
Abstract
ATPases associated with diverse cellular activities (AAA+ proteins) are a superfamily of proteins found throughout all domains of life. The hallmark of this family is a conserved AAA+ domain responsible for a diverse range of cellular activities. Typically, AAA+ proteins transduce chemical energy from the hydrolysis of ATP into mechanical energy through conformational change, which can drive a variety of biological processes. AAA+ proteins operate in a variety of cellular contexts with diverse functions including disassembly of SNARE proteins, protein quality control, DNA replication, ribosome assembly, and viral replication. This breadth of function illustrates both the importance of AAA+ proteins in health and disease and emphasizes the importance of understanding conserved mechanisms of chemo-mechanical energy transduction. This review is divided into three major portions. First, the core AAA+ fold is presented. Next, the seven different clades of AAA+ proteins and structural details and reclassification pertaining to proteins in each clade are described. Finally, two well-known AAA+ proteins, NSF and its close relative p97, are reviewed in detail.
Introduction
Chemo-mechanical energy transduction—in which the energy liberated upon nucleotide hydrolysis is harnessed to perform work—is an essential biochemical feature in all living organisms and viruses. Typically, the energy of the β–γ bond in triphosphate derivatives is released with hydrolysis and subsequently used to drive biological processes. P-loop NTPases are a class of proteins that bind and hydrolyze nucleotides (Saraste et al. Citation1990). P-loop NTPases encapsulate broad protein superfamilies such as kinases, GTPases, and AAA+ proteins, and are thought to predate the last universal common ancestor (LUCA) (Kyrpides et al. Citation1999; Leipe et al. Citation2002; Iyer et al. Citation2004). Common to all P-loop NTPases is the presence of two key motifs, the Walker A motif (GxxxxGK(T/S), where x is any amino acid; also referred to as Walker-A P-loop element), and the Walker B motif (hhhhD(D/E) where h is any hydrophobic amino acid) (Walker et al. Citation1982). The Walker A motif primarily binds nucleotide, and the Walker B motif drives hydrolysis by coordinating Mg2+ (D) and water (D/E) (Story and Steitz Citation1992). The P-loop NTPases are divided into two diverse subclasses, the kinase-GTPase and ASCE (additional strand catalytic element) subclasses. This review concerns the AAA+ proteins, which occupy the ASCE subclasses (Leipe et al. Citation2002, Citation2003; Iyer et al. Citation2004; Erzberger and Berger Citation2006) and in many cases perform mechanical work to remodel their substrates.
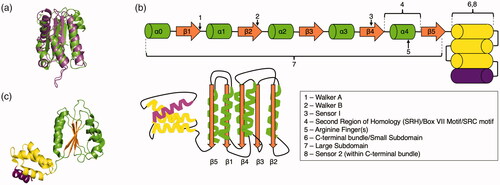
The AAA+ superfamily was first proposed following sequence comparisons of several yeast proteins (e.g. Sec18, an ortholog of NSF, and Cdc48, an ortholog of p97) that cluster together based on a conserved domain (Erdmann et al. Citation1991; Fröhlich et al. Citation1991; Kunau et al. Citation1993). Subsequent work revealed that this AAA+ domain is shared among many more different proteins (Confalonieri and Duguet Citation1995; Beyer Citation1997; Patel and Latterich Citation1998). Key studies of the D2 AAA+ domain of N-ethylmaleimide-sensitive-factor (NSF) (Lenzen et al. Citation1998; Yu et al. Citation1998) and the δ′ subunit from the bacterial clamp loader (Guenther et al. Citation1997) produced the first high-resolution structures of the AAA+ domain (). These structures revealed that, despite little to no sequence conservation, the AAA+ domain maintains a strikingly conserved fold and that these diverse proteins represent a unique protein superfamily (Neuwald et al. Citation1999; Iyer et al. Citation2004; Erzberger and Berger Citation2006).
Figure 1. The core AAA+ architecture. (a) The large subdomains of the first two AAA+ structures (RFC (green, PDB: 1A5T (Guenther et al. Citation1997)) and NSF (purple, PDB: 1NSF (Yu et al. Citation1998))) are overlaid. (b) Cartoon schematic of the AAA+ core domain. α-helices in the large subdomain are colored green, β-sheets are colored orange, and α-helices in the small subdomain are colored yellow. Some AAA+ proteins have either three or four helices in their small subdomain; the fourth helix is colored purple to indicate this. (c) The core AAA+ domain of ATPγS bound E. coli clamp loader complex (PDB: 1XXH (Kazmirski et al. Citation2004)) from the gene HolA is shown. The core β-sheets are colored orange, the surrounding regions of the large subdomain are green, and the small subdomain has the three first helices colored yellow, while the fourth helix is colored purple.
The superfamily of AAA+ proteins is involved in a variety of biochemical systems in all domains of life and viruses (Neuwald et al. Citation1999; Iyer et al. Citation2004; Erzberger and Berger Citation2006; Burroughs et al. Citation2007; Wendler et al. Citation2012; Hilbert et al. Citation2015; Miller and Enemark Citation2016; Puchades et al. Citation2020; Seraphim and Houry Citation2020). For example, in DNA replication (clade 1), a sliding clamp encircles DNA and associates with DNA polymerase at the replication fork to prevent it from falling off; this process is coordinated by a class of AAA+ proteins called clamp loaders (Kelch et al. Citation2012). A separate class of AAA+ proteins (clade 2) called initiators assist in recognizing replication origins and allows for assembly of the replication machinery on the DNA (Duderstadt and Berger Citation2008). AAA+ domains loaded in tandem accomplish the unraveling of folded proteins and even the disassembly of protein complexes, such as ubiquitin-tagged proteins (clade 3) (DeLaBarre and Brunger Citation2003; Banerjee et al. Citation2016; Bodnar et al. Citation2018; Cooney et al. Citation2019; Twomey et al. Citation2019; Pan et al. Citation2021) and SNARE protein complexes (Zhao et al. Citation2015; White et al. Citation2018), respectfully. AAA+ proteins are encoded within viral genomes to assist with their replication (clade 4) (Shen et al. Citation2005; Enemark and Joshua-Tor Citation2006; Santosh et al. Citation2020). AAA+ proteins can also assist in the untangling of protein aggregates prior to destruction by a proteasomal complex (Yedidi et al. Citation2017). AAA+ proteins are the translocation engine that drives protein degradation in several different protease complexes and secretion systems (clade 5) (Sauer and Baker Citation2011; Livneh et al. Citation2016; Ho et al. Citation2018). In addition to DNA polymerase clamp loaders, some members of the AAA+ family bind to and regulate RNA polymerases (clade 6) (Joly et al. Citation2012). Dynein, a widely studied cytoskeletal protein that moves along microtubules in cells by coupling ATP hydrolysis to large conformational changes (clade 7) (Reck-Peterson et al. Citation2018), is also a AAA+ protein (Roberts et al. Citation2009).
AAA+ core domain
This AAA+ core domain consists of an αβα “sandwich” topology (). The central β sheet (β5-β1-β4-β3-β2) is flanked on both sides by α-helices. Linearly, the core domain is ordered as N′-α0-β1-α1-β2-α2-β3-α3-β4-α4-β5-(α helical bundle, 3 or 4 helices)-C′ (). The aforementioned Walker A motif is after strand 1, and the Walker B motif is in strand 3. Strand 4 contains sensor-1, a set of conserved polar residues which mediate ATP hydrolysis in coordination with the Walker B motif. Catalytically inactive AAA+ domains such as the NSF D2 domain lack functional arginine fingers and thus hydrolyze ATP very slowly (Morgan et al. Citation1994; May et al. Citation2001). The arginine finger(s) element is generally between α4 and β5. These residue(s) coordinate ATP from neighboring AAA+ domains and may be involved in communication of the nucleotide state between subunits. The region spanning between β4 and β5 is referred to as the second region of homology (SRH), Box VII motif, or the SRC motif (Neuwald et al. Citation1999; Ogura et al. Citation2004; Erzberger et al. Citation2006) ().
