, the Ras-based screen was conducted in the presence of both Rce1 and Ste24, making it unclear which protease is responsible for cleavage of the top Ras-based sequences. The Ras-based assay also suffers from the same limitation as the a-factor based assay in that incompletely farnesylated sequences could report as false-negatives in the Ras-based assay despite having amino acids compatible for cleavage. For a truly unbiased identification of Rce1 cleavable sequences, it is feasible to expect that a synthetic library representing 8000 farnesylated Cxxx tetrapeptides could be developed to explore specificity without concern for isoprenylation status. Such an approach currently has technical and cost-related challenges.
Figure 6. Weblogo amino acid frequency analysis of sequences identified by a Ras-based strategy (Stein et al. Citation2015). The sequences analyzed were part of a set of sequences (n = 496) having an enrichment score greater than 3, which is suggestive of prenylation. The average enrichment score of the full 8,000 sequence set was 0.80, whereas the average score of the sequences analyzed for frequency analysis was 9.42. Color scheme: Cys is blue; polar charged amino acids are green (Asp, Arg, Glu, His, and Lys); polar uncharged residues are black (Asn, Gln, Ser, Thr, and Tyr); branched chain amino acids are red (Ile, Leu, and Val); and all other residues are purple (Ala, Gly, Met, Phe, Pro, and Trp). (see color version of this figure at www.tandfonline.com/ibmg).

Inhibitors of Rce1
Inhibitors of Rce1 that are not only potent but also selective could be used to further investigate the physiological role of Rce1 in regulating CaaX proteins and downstream effects of its inhibition. There are a number of Rce1 inhibitors reported in the literature (Bergman et al. Citation2011; Schmidt and Dore Citation2011; Dore and Schmidt Citation2013; Mohammed et al. Citation2016) that can be categorized as follows: anti-sense reagents to target the Rce1 mRNA, general protease inhibitors, substrate mimetics, and small molecule inhibitors.
Anti-sense reagents
A cocktail of three siRNA oligonucelotides (19 bases) knocks down human Rce1 expression in human colon carcinoma (HCT-116) cells (Mohammed et al. Citation2016). A 60–75% reduction in Rce1 mRNA levels occurred with a mixture of siRNA oligonucleotides targeting a translated sequence (GUCAUCAAGCGACGCUUCA), the end of the translated region (GCUCCUGACCUAUGCUCCU), and a 3′-untranslated section of the mRNA (CCUCUGCCUCUGAAAAGCU). Treatment of HCT-116 cells expressing GFP tagged H-, N-, or K-Ras (K-Ras4b) with the siRNA cocktail also mislocalized the Ras isoforms. N-Ras was most affected followed by H-Ras, then K-Ras4b. While not directly inhibiting Rce1 activity, siRNA represents a powerful tool to explore the downstream impact of Rce1 function.
General protease inhibitors
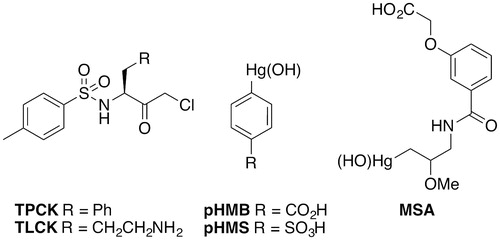
The general inhibitors cover a variety of structures. These include chloromethyl ketones such as Nα-tosyl-l-phenylalanine chloromethyl ketone (TPCK), which is considered a non-specific irreversible inhibitor of serine/cysteine proteases (). This inactivates ScRce1 with an approximate Ki of 1 mM (Chen et al. Citation1996). Interestingly TPCK inhibits ScRce1 in the presence of a Ras reporter, but not in the presence of the a-factor peptide. Therefore, it should be noted that differential inhibition of Rce1 can depend upon the reporter utilized in biochemical activity assays. TPCK has comparable inhibitory activity against Rce1 from species other than yeast, including Arabidopsis thaliana (AtRce1), human (HsRce1), and trypanosome (TbRce1) (Porter et al. Citation2007; Mokry et al. Citation2009). When applied at 250 μM, TPCK inhibited ScRce1, AtRce1, and HsRce1 to 32, 0.17, and 7%, respectively. A similar 200-μM concentration of TPCK inhibited TbRce1 activity to 22% (Mokry et al. Citation2009). A more lipophilic chloromethyl ketone, Nα-tosyl-l-lysine chloromethyl ketone (TLCK), is a trypsin and trypsin-like enzyme inhibitor. By comparison to TPCK, TLCK does not inhibit Rce1 under the same conditions. Other examples of general protease inhibitors reported to inhibit Rce1 include the organomercurial compounds para-hydroxymercuribenzoic acid (pHM), para-hydroxymercuriphenylsulfonic acid (pHMS), and mersalyl acid (MSA) (Dolence et al. Citation2000a). Non-specific organomercurials and other general protease inhibitors have generally not been useful for probing the proteolytic mechanism of Rce1 and have led to various erroneous proposed mechanisms for Rce1.
Figure 7. Structures of general protease inhibitors of Rce1.
Substrate mimetics
A number of inhibitors can be justifiably classified as substrate mimetics based on structural elements that mimic the natural substrate features. The first specific, non-reversible inhibitor of Rce1 was created by modifying TPCK and TLCK with a farnesyl group and replacing the sulfonamide with a tert-butylcarbamate to create BFCCMK (Ki = 30 μM) () (Chen et al. Citation1996). Replacement of the thiofarnesyl unit with a tridecyl group gave a more stable and cell-permeable analog, UM96001 (Chen Citation1998).
Figure 8. Structures of selected halo and acyloxymethyl ketone inhibitors of Rce1.
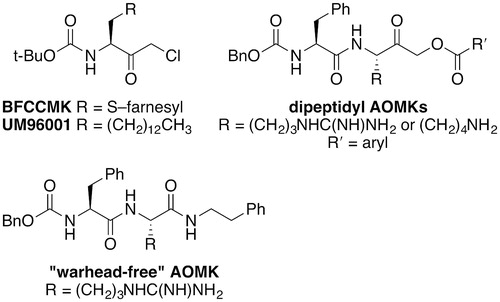
Peptidyl acyloxymethyl ketones (AOMKs) inhibit Rce1 (Porter et al. Citation2007). The peptidyl portion modulates the inhibitory properties of these compounds, but significant improvement of inhibitory properties is not likely achievable through alteration of the peptidyl portion alone (Dechert et al. Citation2010) (). Because AOMKs are known to be irreversible cysteine protease inhibitors, the reactivity of these derivatives can also be modulated by altering the acyloxy leaving group. Various derivatives with differing benzyloxy groups have been evaluated, but AOMKs surveyed had weak inhibitory properties against ScRce1 (>40% ScRce1 activity remaining after 100-μM treatment). A substituted analog that did not contain a leaving group suitable for an irreversible binding mechanism (“warhead-free” AOMK, ) proved to be the best inhibitory compound (∼25% ScRce1 activity remaining after 100-μM treatment). These results suggest that AOMKs do not likely work as mechanism-based inhibitors and are most likely non-covalent, reversible inhibitors of Rce1.
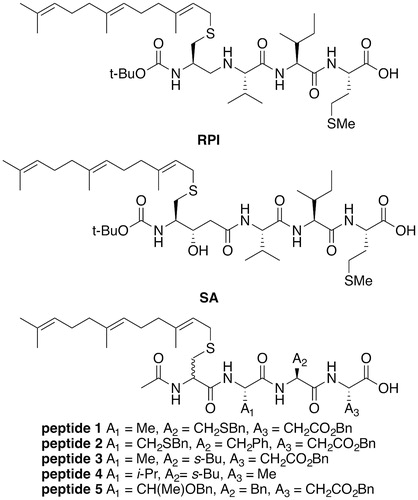
To date, the most potent of all Rce1 inhibitors are the farnesylated peptide mimetics, RPI and a statine analog (SA) () with Ki = 86 and 64 nM, respectively (Ma et al. Citation1993; Chen et al. Citation1996), and an IC50 value of 5 nM for RPI (Otto et al. Citation1999). RPI and SA are farnesylated CaaX motifs with a non-hydrolyzable linkage between cysteine and the aaX sequence of K-Ras (CVIM). SA is most likely a transition state analog inhibitor of Rce1. RPI and SA remain the gold standard in terms of inhibitory activity against HsRce1. In addition to RPI and SA, other tetrapeptide inhibitors (peptides 1–5, ) have been developed. Many of these are less potent by comparison to RPI and SA (ScRce1 IC50 = 3.3–17.6 μM) (Dolence, Dolence, et al. Citation2000; Dolence, Steward, et al. Citation2000; Dolence et al. Citation2001).
Figure 9. Structures of RPI, SA, and selected RPI analogs.
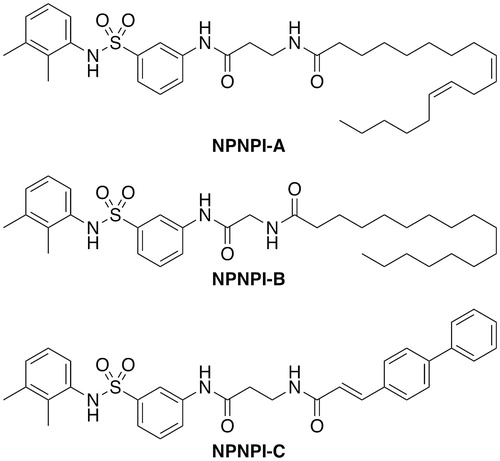
Non-peptidyl, non-prenylic inhibitors (NPNPIs) of the farnesyl transferase also inhibit HsRce1 (Schlitzer et al. Citation2001). The three best compounds NPNPI-A, B, C () displayed moderate inhibitory activity with IC50 values of 14, 7, and 7 µM, respectively. These are bi-substrate analogs that contain a peptidomimetic representing the CaaX motif and a lipophilic non-prenyl moiety representing the farnesyl group. The fatty acid lipid-like structure of these inhibitors invites speculation that these might be promiscuous aggregate-based inhibitors (McGovern et al. Citation2002 2003; Coan and Shoichet Citation2008).
Figure 10. Structures of peptidomimetic inhibitors.
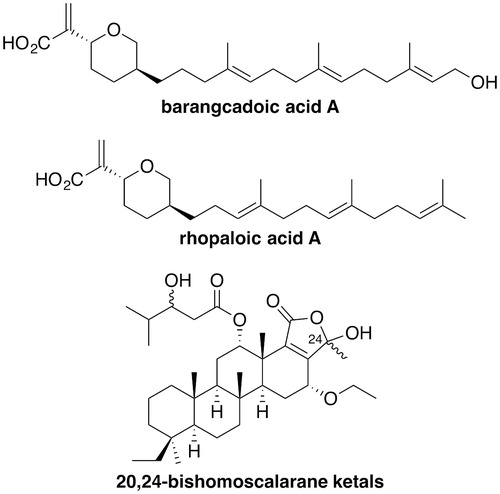
The marine natural products barangcadoic acid A and rhopaloic acids A to G (), isolated from Hippospongia species, are terpenoids with IC50 values of approximately 26 μM in an HsRce1-proteolysis assay (Craig et al. Citation2002). These are the first reported natural product inhibitors of Rce1. Akin to better performing Rce1 inhibitors, these natural products all carry an isoprenoid moiety. Additionally, extracts from the Indonesian sponge Carteriospongia foliascens contained two already known and four previously unreported scalarane-based sesterterpenoids (Williams et al. Citation2009). Of the six compounds, a mixture of two 20,24-bishomoscalarane ketals, differing in the stereochemistry at C-24, inhibited activity of HsRce1 (IC50 = 7.1 μM) despite the fact that these derivatives do not have an isoprenoid group ().
Figure 11. Structures of natural product inhibitors of Rce1.
Small molecule inhibitors
Recent work to identify new inhibitors of Rce1 has focused on the development of non-peptide based structures in an effort to identify small, drug-like molecules. Such compounds would be expected to have improved cell penetration and easier synthetic accessibility compared to peptidomimetic and natural compound-based inhibitors, enabling the use of eukaryotic cell-based assays of Rce1 activity, which would aid investigation into the effects of inhibiting Rce1 at the cellular level.