A distinguishing feature of the AAA+ core domain that separates it from other NTPases is the presence of multiple insertions. The β2 strand insertion is only present in the ASCE subclass (hence the name additional strand catalytic element) (Iyer et al. Citation2004). The α0 helix and a conserved region upstream of this helix are only present in the AAA+ protein superfamily. The α0 helix typically contains a conserved glycine and a conserved polar residue that defines the N-terminal portion of this helix (Iyer et al. Citation2004). The C-terminal α helical bundle, which usually has three or four α helices, is also unique to the AAA+ core domain. Within the α helical bundle is sensor-2, a conserved motif that typically contains arginine (or alanine in the classical AAA+ protein clade (Wendler et al. Citation2012), see below), which interacts with ATP and can change conformation depending on whether ATP or ADP is bound. It has been speculated that this bundle may transmit the free energy of ATP hydrolysis to substrate (Iyer et al. Citation2004). In some cases, the sensor-2 element can act as a trans-acting element and interact with a bound ATP molecule from a different AAA+ domain (e.g. MCM (Miller et al. Citation2014)).
Common mechanistic features of AAA+ proteins
The defining feature of a majority of AAA+ proteins is their ability to oligomerize and form ring- or spiral-like complexes (Vale Citation2000). These complexes can act as molecular screws, unwinders, or threaders. Inter-protomer interaction is mediated by a conserved arginine finger(s) as stated above. This element coordinates the triphosphate nucleotide bound to an adjacent AAA+ protomer. This interaction elicits interprotomer joining and enables the detection of the nucleotide state of neighboring protomers (Kim et al. Citation2015). It may thus play a role in passing information through the complex from protomer to protomer in order to coordinate the broader activity of the oligomer. Generally speaking, however, principles governing the oligomerization of AAA+ proteins, inter-protomer communication, and the collective motions connected to function remain to be fully elucidated.
Another common theme throughout AAA+ proteins is stimulated ATPase activity upon substrate binding. For example, NSF shows limited ATPase activity until it forms a complex with two other components (Cipriano et al. Citation2013) (reviewed thoroughly below). p97 shows stimulated ATPase activity only when associated with the substrate that it can unfold (Blythe et al. Citation2017). Mutations to the DNA binding residues of DnaC severely impaired the ATPase activity of the mutants as compared to the WT (Arias-Palomo et al. Citation2019). PAN, the proteasomal ATPase complex in archaea homologous to a portion of the 26S eukaryotic complex, shows increased ATPase activity in the presence of an ssrA-tagged substrate (Kim et al. Citation2015). Tying substrate binding and an elevation in ATPase activity likely allow AAA+ protein complexes to conserve ATP.
Domains N-terminal to the AAA+ domain (“N-domains”) are diverse and primarily function in substrate engagement in many AAA+ families. The N-domains of AAA+ proteins typically vary across different families, owed in part to the many different types of substrates that AAA+ proteins interact with. The N-terminal domain of DnaC has two α helices that bind to DnaB and trap it in an open ring conformation (Chodavarapu et al. Citation2016). Meiotic AAA+ families VPS4, katanin, fidgetin, and spastin all share a similar N-terminal microtubule-binding domain (reviewed in the clade 3 section) that mediates substrate binding (Monroe and Hill Citation2016). The Lon protease N-domain has a specific binding site for a degron signal from a substrate (Wohlever et al. Citation2014). NSF’s N-domains do not bind substrate directly, but instead bind the adaptor proteins SNAPs/Sec17, which in turn bind the SNARE substrate meant for disassembly (Zhao et al. Citation2015). The protein p97/Cdc48 is a close relative of the NSF/Sec18 family and shares a similar N-domain that also engages with an adaptor/substrate complex (Cooney et al. Citation2019; Twomey et al. Citation2019; Pan et al. Citation2021).
The seven clades of AAA+ proteins
Nearly all AAA+ proteins share the common elements in the AAA+ core domain (). The proteins that share this common blueprint can be further subdivided into seven different clades, each with unique structural elements (Iyer et al. Citation2004; Erzberger and Berger Citation2006) (). This section will cover the phylogeny of known AAA+ proteins within each clade and focus on reviewing structural details of proteins in each clade (). It also includes the categorization of proteins within clade 3, the classical AAA+ proteins.
Figure 2. Schematic of the seven clades. The coloring scheme for the core domain is the same as above. Modifications to the core fold by insertions in the clade are graphically noted. Cylindrical-shaped insertions represent α-helices and two-sided arrows connected with a black connector represent β-hairpins. Clade 2 insertions are colored red,clade 3 insertions are colored purple, clades 4–7 insertions are colored blue, clades 6 and 7 specific insertions are colored yellow, and clade 7 specific insertions are colored gray.
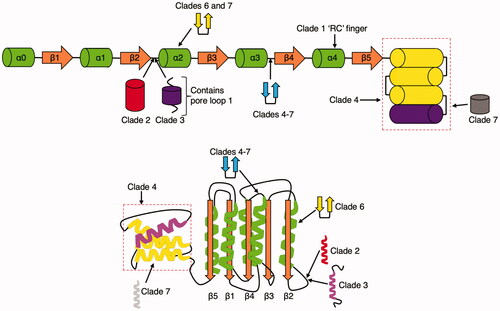
Table 1. Phylogeny, Biological functions, and Substrates for AAA+ proteins classified by clade.
Clade 1: Clamp loader clade
Clade 1 does not deviate from the AAA+ core domain structurally. The arginine finger is immediately followed by a cysteine residue (Iyer et al. Citation2004)
DNA replication in all cells requires the use of clamps—ring-shaped proteins that keep the DNA polymerase closely bound to the DNA (Hedglin et al. Citation2013). The clamp completely encircles the double-stranded DNA to serve as a sliding platform upon which DNA polymerase and other replication factors attach and associate, in addition to the DNA itself (Fernandez-Leiro et al. Citation2015). This complex of clamp and factors translocates along with the DNA and can even bypass DNA-protein crosslinks on the translocation strand (Sparks et al. Citation2019). Association of DNA polymerase with a clamp increases both the rate of nucleotide incorporation and the processivity (i.e. the number of nucleotides added in a single association), underscoring its essential function in DNA replication in all three domains of life (Maki and Kornberg Citation1985; O'Donnell and Kornberg Citation1985; Mok and Marians Citation1987; McInerney et al. Citation2007; Kelch et al. Citation2012). Equally important to the process of replication is its counterpart the clamp loader, an AAA+ protein that fastens this ring to the DNA (Duderstadt and Berger Citation2008). The clamp loader is a molecular switch controlled by ATP binding and hydrolysis (Goedken et al. Citation2004). ATP-bound clamp loader has a high affinity for the clamp, and when bound to ATP (Pietroni et al. Citation1997; Turner et al. Citation1999), DNA, and the clamp, its ATPase activity is stimulated (Jarvis et al. Citation1989; Ason et al. Citation2000). The affinity of the clamp loader for ADP is lower, so following hydrolysis, ADP is released, and the clamp and DNA are ejected (Kelch Citation2016).
Deviating from the “traditional” homohexameric arrangement of AAA+ proteins, clamp loaders are heteropentameric with a larger gap between the first and fifth subunit () (Jeruzalmi et al. Citation2001). This suggests that DNA is loaded through this gap. Following the previous revision of notation (Kelch et al. Citation2012), subunits are referred to as A through E.
Figure 3. Representative clade 1 structure. (a) Top view of the T4 bacteriophage clamp loader (PDB: 3U60 (Kelch et al. Citation2011)). The clamp is shown as a gray surface representation while the clamp loader protomers in the A (gp62), B (gp44), C (gp44), D (gp44), E (gp44) position are shown as surface representations colored green, red, pink, yellow, and blue, respectively. The DNA substrate is shown as a ball and stick representation. (b) Sideview of T4 bacteriophage clamp loader.
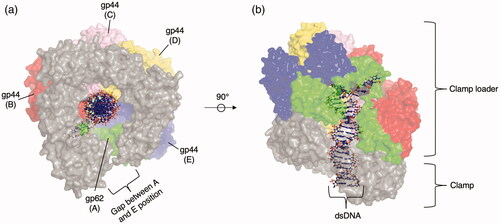
Within clade 1, there are three major phylogenetic families: the bacterial family, the RFC family, and the WHIP family (Iyer et al. Citation2004). T4 bacteriophage clamp loader () is a well-studied member of clade 1. It is closest to members of the eukaryotic RFC family in structure and sequence (Kelch et al. Citation2012), although it is difficult to place phylogenetically because the T4 bacteriophage is composed of genes from both eukaryotic and bacterial sources. The protein gp62 occupies the A position and has four identical gp44 subunits at positions B, C, D, and E. gp62 binds two clamp subunits—unlike gp44, which binds one (Kelch et al. Citation2011). High throughput mutagenesis of the T4 bacteriophage clamp loader revealed that regions not involved in catalysis or binding could tolerate mutations (Subramanian et al. Citation2021). The exception to this trend was a mutationally sensitive glutamine residue (Gln 118) that is spatially distant from both catalytic and interfacial sites. Hydrogen bonds formed by this residue appear to rigidly fasten two helices in the AAA+ core domain and connect DNA bound in the central channel of the clamp loader to the nucleotide at the catalytic site. This glutamine-mediated hydrogen bonding network is present in AAA+ proteins outside of Clade 1 and may thus be an important general feature of these proteins as it appears to link domains responsible for ATP binding and/or hydrolysis across neighboring protomers (Subramanian et al. Citation2021).
The bacterial family contains a zinc cluster insertion downstream of the β1 strand (Guenther et al. Citation1997; Iyer et al. Citation2004) and consists of two lineages that likely arose through ancient gene duplication, exemplified by the E. coli proteins HolA (Dong et al. Citation1993) (δ subunit) and DnaX (Kodaira et al. Citation1983; Mullin et al. Citation1983) (γ and τ subunits)/HolB (Carter et al. Citation1993; Dong et al. Citation1993) (δ′ subunit). Positions A and E are occupied by δ and δ′ subunits, respectively, and lack ATPase activity. Positions B, C, and D are occupied by γ or τ subunits. The γ subunit is a truncated version of the τ subunit that is typically generated by Programmed −1 Ribosomal Frameshifting (−1 PRF) (Blinkowa and Walker Citation1990; Flower and McHenry Citation1990; Tsuchihashi and Kornberg Citation1990), the most well-known translational mechanism among the recoding phenomena (Baranov et al. Citation2015; Atkins et al. Citation2016; Khan et al. Citation2020) that is employed in many viruses (Jacks and Varmus Citation1985; Jacks et al. Citation1988; Moomau et al. Citation2016; Kendra et al. Citation2017) and bacteria (Meydan et al. Citation2017) but not currently discovered in cellular genes in vertebrates (Khan et al. Citation2019).
The RFC family (replication factor C) is found in archaea and eukaryotes with two lineages, which also may have arose through ancient gene duplication (Iyer et al. Citation2004). In the RFC family, eukaryotic proteins RFC1-5 occupy positions A through E. RFC1 occupies position A (active ATPase) and has an extra A′ domain that bridges the gap between the A and E domain (Bowman et al. Citation2004). The other proteins sit as follows: RFC4 at B, RFC3 at C, RFC2 at D, and RFC5 at E (which lacks ATPase activity). The archaeal RFC family has one unique, active ATPase at the A position, and four identical ATPase subunits at the B, C, D, and E positions (Miyata et al. Citation2005). The collar region is a separate domain that C-terminally organizes into a planar ring. Mapping common cancer mutations onto human RFC showed a significant accumulation of mutations in the collar region (Gaubitz et al. Citation2020), which is thought to seed oligomerization.
The WHIP (Werner helicase interacting protein) family is present in eukaryotes and most prokaryotes (Iyer et al. Citation2004). WHIP family proteins contain a ubiquitin-binding zinc finger (UBZ) domain (Yoshimura et al. Citation2017). Gel-filtration chromatography suggests that Human Werner helicase interaction protein 1 oligomerizes into an octamer, but this has not been confirmed by structural studies (Tsurimoto et al. Citation2005). The Werner helicase, its interaction partner, is mutated in patients with Werner’s Syndrome, a disorder characterized by premature aging (Yoshimura et al. Citation2017).
Clade 2: Initiator clade
Clade 2 is characterized by the insertion of an α helical element after β2 and before α2 (Iyer et al. Citation2004; Erzberger and Berger Citation2006; Duderstadt and Berger Citation2008) ().
Replication of DNA in all cells is initiated at the origin of replication. AAA+ initiators can form a spiral corkscrew around the double-stranded DNA, which then recruits helicases and other proteins to either begin the replicative process or to perform other roles in the assembly of the full replication complex on the origin of replication. Bacteria and archaea frequently employ a single origin of replication in the genome, whereas eukaryotes can range anywhere from the hundreds to tens of thousands (Leonard and Méchali Citation2013).
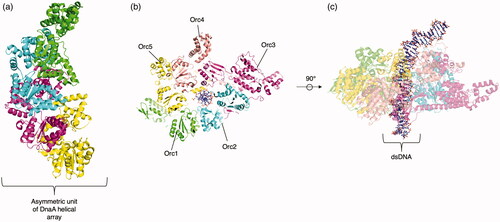
There are two families within the initiator clade, the bacterial DnaA family and the archaeo-eukaryotic CDC6/ORC family () (Iyer et al. Citation2004). The bacterial DnaA family is comprised of two orthologous lineages, DnaA and DnaC. DnaA is present in all known bacteria and forms an oligomeric complex around the origin of replication, which then recruits DnaB, a helicase, to unwind the DNA. DnaA (and its archaeo-eukaryotic counterpart ORC) forms an open, spiral heteroligomer around the DNA () (Erzberger et al. Citation2006). It has a C-terminal domain IV that mediates its interaction with the DNA itself. Many DnaA monomers form a right-handed filamentous structure composed of a tetrameric asymmetric unit.
Figure 4. Representative clade 2 structures. (a) The asymmetric unit (consisting of four identical protomers) of the DnaA crystal structure without DNA bound to AMPPCP (PDB: 2HCB (Erzberger et al. Citation2006)). (b) Top view of the ORC complex (PDB: 5ZR1 (Li et al. Citation2018), trimmed to only include AAA+ containing ORC1-5). Orc1, Orc2, Orc3, Orc4, and Orc5 are colored green, teal, purple, peach, and yellow respectively. The DNA substrate, centered in the pore, is shown with a ball and stick representation. (c) Side view of the ORC complex.
DnaC (also called DnaI in Gram-positive bacteria) is thought to have arisen through duplication of DnaA and is not widely present in bacteria. Six DnaC protomers assist with the loading of DnaB helicase onto the DNA by “cracking” open the DnaB hexamer (with its extended N-terminal domain) (Wahle et al. Citation1989; Arias-Palomo et al. Citation2019; Nagata et al. Citation2020). Upon DNA binding, DnaC hydrolyzes ATP, allowing DnaB to rearrange into a closed ring formation around the DNA.