The first set of small molecule Rce1 inhibitors reported in the literature were identified as inhibitors of ScRce1 in a medium throughput screening campaign using a National Cancer Institute (NCI) library of approximately 2000 compounds (Manandhar et al. Citation2007). A final set of nine compounds demonstrated IC50 values of 6–35 µM against ScRce1. The specificity of the compounds was evaluated in part by examining their inhibitory properties toward other proteases. All of the compounds inhibited ScSte24 (IC50 = 8–28 μM), but had little or no activity on trypsin at ∼30 μM. For HsRce1 and TbRce1, IC50 values of 0.4–46 and 0.25–177 μM were observed, respectively (Mokry et al. Citation2009). Four of the compounds were complexes with toxic metals (mercury, palladium, or copper), and one was santowhite powder, an antioxidant added to rubber and latex. The additional four hits were NSC609974, NSC73101, NSC1011, and NSC270718 (). In a subsequent yeast study, NSC1011 and NSC73101 disrupted the proper membrane localization of a GFP-Ras2p reporter () (Manandhar et al. Citation2010). In yeast expressing TbRce1 instead of ScRce1, six of the nine compounds disrupted GFP-Ras2p membrane localization, including NSC1011. No species-specific selectivity was observed for the trypanosomal protein over the human ortholog, however, which diminishes enthusiasm for selective targeting of TbRce1 in the search for therapeutic compounds.
Figure 12. Selected examples of Rce1 inhibitors identified in a medium throughput screen.
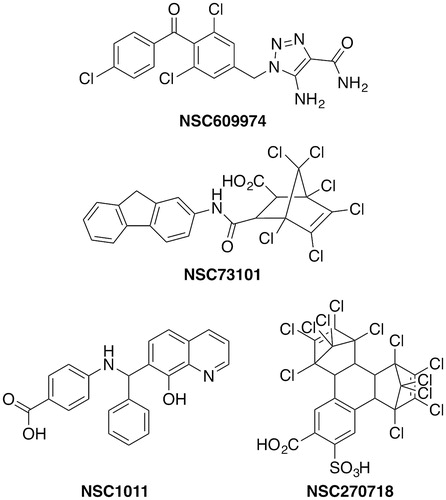
NSC609974, NSC73101, and NSC270718 were active in other screening programs, suggesting that these might be pan-assay interference compounds (PAINS) (Baell and Holloway Citation2010; Thorne et al. Citation2010; Baell et al. Citation2013; Baell and Walters Citation2014; Baell Citation2016). NSC 609974, also known as carboxyamidotriazole (CAI) or L651582, inhibits several calcium-mediated signal transduction pathways (Kohn and Liotta Citation1990; Felder et al. Citation1991; Kohn et al. Citation1992). It is an inhibitor of receptor-operated non-voltage-gated and voltage-gated calcium influx (Felder et al. Citation1991; Hupe et al. Citation1991), calcium-dependent arachidonic acid release (Clark et al. Citation1991; Felder et al. Citation1991), and tyrosine phosphorylation of phospholipase Cγ (PLCγ) (Gusovsky et al. Citation1993), and it has been used to study how calcium influx modulates the expression of matrix metalloproteinase-2 (MMP2) (Kohn et al. Citation1994b). NSC609974 and its orotate salt possess anti-proliferative, anti-angiogenic, and anti-metastatic properties (Kohn et al. Citation1992; Kohn, Felder, et al. Citation1994a; Mignen et al. Citation2005; Guo et al. Citation2006; Corrado et al. Citation2012) and have been in clinical trials for cancer (Grover et al. Citation2007; Taylor et al. Citation2015; ClinicalTrials.gov Citation2017). It is tempting to speculate that the anti-oncogenic activity and perhaps some of the adverse side effects could be due in part to the inhibition of Rce1.
NSC73101 was identified in a screen for inhibitors of poxvirus type I topoisomerase, but it was not the most promising inhibitor and was not pursued further (Bond et al. Citation2006) It was also identified in other screens as an inhibitor of Vibrio cholera biofilm formation (Peach et al. Citation2011), an inhibitor of the manganese/magnesium-dependent phosphatase PP2Cα (Sierecki and Newton Citation2014), and an irreversible inhibitor of replication protein A, an essential DNA repair protein (Gavande et al. Citation2016).
NSC270718 was independently found in multiple HTS and virtual ligand docking screens to be a moderate inhibitor of angiogenin (Ki = 75 μM), an inducer of new blood vessel growth (Jenkins et al. Citation2002; Kao et al. Citation2002); the human mitotic kinesin Eg5 (DeBonis et al. Citation2004); DNA synthesis specific to Kaposi’s sarcoma-associated herpes virus (KSHV) (IC50 = 2.38 μM) (Dorjsuren et al. Citation2006); the soluble form of adenylyl cyclase (Schlicker et al. Citation2008), and the ATP-dependent MurD ligase (IC50 = 34 μM in biochemical assays), an enzyme involved in the biosynthesis of an essential peptidoglycan component of the bacterial cell wall (Turk et al. Citation2009). It was also identified in the same screen against poxvirus type I topoisomerase that NSC73101 was found (Bond et al. Citation2006).
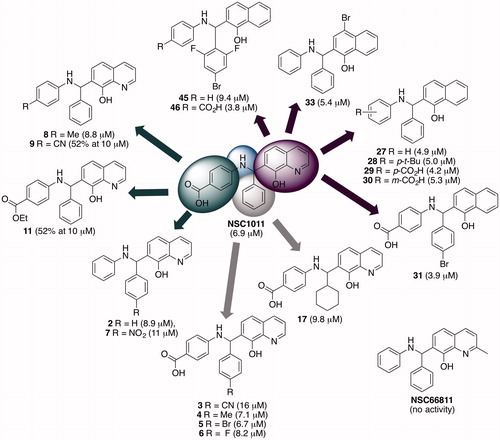
Other biological activities for NSC1011 have not been reported, but its structural analog, NSC66811 () inhibits the interaction between murine double minute 2 (MDM2) and p53 tumor suppressor with a Ki = 120 nM (Lu et al. Citation2006). The oncogene MDM2 is the cellular inhibitor of p53, which is an essential component of cell cycle regulation. In part because of this relatively higher potency against HsRce1, NSC1011 was taken forward into an SAR study in an attempt to improve upon potency and selectivity toward HsRce1 (Mohammed et al. Citation2016). A small library of approximately 56 new compounds was synthesized around the core 8-hydroxychloroquine scaffold () and tested against HsRce1 using the in vitro IQF proteolysis assay. Many of the derivatives failed to improve upon potency, although substitutions on the benzoic acid and phenyl rings were well-tolerated. Changing the hydroxyquinoline ring to a 1-napthol moiety showed a modest improvement in IC50 against HsRce1 from 6.9 µM for NSC1011 to 4.2, 3.9, and 3.8 µM for compounds 29, 31, and 46, respectively (). The hydroxyl group on the quinoline was essential for biological activity. The most active compounds were selective for HsRce1 versus HsSte24, and a representative subset of NSC1011 analogs did not inhibit human farnesyl transferase. NSC66811 was not active against HsRce1.
Figure 13. Examples of NSC1011 derivatives that remained active against HsRce1. Numbers in parentheses are IC50 values or the percent activity of Rce1 after 10 μM treatment as judged using the Rce1 IQF in vitro proteolysis assay (Mohammed et al. Citation2016). (see color version of this figure at www.tandfonline.com/ibmg).
The effectiveness of NSC1011-based compounds to work in vivo was demonstrated through a Ras mislocalization assay. NSC1011 and compounds 2, 5, 6, and 17 were effective at mislocalizing Ras to the cytosol in a human colorectal carcinoma cell line (HCT-116). Interestingly, the Rce1 inhibitors mislocalized N-Ras to the greatest extent, followed by H-Ras, then K-Ras4b. A similar result was found using Rce1 siRNA knockdowns (Mohammed et al. Citation2016). Curiously, NSC1011 and its analogs were more effective than an FTI at mislocalizing K-Ras4b, which is significant, because activating K-Ras mutations are more frequently observed in cancer compared to those of H- and N-Ras (Prior et al. Citation2012).
Perspectives
Rce1 has been studied for more than two decades. A great deal of progress has been made in terms of elucidating the precise involvement of Rce1 in CaaX protein maturation, yet much remains to be discovered regarding its cleavage specificity and impact of chemical inhibition, especially in vivo. Efforts to date have focused on three main areas: genetic inhibition to identify the impact of down regulation of Rce1, structural elucidation of a distantly related archeal ortholog, and development of chemical probes for its inhibition. Future inhibitor development is certainly currently limited by the lack of structural data of a eukaryotic enzyme. This has made it difficult to understand the mechanistic properties of existing inhibitors and has delayed structure-based design of inhibitors that may have better inhibitory properties than those currently available.
Whereas the current most potent inhibitor of Rce1 is a farnesylated tetrapeptide (i.e. a likely transition state substrate mimetic), it is cell impermeable and not useful to investigate the effects of Rce1 inhibition in vivo. A crystal structure involving the complex of SA with Rce1 should be a high priority. If it is a transition state inhibitor, then the hydroxyl group should displace the catalytic water molecule in the active site. The activity of the hydroxy epimer of SA against Rce1 would also be of interest to give insight into the orientation of the substrate in the active site. Attempts to overcome the issue of membrane impermeability of RPI and SA through development of small molecules are beginning, but these compounds have so far fallen short of expected standards for detailed animal studies. Nevertheless, the small molecule inhibitors identified thus far with activity in both in vitro and cell-based assays suggest that potent and selective inhibitors might yet be developed. New scaffolds may derive from additional HTS screening and possibly through computational methods for structure-guided design. Regardless of how developed, understanding the inhibitory mechanisms of these new compounds will undoubtedly require new structures of Rce1 orthologs, especially a eukaryotic one, and preferably that of the human enzyme.
Once appropriate inhibitory compounds are developed, it will be interesting to investigate whether Rce1 inhibitors alone or in combination with FTIs will be more effective than FTIs alone at modulating Ras activity. It is intriguing to speculate that modulation of K-Ras4b activity, in particular, might be more effective when FTIs are applied in combination with Rce1 inhibitors. It is well documented that alternative geranylgeranylation of K-Ras4b occurs in the presence of FTIs. Concomitant use of FTIs and Rce1 inhibitors could thus help limit the residual function associated with geranylgeranylated K-Ras4b. Inhibitors, regardless of their impact on Ras activity, may also have overlooked utility in other disease situations. Many CaaX-type isoprenylated proteins exist, and they are involved in many cellular processes. It is not yet clear, however, which CaaX proteins are cleaved by Rce1 as opposed to Ste24. Understanding the specificities and mechanisms of both enzymes is thus intertwined for developing accurate substrate profiles, which will be needed to better understand the potential biological impact of Rce1 inhibition.
Acknowledgements
We thank Drs. Louise Ashall and Emily R. Hildebrandt for their comments on this manuscript.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Baell JB. 2016. Feeling nature’s PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS). J Nat Prod. 79:616–628.
- Baell JB, Ferrins L, Falk H, Nikolakopoulos G. 2013. PAINS: relevance to tool compound discovery and fragment-based screening. Aust J Chem. 66:1483–1494.
- Baell JB, Holloway GA. 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 53:2719–2740.
- Baell JB, Walters MA. 2014. Chemistry: chemical con artists foil drug discovery. Nature. 513:481–483.
- Baker RP, Young K, Feng L, Shi Y, Urban S. 2007. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci USA. 104:8257–8262.
- Ben-Shem A, Fass D, Bibi E. 2007. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci USA. 104:462–466.
- Bergman JA, Majmudar JD, Gibbs RA, Hrycyna CA. 2011. Inhibitors of postprenylation CAAX processing enzymes. In: Tamanoi F, Hrycyna CA, Bergo M, editors. The enzymes, Vol. 30B. New York: Elsevier/Academic Press; p. 205–229.
- Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. 2002. Absence of the CAAX endoprotease Rce1: effects on cell growth and transformation. Mol Cell Biol. 22:171–181.
- Bergo MO, Lieu HD, Gavino BJ, Ambroziak P, Otto JC, Casey PJ, Walker QM, Young SG. 2004. On the physiological importance of endoproteolysis of CAAX proteins: heart-specific Rce1 knockout mice develop a lethal cardiomyopathy. J Biol Chem. 279:4729–4736.