The ORC/CDC6 family is present in all eukaryotes and almost all archaea. In eukaryotes, AAA+ initiator proteins are involved with first forming the ORC complex on the DNA and then forming the pre-replication complex after the recruitment of the additional AAA+ initiator CDC6 and, subsequently, the clade 6 AAA+ mini-chromosome maintenance complex (MCM) of MCM2-7 (Fragkos et al. Citation2015).
In the ORC family, five different subunits of Orc (Orc1–5 in S. cerevisiae) form a spiral around the DNA () (Li et al. Citation2018). Orc6 does not directly interact with the DNA but contacts Orc3, Orc2, and Orc5. Orc6 also has little homology to ORC1–5 and its function varies among eukaryotic organisms. In the spiral around the DNA, the order is Orc1, Orc4, Orc5, Orc3, and Orc2 with a gap between Orc1 and Orc2. This gap is partially filled by a winged helix domain (WHD) of the Orc1–5 proteins, which is a nucleic acid binding structural element. After the ORC complex is formed, cdc6 likely docks onto the complex by passing through this gap. Only Orc1, Orc4, Orc5 contain functional ATPase AAA+ domains while Orc2 and Orc3 do not. Mutations to human Orc proteins cause a wide range of diseases such as Meier–Gorlin syndrome (Jackson et al. Citation2014).
Archaea also employ the ORC/CDC6 family. Proteins in archaea that have homology to eukaryotic Cdc6 and Orc1 are called either Orc1 or Cdc6 proteins in archaea, despite harboring homology to both (Arora et al. Citation2014). Archaeal Orc1/Cdc6 proteins form monomers or complexes at the origin of replication (Ausiannikava and Allers Citation2017).
The additional α-helix characteristic of clade 2 (), also called an initiator-specific motif (ISM), differs between bacteria and archaea-eukaryotes as well. In DnaA, the ISM coordinates the DNA (Duderstadt et al. Citation2011) and forces the neighboring DnaA subunits out of plane with respect to each other to form a right-handed helix (Erzberger et al. Citation2006). In the archaea-eukaryotic ORC/CDC6 family, the ORC ISM typically contacts the DNA (Li et al. Citation2018) also but does not promote a radical spiraling of subunits of the DNA due to its topology (Costa et al. Citation2013).
Clade 3: classical clade
A flexible linker and short α-helix are inserted after β2 and before α2 (). This flexible linker is typically referred to as pore loop 1. Pore loop 1 typically contains one or more aromatic residues that interact with the substrate. There is a less conserved pore loop 2 portion in some classical AAA+ proteins, which also binds to substrate in the pore (Puchades et al. Citation2020). Members of this clade lack a conserved arginine at sensor-2 and are instead typically an alanine. There is often a conserved glycine N-terminal to the arginine finger and there are typically two conserved arginine residues in the SRH instead of the single residue seen in other clades (Iyer et al. Citation2004).
The classical clade encompasses the widest breadth of AAA+ proteins. Members of this clade usually form homo-oligomeric complexes that process and remodel nucleic acids, proteins, and protein complexes (YME1 in and NSF in ). With regard to protein remodeling, these roles often involve key aspects of disaggregation and degradation. A wide variety of families that occupy diverse functions in this clade exist. These families have differing, seemingly modular arrangements of multiple AAA+ domains and/or accessory domains.
Figure 5. Representative clade 3 structure. (a) YME1 (PDB: 6AZ0 (Puchades et al. Citation2017)) bound to substrate. Identical protomers are colored differently, with the substrate represented as a purple colored surface. (b) YME1 staircase threading of the substrate (purple) by tyrosine residues (shown as ball and stick) present in the pore loop of the four engaged protomers. (c) Cartoon schematic of only the four engaged YME1 pore loops to the substrate (purple).
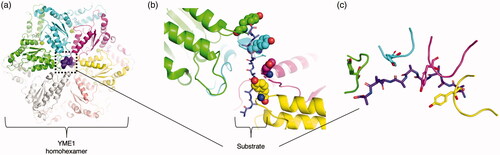
While there is much diversity within clade 3, a common thread among the classic AAA+ proteins with niche functions is the presence of a conserved pore loop 1 (Puchades et al. Citation2020) (). The pore loop 1 typically contains an aromatic residue that acts like the teeth of a gear to exert axial force on the substrate through the hexamer pore in a somewhat nonspecific manner. A less-conserved pore loop 2 present in some proteins has also been implicated in both substrate recognition and in forming a second “conveyor belt” of residues to further aid in the translocation of substrate (Han et al. Citation2017).
Due to the breadth of functional diversity, the classical clade is typically subdivided into two different categories: type I and type II (Saffert et al. Citation2017; Banchenko et al. Citation2019). Sometimes, a third “AAA+ protease” category is employed as well (Puchades et al. Citation2020). Here, we summarize the definitions and categories of these types within the classical clade to provide a straightforward classification scheme. The type I category will include classical clade AAA+ proteins with only a single AAA+ domain. The type II category will include classical AAA+ proteins with two (or more, if found to exist in the classical clade) AAA+ domains within the same protein chain. The presence of accessory or proteasomal domains will not impact the classification of these classical AAA+ proteins.
Type I classical AAA+ proteins include the eukaryotic katanin, fidgetin, spastin, and VPS4 protein families (VPS4 is found in some archaeal phyla as well (Iyer et al. Citation2004)). These four families, within the Type I ATPases, are also referred to as Meiotic AAA+ proteins (Monroe & Hill Citation2016). The N-terminal domains of katanin, spastin, and fidgetin function in the recognition of tubulin polymers and help to recruit other enzymes involved in cytoskeletal remodeling. VPS4 disassembles ESCRT-III polymers, assisting in the membrane-remodeling ESCRT pathway and is stimulated to hydrolyze ATP in the presence of ESCRT-III (Merrill and Hanson Citation2010). VPS4’s catalytic activity is directly responsible for membrane scission (Schöneberg et al. Citation2018). All four families share an N-terminal Microtubule Interacting and Trafficking (MIT) domain. C-terminal to the MIT domain and N-terminal to the AAA+ domain, spastin contains a microtubule-binding domain (MTBD) necessary for microtubule binding. Katanin functions in microtubule severing (Lindeboom et al. Citation2013) and is a heterodimer of two unrelated proteins, p60 (AAA+ domain-containing) and p80. p80 enhances the in vitro ATPase and microtubule severing activity of p60. Fidgetin also has the ability to cleave microtubules. Only eukaryotic VPS4, but not archaeal VPS4 or other members of the Type I ATPase category, has a unique insertion into the AAA+ domain called the β-domain. The β-domain packs against the AAA+ β-sheet in an antiparallel orientation and binds a VPS4 activator. Type I ATPases oligomerize into hexamers around their substrate. High-resolution structures of hexameric spastin, katanin, and VPS4 confirm this (Caillat et al. Citation2015; Monroe et al. Citation2017; Sun et al. Citation2017; Zehr et al. Citation2017; Sandate et al. Citation2019). Mutations in spastin and katanin are associated with Hereditary Spastic Paraplegia, a disease showing abnormal microtubule arrangement and amount (Ghosh et al. Citation2012). These protein families are considered drug targets (Cupido et al. Citation2019; Citation2021).
The eukaryotic Bcs1 is another Type I AAA+ protein, with an N-terminal domain that anchors it to the inner membrane of the mitochondria. Bcs1 is essential for the transport of RIP1 across the inner membrane and assembly into complex III of the electron transport chain (Wagener et al. Citation2011). Surprisingly, Bcs1 has been observed in a heptameric state in several structural studies (Kater et al. Citation2020; Tang et al. Citation2020).