- Berndt N, Hamilton AD, Sebti SM. 2011. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 11:775–791.
- Bond A, Reichert Z, Stivers JT. 2006. Novel and specific inhibitors of a poxvirus type I topoisomerase. Mol Pharmacol. 69:547–557.
- Boyartchuk VL, Ashby MN, Rine J. 1997. Modulation of ras and a-factor function by carboxyl-terminal proteolysis. Science. 275:1796–1800.
- Boyartchuk VL, Rine J. 1998. Roles of prenyl protein proteases in maturation of Saccharomyces cerevisiae a-factor. Genetics. 150:95–101.
- Bracha-Drori K, Shichrur K, Lubetzky TC, Yalovsky S. 2008. Functional analysis of Arabidopsis postprenylation CaaX processing enzymes and their function in subcellular protein targeting. Plant Physiol. 148:119–131.
- Burrows JF, Kelvin AA, McFarlane C, Burden RE, McGrattan MJ, De la Vega M, Govender U, Quinn DJ, Dib K, Gadina M, et al. 2009. USP17 regulates Ras activation and cell proliferation by blocking RCE1 activity. J Biol Chem. 284:9587–9595.
- Cadiñanos J, Schmidt WK, Fueyo A, Varela I, López-Otín C, Freije JMP. 2003a. Identification, functional expression and enzymic analysis of two distinct CaaX proteases from Caenorhabditis elegans. Biochem J. 370:1047–1054.
- Cadiñanos J, Varela I, Mandel DA, Schmidt WK, Diaz-Perales A, Lopez-Otin C, Freije JMP. 2003b. AtFACE-2, a functional prenylated protein protease from Arabidopsis thaliana related to mammalian Ras-converting enzymes. J Biol Chem. 278:42091–42097.
- Chen Y. 1998. Inhibition of K-ras-transformed rodent and human cancer cell growth via induction of apoptosis by irreversible inhibitors of ras endoprotease. Cancer Lett. 131:191–200.
- Chen Y. 1999. Selective inhibition of ras-transformed cell growth by a novel fatty acid-based chloromethyl ketone designed to target ras endoprotease. Ann NY Acad Sci. 886:103–108.
- Chen Y, Ma Y-T, Rando RR. 1996. Solubilization, partial purification, and affinity labeling of the membrane-bound isoprenylated protein endoprotease. Biochemistry. 35:3227–3237.
- Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE, Philips MR. 1999. Endomembrane trafficking of Ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 98:69–80.
- Christiansen JR, Kolandaivelu S, Bergo MO, Ramamurthy V. 2011. RAS-converting enzyme 1-mediated endoproteolysis is required for trafficking of rod phosphodiesterase 6 to photoreceptor outer segments. Proc Natl Acad Sci USA. 108:8862–8866.
- Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. 1991. A novel arachidonic acid-selective cytosolic PLA2 contains a calcium-dependent translocation domain with homology to PKC and GAP. Cell. 65:1043–1051.
- ClinicalTrials.gov. 2017. [accessed 2017 Nov 6]. https://clinicaltrials.gov/ct2/results?cond=&term=Carboxyamidotriazole&cntry1=&state1=&Search=Search
- Coan KED, Shoichet BK. 2008. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J Am Chem Soc. 130:9606–9612.
- Corrado C, Flugy AM, Taverna S, Raimondo S, Guggino G, Karmali R, De Leo G, Alessandro R. 2012. Carboxyamidotriazole-orotate inhibits the growth of imatinib-resistant chronic myeloid leukaemia cells and modulates exosomes-stimulated angiogenesis. PLoS One. 7:e42310.
- Cox AD, Der CJ, Philips MR. 2015. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 21:1819–1827.
- Craig KS, Williams DE, Hollander I, Frommer E, Mallon R, Collins K, Wojciechowicz D, Tahir A, Van Soest R, Andersen RJ. 2002. Novel sesterterpenoid and norsesterterpenoid RCE-protease inhibitors isolated from the marine sponge Hippospongia sp. Tetrahedr Lett. 43:4801–4804.
- DeBonis S, Skoufias DA, Lebeau L, Lopez R, Robin G, Margolis RL, Wade RH, Kozielski F. 2004. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol Cancer Ther. 3:1079–1090.
- Dechert A-M, MacNamara JP, Breevoort SR, Hildebrandt ER, Hembree NW, Rea AC, McLain DE, Porter SB, Schmidt WK, Dore TM. 2010. Modulation of the inhibitor properties of (acyloxy)methyl ketones toward the CaaX proteases. Bioorg Med Chem. 18:6230–6237.
- Dolence EK, Dolence JM, Poulter CD. 2000. Solid-phase synthesis of a farnesylated CaaX peptide library: inhibitors of the Ras CaaX endoprotease. J Comb Chem. 2:522–536.
- Dolence EK, Dolence JM, Poulter CD. 2001. Solid-phase synthesis of a radiolabeled, biotinylated, and farnesylated Ca1a2X peptide substrate for Ras- and a-mating factor converting enzyme. Bioconj Chem. 12:35–43.
- Dolence JM, Steward LE, Dolence EK, Wong DH, Poulter CD. 2000. Studies with recombinant Saccharomyces cerevisiae CaaX prenyl protease Rce1p. Biochemistry. 39:4096–4104.
- Dore TM, Schmidt WK. (2013). Isoprenylated protein peptidase Rce1p. In: Rawlings ND, Salvesen GS, editors. Handbook of proteolytic enzymes. 3rd ed., Vol. 2. Oxford, UK: Elsevier; p. 1720–1726.
- Dorjsuren D, Burnette A, Gray GN, Chen X, Zhu W, Roberts PE, Currens MJ, Shoemaker RH, Ricciardi RP, Sei S. 2006. Chemical library screen for novel inhibitors of Kaposi’s sarcoma-associated herpesvirus processive DNA synthesis. Antiviral Res. 69:9–23.
- Ellermeier CD, Losick R. 2006. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 20:1911–1922.
- Esher SK, Ost KS, Kozubowski L, Yang D-H, Kim MS, Bahn Y-S, Alspaugh JA, Nichols CB. 2016. Relative contributions of prenylation and postprenylation processing in Cryptococcus neoformans pathogenesis. mSphere. 1:e00084–15.
- Felder CC, Ma AL, Liotta LA, Kohn EC. 1991. The antiproliferative and antimetastatic compound L651582 inhibits muscarinic acetylcholine receptor-stimulated calcium influx and arachidonic acid release. J Pharmacol Exp Ther. 257:967–971.
- Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. 2007. Structure of a site-2 protease family intramembrane metalloprotease. Science. 318:1608–1612.
- Frankel MB, Wojcik BM, DeDent AC, Missiakas DM, Schneewind O. 2010. ABI domain-containing proteins contribute to surface protein display and cell division in Staphylococcus aureus. Mol Microbiol. 78:238–252.
- Fueller F, Bergo MO, Young SG, Aktories K, Schmidt G. 2006. Endoproteolytic processing of RhoA by Rce1 is required for the cleavage of RhoA by Yersinia enterocolitica outer protein T. Infect Immun. 74:1712–1717.
- Fujinaga M, Cherney MM, Oyama H, Oda K, James MNG. 2004. The molecular structure and catalytic mechanism of a novel carboxyl peptidase from Scytalidium lignicolum. Proc Natl Acad Sci USA. 101:3364–3369.
- Gao J, Liao J, Yang G-Y. 2009. CAAX-box protein, prenylation process and carcinogenesis. Am J Transl Res. 1:312–325.
- Gavande NS, VanderVere-Carozza PS, Pawelczak KS, Turchi JJ. (2016). Targeting the nucleotide excision repair pathway for therapeutic applications. In: Kelley MR, Fishel ML, editors. DNA repair in cancer therapy. 2nd ed. London: Academic Press; p. 135–150.
- Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, Waldmann H. 2006. Therapeutic intervention based on protein prenylation and associated modifications. Nat Chem Biol. 2:518–528.
- Gelb MH, Van Voorhis WC, Buckner FS, Yokoyama K, Eastman R, Carpenter EP, Panethymitaki C, Brown KA, Smith DF. 2003. Protein farnesyl and N-myristoyl transferases: piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics. Mol Biochem Parasitol. 126:155–163.
- Gentry LR, Nishimura A, Cox AD, Martin TD, Tsygankov D, Nishida M, Elston TC, Der CJ. 2015. Divergent roles of CAAX motif-signaled posttranslational modifications in the regulation and subcellular localization of Ral GTPases. J Biol Chem. 290:22851–22861.
- Gillespie JR, Yokoyama K, Lu K, Eastman RT, Bollinger JG, Van Voorhis WC, Gelb MH, Buckner FS. 2007. C-terminal proteolysis of prenylated proteins in trypanosomatids and RNA interference of enzymes required for the post-translational processing pathway of farnesylated proteins. Mol Biochem Parasitol. 153:115–124.
- Grover GJ, Kelly J, Moore G, JAcoby H, Karmali RA, Gorman GS. 2007. Comparative pharmacokinetic profile of carboxyamidotriazole and carboxyamidotriazoleorotate. Cancer Ther. 5:437–442.
- Guo L, Li ZS, Wang HL, Ye CY, Zhang DC. 2006. Carboxyamidotriazole inhibits proliferation of human breast cancer cells via G2/M cell cycle arrest and apoptosis. Eur J Pharmacol. 538:15–22.
- Gusovsky F, Lueders JE, Kohn EC, Felder CC. 1993. Muscarinic receptor-mediated tyrosine phosphorylation of phospholipase C-gamma. An alternative mechanism for cholinergic-induced phosphoinositide breakdown. J Biol Chem. 268:7768–7772.
- Hancock JF, Cadwallader K, Paterson H, Marshall CJ. 1991. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO J. 10:4033–4039.
- Hancock JF, Peterson H, Marshall CJ. 1990. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 63:133–139.
- Hanker AB, Mitin N, Wilder RS, Henske EP, Tamanoi F, Cox AD, Der CJ. 2010. Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene. 29:380–391.
- Hildebrandt ER, Arachea BT, Wiener MC, Schmidt WK. 2016a. Ste24p mediates proteolysis of both isoprenylated and non-prenylated oligopeptides. J Biol Chem. 291:14185–14198.
- Hildebrandt ER, Cheng M, Zhao P, Kim JH, Wells L, Schmidt WK. 2016b. A shunt pathway limits the CaaX processing of Hsp40 Ydj1p and regulates Ydj1p-dependent phenotypes. eLife 5:e15899.
- Hildebrandt ER, Davis DM, Deaton J, Krishnankutty RK, Lilla E, Schmidt WK. 2013. Topology of the yeast Ras converting enzyme as inferred from cysteine accessibility studies. Biochemistry. 52:6601–6614.
- Hollander I, Frommer E, Mallon R. 2000. Human Ras-converting enzyme (hRCE1) endoproteolytic activity on K-Ras-derived peptides. Anal Biochem. 286:129–137.
- Hollander IJ, Frommer E, Aulabaugh A, Mallon R. 2003. Human Ras converting enzyme endoproteolytic specificity at the P2’ and P3’ positions of K-Ras-derived peptides. Biochim Biophys Acta. 1649:24–29.
- Huang L, Li M, Wang D, He J, Wu W, Zeng Q, Li J, Xiao M, Hu J, He Y, et al. 2016. Overexpressed Rce1 is positively correlated with tumor progression and predicts poor prognosis in prostate cancer. Hum Pathol. 47:109–114.
- Hupe DJ, Boltz R, Cohen CJ, Felix J, Ham E, Miller D, Soderman D, Van Skiver D. 1991. The inhibition of receptor-mediated and voltage-dependent calcium entry by the antiproliferative L-651,582. J Biol Chem. 266:10136–10142.
- Jaworski J, Govender U, McFarlane C, de la Vega M, Greene MK, Rawlings ND, Johnston JA, Scott CJ, Burrows JF. 2014. A novel RCE1 isoform is required for H-Ras plasma membrane localization and is regulated by USP17. Biochem J. 457:289–300.