The TIP49 family is an archaeo-eukaryotic family involved in transcription that is stimulated by DNA (Iyer et al. Citation2004). TIP49 family proteins have a large insertion between the Walker A and B motifs, containing an oligosaccharide/oligonucleotide binding (OB) domain (Petukhov et al. Citation2012). Confusingly, the RuvB-like 1 (RUVBL1, also known as pontin) and RuvB-like 2 (RUVBL2, also known as reptin) eukaryotic proteins are classified in the TIP49 family despite the actual RuvB protein belonging in clade 5 (discussed below) and lacking the PS1βH element that characterizes clade 5 AAA+ proteins. RUVBL1 and RUVBL2 have been implicated in a wide variety of molecular processes (Mao and Houry Citation2017). RUVBL1 and RUVBL2 form a heterohexamer with alternating RUVBL1 and RUVBL2 units in the larger human INO80 chromatin complex (Aramayo et al. Citation2018). Activities of the two range from transcriptional regulation, chromatin remodeling, DNA damage signaling and repair, assembly of macromolecular complexes (such as γTuRC (Zimmermann et al. Citation2020)), cell cycle, and motility. These proteins are overexpressed in many cancer types, which is perhaps unsurprising given their role in so many cellular processes involved in cellular replication; identification of inhibitors is ongoing (Nano et al. Citation2020). Because members of the TIP49 family are classical AAA+ proteins with a single AAA+ domain, they are here designated as Type I ATPases.
The prokaryo-eukaryotic AFG1 was originally discovered as being an AAA+ domain containing protein with homology to NSF/Sec18, Cdc48/p97, and other AAA+ proteins (Lee and Wickner Citation1992). For this reason, the AFG1 family has been typically placed closely to the NSF and p97 families. It likely made its way to eukaryotes via the proto-mitochondrion. Loss of functional Afg1 leads to progressive mitochondrial failure and impaired oxidative stress tolerance in eukaryotes (Germany et al. Citation2018). Despite a lack of structural studies, it appears that AFG1 only contains one AAA+ domain and is thus more appropriately suited for the Type I category.
Type I AAA+ proteins can also have proteases fused to them. For example, the FtsH family of proteases has a metalloprotease fused C-terminally to a AAA+ domain (Iyer et al. Citation2004). This family is pan-bacterial and present in some eukaryotes via proto-mitochondrion transfer. Examples of proteins in this family include YME1 and AFG3L2 (Puchades et al. Citation2019; Citation2020). YME1 forms a spiral-shaped hexameric AAA+ ring that passes substrate into a hexameric protease for degradation. Four subunits appear to engage the substrate, while the top and bottom protomers along the spiral are partially or completely disengaged () (Puchades et al. Citation2017). AFG3L2 also forms a spiral when engaged to substrate (Puchades et al. Citation2019) and has specificity for certain degron sequences (Ding et al. Citation2018). Mutations in YME1 and AFG3L2, both which are involved in the maintenance of the mitochondrial proteome, can cause autosomal dominant spinocerebellar ataxias (Di Bella et al. Citation2010).
Type I AAA+ proteins can also function in proteasomal processing without being fused to a protease, such as the RPT family. The base of the eukaryotic 26S proteasome consists of a ring of Rpt1-2-6-3-4-5, all AAA+ proteins (Beckwith et al. Citation2013). The archaeal proteosome also employs a AAA+ ring system, PAN, with a similar functional purpose (Majumder et al. Citation2019). This AAA+ ring is the engine of translocation in the 26S proteasome (Dong et al. Citation2019). We direct the reader to thorough reviews on the 26S proteasome (Livneh et al. Citation2016; Bard et al. Citation2018).
Type II ATPases are AAA+ proteins that contain tandem AAA+ domains which arose through duplication or fusion events. Families in this category include the eukaryotic NSF, archaeo-eukaryotic CDC48, eukaryotic Drg1, eukaryotic Rix7, eukaryotic Pex1/6, the eukaryotic ATAD, and the ClpAB families (N-terminal domain). The NSF family is most likely derived from the CDC48 family as eukaryotic vesicle trafficking evolved and grew in complexity (Iyer et al. Citation2004). NSF is thoroughly reviewed below.
The Cdc48 family, members of which are composed of an N-domain and two AAA+ domains like NSF, is involved in extracting and unfolding peptides targeted by ubiquitination. It appears to actively use both ATPase rings in the threading and unfolding of substrate () (Bodnar et al. Citation2018). Mutations in p97, the human protein in the CDC48 family, cause a variety of diseases such as multisystem proteinopathy (Tang and Xia Citation2016).
The Drg1, Rix7, and Rea1/Midasin (clade 7, discussed below) protein families are all involved in different maturation steps of the pre-60S ribosomal particles (Prattes et al. Citation2019). Drg1 is a eukaryotic family that is a type II AAA+ protein. It appears to have branched off from the Cdc48 family, as it has similarity in its N-domain, D1, and D2 domain. Structure prediction suggests that the Drg1 N-domain folds into two subdomains, as do Cdc48 and other related classical AAA+ proteins (Prattes et al. Citation2019).
Rix7 is a eukaryotic type II AAA+ protein that also appears to have branched off of the Cdc48 family earlier on in eukaryotic evolution, but unlike Drg1, it has a unique N-terminal domain from Cdc48’s (Prattes et al. Citation2019). Members of the Rix7 family have a 40–50 amino acid insertion after α7 in D1 and a 10–35 amino acid insertion after α7 in D2 (Prattes et al. Citation2019). Rix7 was found to adopt an asymmetric spiral in complex with an unknown protein substrate; both D1 and D2 were found engaged to the substrate (Lo et al. Citation2019) but no clear N-terminal domain density was present.
The Pex1/6 family forms a heterohexameric complex of Pex1 and Pex6 that assists in peroxisome biogenesis. The D2 domains of Pex1 and Pex6 are strongly conserved, in contrast to their partially functional or nonfunctional D1 domains. Pex1/6 forms a unique, somewhat triangular hexamer due to the asymmetric arrangement of the N-terminal domains of Pex1 and Pex6 (Blok et al. Citation2015; Ciniawsky et al. Citation2015; Gardner et al. Citation2015).
The ATAD, 1 through 5, (ATPase family AAA domain-containing family) is a protein family containing the AAA+ domain that is conserved in eukaryotes (Cattaneo et al. Citation2014). ATAD1 (Msp1 in yeast) consists of a transmembrane domain, linker domain, and the AAA+ domain (Wang et al. Citation2020) and is thus considered a type I classical AAA+ protein. ATAD2 (Yta7 in yeast) is comprised of an N-domain, one functional AAA+ domain (D1), one nonfunctional AAA+ domain (D2), and a C-terminal bromodomain (Cho et al. Citation2019). ATAD2 is classified as a type II classical AAA+ protein due to the presence of two AAA+ domains. Thus, the ATAD family contains both Type I and Type II classical AAA+ proteins.
Msp1, ortholog of ATAD1, has been shown to processively thread substrate through its central pore (Castanzo et al. Citation2020). It contains a transmembrane domain and primarily functions in the quality control of proteins anchored in membranes by removing them and initiating their degradation (Okreglak & Walter Citation2014). Structural analysis reveals two sets of pore loops (pore loops 1 and 2) that directly engage the substrate. A pore loop 3 exists which does not contact the substrate directly but instead functions in stabilizing the interactions with the substrate (Wang et al. Citation2020).
ATAD2 primarily functions in the context of chromatin dynamics, where it acts as a histone chaperone that regulates their interaction with DNA. The D2 domain is functionally inactive and lacks both Walker A and B motifs. The bromodomain (BRD) functions in the recognition of acetylated lysines, a common modification of the N-termini of histones (Fujisawa and Filippakopoulos Citation2017). In humans, ATAD2 has been identified as an oncogene overexpressed in multiple cancers, and efforts have been made to target the ATAD2 BRD with small-molecule inhibitors (Hussain et al. Citation2018). Interestingly, cryo-EM structural analysis of a yeast ortholog bound to a substrate (Cho et al. Citation2019) revealed that the complex is able to oligomerize without the presence of nucleotide, unlike many other AAA+ oligomers. This is due to the unique insertion of a helix-turn-helix “knob” that reaches across to a “linker arm” on a neighboring protomer. Affinity of chaperone to substrate does not change in the presence of nucleotide, but the deposition of chaperone on DNA is nevertheless ATP hydrolysis dependent.