- Jenkins JL, Kao RYT, Shapiro R. 2002. Virtual screening to enrich hit lists from high-throughput screening: a case study on small-molecule inhibitors of angiogenin. Proteins Struct Funct Genet. 50:81–93.
- Kao RYT, Jenkins JL, Olson KA, Key ME, Fett JW, Shapiro R. 2002. A small-molecule inhibitor of the ribonucleolytic activity of human angiogenin that possesses antitumor activity. Proc Natl Acad Sci USA. 99:10066–10071.
- Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, Casey PJ, Young SG. 1999. Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J Biol Chem. 274:8383–8390.
- Kjos M, Snipen L, Salehian Z, Nes IF, Diep DB. 2010. The Abi proteins and their involvement in bacteriocin self-immunity. J Bacteriol. 192:2068–2076.
- Kohn EC, Felder CC, Jacobs W, Holmes KA, Day A, Freer R, Liotta LA. 1994a. Structure-function analysis of signal and growth inhibition by carboxyamido-triazole, CAI. Cancer Res. 54:935–942.
- Kohn EC, Jacobs W, Kim YS, Alessandro R, Stetler-Stevenson WG, Liotta LA. 1994b. Calcium influx modulates expression of matrix metalloproteinase-2 (72-kDa type IV collagenase, gelatinase A). J Biol Chem. 269:21505–21511.
- Kohn EC, Liotta LA. 1990. L651582: a novel antiproliferative and antimetastasis agent. J Natl Cancer Inst. 82:54–60.
- Kohn EC, Sandeen MA, Liotta LA. 1992. In vivo efficacy of a novel inhibitor of selected signal transduction pathways including calcium, arachidonate, and inositol phosphates. Cancer Res. 52:3208–3212.
- Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. 2007. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 6:541–555.
- Krishnankutty RK, Kukday SS, Castleberry AJ, Breevoort SR, Schmidt WK. 2009. Proteolytic processing of certain CaaX motifs can occur in the absence of the Rce1p and Ste24p CaaX proteases. Yeast. 26:451–463.
- Li J, Wang D, Liu J, Qin Y, Huang L, Zeng Q, Xiao M, Hu J, Yang Q, He J, et al 2017. Rce1 expression in renal cell carcinoma and its regulatory effect on 786-O cell apoptosis through endoplasmic reticulum stress. Acta Biochim Biophys Sin. 49:254–261.
- Lu Y, Nikolovska-Coleska Z, Fang X, Gao W, Shangary S, Qiu S, Qin D, Wang S. 2006. Discovery of a nanomolar inhibitor of the human murine double minute 2 (MDM2)-p53 interaction through an integrated, virtual database screening strategy. J Med Chem. 49:3759–3762.
- Ma YT, Gilbert BA, Rando RR. 1993. Inhibitors of the isoprenylated protein endoprotease. Biochemistry. 32:2386–2393.
- Manandhar SP, Hildebrandt ER, Jacobsen WH, Santangelo GM, Schmidt WK. 2010. Chemical inhibition of CaaX protease activity disrupts yeast Ras localization. Yeast. 27:327–343.
- Manandhar SP, Hildebrandt ER, Schmidt WK. 2007. Small-molecule inhibitors of the Rce1p CaaX protease. J Biomol Screen. 12:983–993.
- Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, Reilly NO, Hanrahan SJ, Thompson AJ, Cronin N, et al. 2013. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature. 504:301–305.
- McGovern SL, Caselli E, Grigorieff N, Shoichet BK. 2002. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 45:1712–1722.
- McGovern SL, Helfand BT, Feng B, Shoichet BK. 2003. A specific mechanism of nonspecific inhibition. J Med Chem. 46:4265–4272.
- Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, Wright L, Young SG, Philips M. 2005. Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol Biol Cell. 16:1606–1616.
- Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, Philips MR. 2001. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 152:111–126.
- Mignen O, Brink C, Enfissi A, Nadkarni A, Shuttleworth TJ, Giovannucci DR, Capiod T. 2005. Carboxyamidotriazole-induced inhibition of mitochondrial calcium import blocks capacitative calcium entry and cell proliferation in HEK-293 cells. J Cell Sci. 118:5615–5623.
- Mohammed I, Hampton SE, Ashall L, Hildebrandt ER, Kutlik RA, Manandhar SP, Floyd BJ, Smith HE, Dozier JK, Distefano MD, et al. 2016. 8-Hydroxyquinoline-based inhibitors of the Rce1 protease disrupt Ras membrane localization in human cells. Bioorg Med Chem. 24:160–178.
- Mokry DZ, Manandhar SP, Chicola KA, Santangelo GM, Schmidt WK. 2009. Heterologous expression studies of Saccharomyces cerevisiae reveal two distinct trypanosomatid CaaX protease activities and identify their potential targets. Eukaryot Cell. 8:1891–1900.
- Monne M, Nilsson I, Elofsson A, von Heijne G. 1999. Turns in transmembrane helices: determination of the minimal length of a “Helical Hairpin” and derivation of a fine-grained turn propensity scale. J Mol Biol. 293:807–814.
- Norholm MHH, Shulga YV, Aoki S, Epand RM, von Heijne G. 2011. Flanking residues help determine whether a hydrophobic segment adopts a monotopic or bitopic topology in the endoplasmic reticulum membrane. J Biol Chem. 286:25284–25290.
- Otto JC, Kim E, Young SG, Casey PJ. 1999. Cloning and characterization of a mammalian prenyl protein-specific protease. J Biol Chem. 274:8379–8382.
- Peach KC, Bray WM, Shikuma NJ, Gassner NC, Lokey RS, Yildiz FH, Linington RG. 2011. An image-based 384-well high-throughput screening method for the discovery of biofilm inhibitors in Vibrio cholerae. Mol BioSyst. 7:1176–1184.
- Pei J, Grishin NV. 2001. Type II CAAX prenyl endopeptidases belong to a novel superfamily of putative membrane-bound metalloproteases. Trends Biochem Sci. 26:275–277.
- Pei J, Mitchell DA, Dixon JE, Grishin NV. 2011. Expansion of type II CAAX proteases reveals evolutionary origin of γ-secretase subunit APH-1. J Mol Biol. 410:18–26.