The ClpAB ATPase family is composed of tandem AAA+ domains. The N-terminal AAA+ domain is from the classical clade, but the C-terminal domain is related to clade 5 (HCLR). ClpAB proteins are found in bacteria and eukaryotes, which likely arose from acquisition from the proto-mitochondrion (Iyer et al. Citation2004). The ClpAB family is discussed in the clade 5 section of the review.
Clades 4–7: pre-sensor-1 β-hairpin (PS1βH) superclade
All four subsequent clades share a conserved insertion between α3 and β4, the strand containing the sensor-1 motif. This insert forms a β-hairpin that projects out of the AAA+ core () (Erzberger and Berger Citation2006).
Clade 4: superfamily III (SF3) helicase clade
In addition to the PS1βH insertion, the C-terminal alpha helical bundle is replaced with a unique arrangement of C-terminal elements.
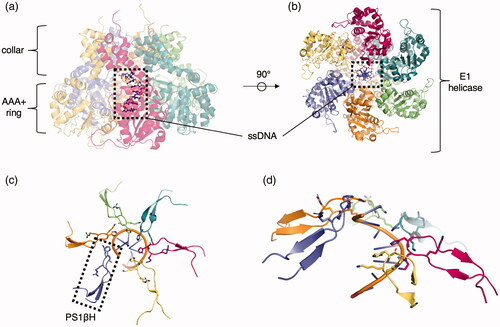
The PS1βH is present in the central channel of the oligomer and interacts with a nucleic acid substrate (a representative example is shown in ). The SF3 helicases are found in RNA and DNA viruses but not in cellular genomes, aside from viral remnants. These initiators form closed hexameric rings around the DNA. In the SV40 initiation complex, His513 and Phe459 of the β-hairpin, along with Lys512 and Lys516, interact with and help separate the dsDNA. These and other residues allow for proper DNA melting and processive unwinding (Shen et al. Citation2005). Similarly, the E1 protein of papillomavirus is a clade 4 helicase. It engages DNA through the use of Lys506, coordinating the ssDNA phosphate, the main-chain amide of His507 (both of which are found in the β-hairpin insert) and several other residues () (Enemark and Joshua-Tor Citation2006). Interestingly, the REP68 complex in adeno-associated virus forms a heptameric AAA+ ring structure (Santosh et al. Citation2020). Under different conditions, depending on the presence of substrate and type of nucleotide, the AAA+ ring (referred to as the SF3 helicase domain) could transition into a hexameric form as well.
Figure 6. Representative clade 4 structure. (a) Side view of papillomavirus E1 hexameric helicase bound to MgADP and ssDNA (PDB: 2GXA (Enemark & Joshua-Tor Citation2006)). Each identical protomer is uniquely colored with ssDNA (ball and stick) centered in the pore. (b) Top view of the helicase complex with ssDNA in the pore (ball and stick). (c) Engagement of the K506 and H507 from the PS1βH insertion to the ssDNA (cartoon). (d) Side view of the staircase engagement of the six protomers via the PS1βH to the ssDNA (cartoon).
Clade 5: HslU/ClpAB/lon/RuvB (HCLR) clade
There are no additional, distinctive structural features aside from the PS1βH ().
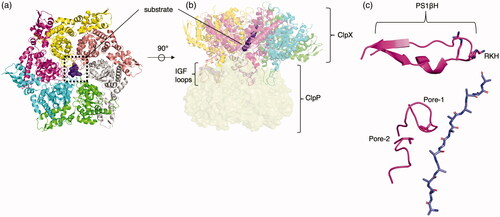
The HCLR clade is comprised of AAA+ proteins that act as chaperones and proteases. The HCLR clade is comprised of four major families: HslU, ClpAB (C-terminal domain), Lon family, and RuvB family. The HslU (heat shock locus U) family (also called the Hsp100 or Clp family) is a bacterial family that has two orthologous lineages, HslU and ClpX. Both HslU and ClpX proteins have also made their way into eukaryotic organisms, likely via acquisition from the proto-mitochondrion. HslU (sometimes called ClpY) forms a hexamer and coordinates itself with proteolytic HslV (also hexameric, sometimes called ClpQ) to form a bacterial protease(Sousa et al. Citation2000). Two HslU hexamers sandwich two HsIV hexamers to form a cylindrical hetero-24mer (Bochtler et al. Citation2000). ClpX forms a hexamer as well (Glynn et al. Citation2009) and associates with its conjugate protease ClpP (Wang et al. Citation1997) to form the ClpXP protease (Gatsogiannis et al. Citation2019; Fei et al. Citation2020; Ripstein et al. Citation2020), which recognizes and degrades proteins that have a degradation signal in a multistep binding and engagement process (Saunders et al. Citation2020). Some of these degradation signals are added to the C-terminus of an incomplete protein from a stalled ribosome, which directs the incomplete protein to ClpXP for degradation (Flynn et al. Citation2001). ClpX on its own forms an asymmetric homohexamer similar to HslU but docks onto ClpP, which forms a homoheptameric complex. ClpX binds substrate through via pore-1, pore-2 and RKH loops (). The RKH loop is part of the β-hairpin loop. ClpX docks into ClpP using flexible IGF loops that fit into hydrophobic pockets of ClpP. Structures showing ClpXP forming a “double capped” complex with two ClpX hexamers flanking a double-ringed heptameric ClpP have also been found, analogous to HslUV. The ClpXP is a principal player in cellular homeostasis (Bhandari et al. Citation2018) and has recently been leveraged as a therapeutic target against cancer (Ishizawa et al. Citation2019). Mitochondrial ClpX activates 5-aminoleuvilinic acid synthase not by proteolysis but by remodeling the peptide substrate, demonstrating that ClpX can function independently of ClpP as an unfoldase. (Kardon et al. Citation2020).
Figure 7. Representative clade 5 structure. (a) Top view of ClpXP engaged to substrate (PDB: 6PP5 (Fei et al. Citation2020)) trimmed to include ClpX and substrate (purple surface). Each identical protomer is uniquely colored. (b) Side view of complex with flexible IGF loops used to dock into ClpP (yellow, transparent surface) hanging from the bottom of each protomer. (c) Engagement of a single protomer (pink) to substrate via, from top to bottom, RKH loop, pore-1-loop (tyrosine), and pore-2-loop (valine).
The second tandem AAA+ domain of the ClpAB proteins (C-terminal domain) and other related proteins make up another bacterio-eukaryotic family. Unlike ClpX, ClpA is a double-ringed AAA+ protein that associates with ClpP but still maintains a hexamer-heptamer mismatch interaction. ClpA is found gram-negative bacteria, while close AAA+ relative ClpC, a functional ortholog, is found in gram-positive bacteria and cyanobacteria (Hamon et al. Citation2015). The first domain of ClpA is referred to as D1 and the second domain is referred to as D2, a common naming convention applied to AAA+ proteins with two connected ATPase domains. ClpP consists of two heptameric rings stacked together. Together, ClpA and ClpP form either a ClpAP 20-mer or 26-mer, with ClpA docking with ClpP via flexible IGL loops on one or both axial faces of ClpP. ClpA’s IGL loops fill 6 out of the 7 hydrophobic pockets of ClpP. Pocket switching of the IGL loops appears to be associated with ClpA rotation, suggesting that ClpA uses ClpP as a surface to push against and rotate about during translocation (Lopez et al. Citation2020). Like ClpA, ClpC also contains tandem AAA+ domains, with the first belonging to the classical clade and the second belonging to the HCLR clade. It also associates with ClpP to form a proteolytic complex but also requires the adaptor protein MecA (Wang et al. Citation2011). ClpE is also a double-ringed AAA+ closely related to ClpA and ClpC and presumably functions in a similar manner to form a ClpEP proteolytic complex (Kress et al. Citation2009). ClpL, a close relative to ClpC, is found mostly in gram-positive bacteria and requires no other proteins to function as a chaperone (Park et al. Citation2015). Curiously, it forms a tetradecameric complex of two heptameric rings with unfoldase activity (Kim et al. Citation2020). ClpD, another close relative of ClpC, is found in bacteria and eukaryotes—specifically, in plants, where it localizes to the chloroplast and presumably also associates with ClpP (Singh and Grover Citation2010).
The directionality of substrate loading (i.e. N-terminus or C-terminus inserted into pore) is key to determining the efficiency of energy coupling to unfolding (Olivares et al. Citation2017). Single-molecule optical trap experiments for both ClpAP and ClpXP suggested that titin is unfolded more quickly when the N-terminus is loaded versus the C-terminus (controlled by location of the degron signal) (Olivares et al. Citation2017) and that local mechanical stability at the position proximal to the enzymatic complex is rate limiting (Cordova et al. Citation2014; Olivares et al. Citation2017). The double-ringed ClpA unfolded some substrates faster than the single ringed ClpX, while taking smaller 1–2 nm step sizes as opposed to 1–4 nm by ClpX despite this similarity in energy coupling with regards to N-terminus or C-terminus loading (Olivares et al. Citation2014).
ClpB, with ortholog Hsp104/Skd3 in eukaryotes, is a disaggregase that associates with DnaK/Hsp70 to unravel proteins (Deville et al. Citation2019). It consists of a two AAA+ domains, with an M-domain between them. ClpB does not associate with ClpP and does not form a proteolytic complex. The coiled-coil M-domain is thought to regulate ClpB activity by encircling the first AAA+ domain. The first AAA+ domain has a tyrosine residue on its pore loop (because the first AAA+ domain belongs to the classical clade) and a secondary, less-conserved loop that appears to interact with substrate as well. Although the second AAA+ domain seems to be more important to disaggregase activity than the first, the PS1βH does not engage substrate; instead, a small tyrosine-containing motif interrupts the alpha-helical element between β2 and β3 and interacts with substrate. As with many AAA+ proteins, mutation of ClpB leads to many diseases, ranging from brain atrophy (Wortmann et al. Citation2015) to aciduria (Kiykim et al. Citation2016). It has been suggested that Hsp104 could be repurposed to combat neurodegenerative disease caused by protein aggregation (March et al. Citation2019) considering the fact that the disaggregase activity of Skd3 (ClpB/Hsp104 in humans) is critical in maintaining mitochondrial proteostasis (Cupo and Shorter Citation2020).
Torsin proteins are also members of the ClpAB family because they share homology to the 2nd tandem AAA+ domain of the ClpAB family (Iyer et al. Citation2004). Torsin proteins are primarily limited to metazoans (Rose et al. Citation2015), suggesting that they evolved rapidly from eukaryotic ClpAB into their own subfamily (Iyer et al. Citation2004). TorsinA is ubiquitously expressed in all cell types, and deletion(s) in TorsinA are associated with early-onset torsion dystonia (EOTD) (Rose et al. Citation2015). Torsin localizes to the ER membrane and has been shown to affect lipid content in Drosophila (Grillet et al. Citation2016). Torsin ATPase activity, unlike other AAA+ proteins, relies on cofactors LAP1 and LULL1 (Goodchild and Dauer Citation2005; Brown et al. Citation2014). Structures of TorsinA bound to LULL1 or LAP1 reveal that both of these activators have conserved arginines that activate TorsinA in trans (Sosa et al. Citation2014; Demircioglu et al. Citation2016). Additionally, TorsinA has been found to form helical filaments (Demircioglu et al. Citation2019), reminiscent of the DnaA right-handed superhelix (Erzberger et al. Citation2006).
Lon proteins comprise the third major family in the HCLR clade. There are two major clades within this family, the bacterial Lon lineage (which made its way into eukaryotes via mitochondria) and the archaeal Lon lineage (Iyer et al. Citation2004). The bacterial Lon proteins consist of a AAA+ domain with an N-terminal domain that functions in the recognition of substrate and a C-terminal protease domain (Vieux et al. Citation2013). Like other AAA+ proteins, four Lon protomers bind the substrate while the top and bottom protomer occupy do not directly bind the substrate and have a gap between the two protomers, also called the seam region (Zhang et al. Citation2020). When not bound to substrate, bacterial Lon adopts a “locked,” left-handed spiral that is ADP bound. When bound to substrate, Lon adopts a “closed,” right-handed spiral along the translocating peptide. PS1βH serves to stabilize substrate-engaged organization of the pore loops, playing a critical role in substrate processing (Shin et al. Citation2020).
The RuvB family associates with double-stranded DNA and assists in recombination in bacteria. RuvB, in concert with RuvA and RuvC, processes Holliday junctions. RuvB appears to form a hexamer that surrounds the DNA. The beta hairpin of RuvB interacts with RuvA (Yamada et al. Citation2002).
Clade 6: H2 insert clade
In addition to the PS1βH, there is another β-hairpin insertion in α2 (Erzberger and Berger Citation2006).
There are two families that make up clade 6: the archaeo-prokaryotic-eukaryotic McrB family and a prokaryotic family of σ54 -related transcription factors (Iyer et al. Citation2004). The McrB family uses GTP instead of ATP, unlike most AAA+ proteins (Nirwan, Singh, et al. Citation2019). They are sporadically distributed throughout bacterial, archaeal, and animal phylogeny. In bacteria and archaea, McrB is encoded on a mobile operon that encodes a modification-dependent restriction enzyme system. The N-terminal domain of McrB forms a DNA-binding domain that precedes the AAA+ domain. McrB then associates with the nucleolytic McrC to form the McrBC complex (), which can recognize and cleave DNA. McrB can form both homohexamers and homoheptamers (Panne et al. Citation2001). In a structure of McrB in complex with McrC, McrB forms an hexameric oligomer (Nirwan, Itoh, et al. Citation2019). Two hexameric rings of McrB are linked to McrC to form a 14-mer structure. Furthermore, this complex with hexameric McrB is catalytically active in cleaving DNA and consuming GTP. Additionally, GTPase activity of McrB was stimulated upon the addition of McrC, following the theme of stimulated ATP/GTPase activity in the presence of substrate or a substrate-interacting domain. McrB-related proteins in eukaryotes were likely acquired through horizontal gene transfer. A eukaryotic member, Unc53/Nav2/HELAD1, contains an N-terminal calponin-homology (CH) domain that precedes the AAA+ domain (Muley et al. Citation2008). This protein is required for the process of axonal elongations it facilitates interactions between microtubules and neurofilaments, and it exhibits 3′ to 5′ helicase activity and exonuclease activity in vitro (Ishiguro et al. Citation2002).
Figure 8. Representative clade 6 structures. (a) Top view of hetero 14-mer McrBC complex, two McrB hexamers (green and red surface) and two McrC (gray and purple) chains (PDB: 6HZ5 (Nirwan, Itoh, et al. Citation2019)). The two McrC chains (purple and gray) form a dimeric bridge between the two McrB hexamers that penetrates the center of the pore. (b) Side view of McrBC complex. (c) Top view of the NtrC1 heptamer (PDB: 3M0E (Chen et al. Citation2010)). Each identical protomer is uniquely colored. (d) Sideview of the NtrC1 heptamer.