- Plummer LJ, Hildebrandt ER, Porter SB, Rogers VA, McCracken J, Schmidt WK. 2006. Mutational analysis of the Ras converting enzyme reveals a requirement for glutamate and histidine residues. J Biol Chem. 281:4596–4605.
- Porter SB, Hildebrandt ER, Breevoort SR, Mokry DZ, Dore TM, Schmidt WK. 2007. Inhibition of the CaaX proteases Rce1p and Ste24p by peptidyl (acyloxy)methyl ketones. Biochim Biophys Acta. 1773:853–862.
- Price CTD, Al-Quadan T, Santic M, Jones SC, Abu Kwaik Y. 2010. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. J Exp Med. 207:1713–1726.
- Prior IA, Lewis PD, Mattos C. 2012. A comprehensive survey of Ras mutations in cancer. Cancer Res. 72:2457–2467.
- Pryor EE, Jr., Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, Zhu G, Malkowski MG, Wiener MC, Dumont ME. 2013. Structure of the integral membrane protein CAAX protease Ste24p. Science (Washington, DC). 339:1600–1604.
- Quigley A, Dong YY, Pike ACW, Dong L, Shrestha L, Berridge G, Stansfeld PJ, Sansom MS, Edwards AM, Bountra C, et al. 2013. The structural basis of ZMPSTE24-dependent laminopathies. Science (Washington, DC). 339:1604–1607.
- Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, Cox AD, Wilson O, Kirschmeier P, Der CJ. 2008. Rho family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J Biol Chem. 283:25150–25163.
- Schlicker C, Rauch A, Hess KC, Kachholz B, Levin LR, Buck J, Steegborn C. 2008. Structure-based development of novel adenylyl cyclase inhibitors. J Med Chem. 51:4456–4464.
- Schlitzer M, Winter-Vann A, Casey PJ. 2001. Non-peptidic, non-prenylic inhibitors of the prenyl protein-specific protease Rce1. Bioorg Med Chem Lett. 11:425–427.
- Schmidt WK, Dore TM. 2011. The Ras converting enzyme (Rce1p): orthologs, enzymology, and inhibitors. In: Tamanoi F, Hrycyna CA, Bergo M, editors. The enzymes, Vol. 30B. New York: Elsevier/Academic Press; p. 231–258.
- Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. 1998. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc Natl Acad Sci USA. 95:11175–11180.
- Shi B, Zhou X, Liang M, Luo Y, He L, Jiang P. 2017. Reduced RCE1 expression predicts poor prognosis of colorectal carcinoma. BMC Cancer. 17:414.
- Sierecki E, Newton AC. 2014. Biochemical characterization of the phosphatase domain of the tumor suppressor PH domain leucine-rich repeat protein phosphatase. Biochemistry. 53:3971–3981.
- Silvius JR. 2002. Mechanisms of Ras protein targeting in mammalian cells. J Membr Biol. 190:83–92.
- Stein V, Kubala MH, Steen J, Grimmond SM, Alexandrov K. 2015. Towards the systematic mapping and engineering of the protein prenylation machinery in Saccharomyces cerevisiae. PLoS One. 10:e0120716.
- Tam A, Nouvet FJ, Fujimura-Kamada K, Slunt H, Sisodia SS, Michaelis S. 1998. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J Cell Biol. 142:635–649.
- Tam A, Schmidt WK, Michaelis S. 2001. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J Biol Chem. 276:46798–46806.
- Taylor MH, Sandler A, Urba WJ, Omuro AMP, Gorman GS, Karmali RA. 2015. Effect of carboxyamidotriazole orotate, a modulator of calcium-dependent signaling pathways, on advanced solid tumors. J Cancer Ther. 6:322–333.
- Thorne N, Auld DS, Inglese J. 2010. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr Opin Chem Biol. 14:315–324.
- Tissue Expression of RCE1, The Human Protein Atlas. 2017. [accessed 2017 Nov 12]. https://www.proteinatlas.org/ENSG00000173653-RCE1/tissue
- Trueblood CE, Boyartchuk VL, Picologlou EA, Rozema D, Poulter CD, Rine J. 2000. The CaaX proteases, Afc1p and Rce1p, have overlapping but distinct substrate specificities. Mol Cell Biol. 20:4381–4392.
- Turk S, Kovac A, Boniface A, Bostock JM, Chopra I, Blanot D, Gobec S. 2009. Discovery of new inhibitors of the bacterial peptidoglycan biosynthesis enzymes MurD and MurF by structure-based virtual screening. Bioorg Med Chem. 17:1884–1889.
- Uhlen M, Fagerberg L, Hallstroem BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. 2015. Proteomics. Tissue-based map of the human proteome. Science (Washington, DC). 347:1260419.
- Vinothkumar KR, Strisovsky K, Andreeva A, Christova Y, Verhelst S, Freeman M. 2010. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 29:3797–3809.
- Wahlstrom AM, Cutts BA, Karlsson C, Andersson KME, Liu M, Sjogren A-KM, Swolin B, Young SG, Bergo MO. 2007. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 109:763–768.
- Wang M, Casey PJ. 2016. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol. 17:110–122.
- Wang Y, Ha Y. 2007. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci USA. 104:2098–2102.
- Williams DE, Hollander I, Feldberg L, Frommer E, Mallon R, Tahir A, van Soest R, Andersen RJ. 2009. Scalarane-based sesterterpenoid RCE-protease inhibitors isolated from the Indonesian marine sponge Carteriospongia foliascens. J Nat Prod. 72:1106–1109.
- Winter-Vann AM, Casey PJ. 2005. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 5:405–412.
- Xiang S, Bai W, Bepler G, Zhang X. 2017. Activation of Ras by post-translational modification. In: Azmi AS, editor. Conquering RAS: from biology to cancer therapy. London: Academic Press; p. 97–118.
- Young SG, Ambroziak P, Kim E, Clarke S. 2001. Postisoprenylation protein processing: CXXX (CaaX) endoproteases and isoprenylcysteinecarboxyl methyltransferase. In: Tamanoi F, Sigman DS, editors. The enzymes, Vol. 22. New York: Academic Press; p. 155–213.
- Young SG, Fong LG, Michaelis S. 2005. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria-new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J Lipid Res. 46:2531–2558.