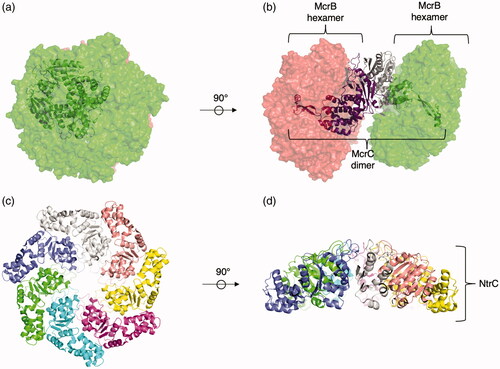
Proteins from the σ54-related transcription factors family assist in transcription by the σ54 RNA polymerase, which is used to transcribe genes involved in the stress response (Yang et al. Citation2015). Proteins in the σ54-related transcription factors family have diversified with various N-terminal fusions connected to the ATPase; these domains allow them to respond to small molecules in bacteria by triggering a conformational change of σ54. NtrC, one such member of this family, controls the transcription of nitrogen-related genes. σ54 requires NtrC to transition from a closed complex to an open complex (Soules et al. Citation2020). NtrC1, a protein in the family, binds to σ54 through a highly conserved GAFTGA motif centered in the α2 insertion (Bush and Dixon Citation2012). NtrC1 is heptameric(Lee et al. Citation2003; Chen et al. Citation2010) under a variety of conditions, with and without mutation to the Walker B site (). Moreso, the heptameric NtrC1 binds σ54. It is not clear if the heptameric state bound to substrate is the active form or a locked form prior to activation. When bound to ATP, a series of conformational changes are associated with the binding of the arginine finger to the γ-phosphate group. Notably, this leads to stabilization of the loop containing the GAFTGA motif used for engagement of RNA polymerase.
Clade 7: PS-II insert clade
In addition to the β-hairpin insertion in α2, an additional α-helix in the C-terminal α helical bundle causes a repositioning of the sensor-2 motif ( and ) (Erzberger and Berger Citation2006).
Figure 9. Representative clade 7 structures. (a) Top view of the MCM complex (PDB: 6MII (Meagher et al. Citation2019)) bound to ssDNA (shown as cartoon). Representative chains are colored uniquely. (b) Large and small subdomains of a single MCM AAA+ domain. Core β-sheets are colored orange, surrounding area of large subdomain are colored green and the small subdomain is colored yellow (first three helices) and purple (fourth helix). The PS1βH is colored blue, the helix-2 insert is colored yellow, and the C-terminal helix insertion is colored gray. (c) Front view of core AAA+ MCM domain. (d) Side view of Dynein chain (PDB: 4RH7 (Schmidt et al. Citation2015)). Dynein has all six of its AAA+ domains and the rest of its domains linked in a single polypeptide. AAA1 is colored red, AAA2 is colored blue, AAA3 is colored orange, AAA4 is colored magenta, AAA5 is colored yellow, AAA6 is colored gray, the stalk is colored pink, and intervening regions are colored green.
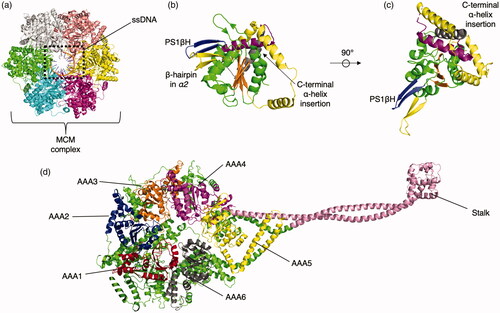
Clade 7 is made up of the following families: MCM Family, MoxR family, Chelatase/YifB family, and Dynein/Midasin/Mysterin family. The archaeo-eukaryotic MCM family has been studied extensively in the context of DNA replication. In both archaea and eukaryotes, MCM is a hexameric ring that serves as the helicase loaded by clade 2 Orc1/Cdc6 or Cdc6 respectively (Duderstadt and Berger Citation2008). The helicase hydrolyzes ATP to perform strand separation at the replication fork, subsequently allowing the replication machinery to continue. In archaea (e.g. Sulfolobus solfataricus), the MCM ring is a homohexamer of six identical MCM proteins and is able to unwind DNA without forming a larger complex in vitro () (Meagher et al. Citation2019). The N-terminal domain of an archaeal MCM subunit binds DNA. The C-terminal domain contains a helix-turn-helix motif, a common DNA-binding motif, and the PS-II AAA+ domain is flanked by these domains in the middle (). Two MCM spiral staircase-shaped hexamers form a double ring in a head-to-head configuration. Both of the hairpins, the H2 insert, and PS1βH, project into the central channel and bind the DNA; one protomer interacts with two nucleotides at a time, the same ratio observed for DnaB. The PS1βH interaction with the DNA is comparable to how papilloma virus E1 from clade 4 interacts with DNA. The PS1βH A431 amide interacts with one phosphate of the ssDNA while the PS1βH K430 forms an ionic interaction with the phosphate immediately 3′. The H2 V377 amide and the side chain of T369 also bind phosphates in the same manner.
The eukaryotic MCM ring consists of the six related proteins, MCM2–7 (Li et al. Citation2015). In eukaryotes, the active replicative helicase consists of Cdc45-MCM2-7-GINS to form the CMG complex. CMG unwinds duplex DNA from 3′–5′. MCM2-7 also forms a double head-to-head configuration of hexameric rings, mediated by the zinc fingers in the N-domains. The eukaryotic MCM proteins differ from the single archaeal MCM protein in that each subunit has N and C terminal extensions and insertions. These extensions contribute to inter-hexamer interactions, such as between the zinc finger of MCM2 of one and β-turn of MCM6 of another, the N-terminal domain MCM5, an N-terminal insertion of MCM7, and lastly, the N-terminal domains of MCM3 and MCM7. The H2I and PS1βH also arrange themselves in a spiral fashion in the eukaryotic variant.
The MoxR family is well-represented in most bacteria and archaea, where they are thought to function as chaperones for metabolic complexes (Iyer et al. Citation2004). For example, in P. denitrificans, norQ codes for a MoxR ATPase that works in inserting an iron cofactor into an oxide reductase (Snider et al. Citation2006). CbbQ, found in some chemoautrophic bateria, forms a hexameric complex to which the CbbO adaptor protein binds (Tsai et al. Citation2020). RavA is another well-characterized protein in the MoxR family that works in concert with von Willebrand factor A (VWA) domain-containing protein ViaA (Jessop et al. Citation2020). RavA is also known to bind to lysine decarboxylase LdcI (El Bakkouri et al. Citation2010; Kandiah et al. Citation2016; Jessop et al. Citation2020). RavA is comprised of a AAA+ domain: a triple helical domain and a LARA domain (LdcI associating domain of RavA). RavA forms a cage with the LdcI decamer to form a larger complex. RavA on its own forms a hexamer, and five of these hexamers arrange themselves around two LdcI decamers to form the RavA-LdcI cage. The LARA domain mediates interaction between RavA and LdcI.
The YifB/chelatase family is a family found exclusively in bacteria. The YifB arm of this family is fused to a Lon protease domain N-terminally and has an insertion of a zinc cluster in the fourth beta strand of the AAA+ core domain (Iyer et al. Citation2004). Members of the chelatase family have two AAA+ domains. In the ChID protein, the first is active, and the second is inactive, similar to the arrangement of NSF’s D1 and D2 domains (Adams & Reid Citation2013). Other magnesium chelatases, such as BchI from R. capsulatus and Ch1I from Synechocystis, can form both hexameric and heptameric rings (Lundqvist et al. Citation2010).
Finally, the dynein/midasin/mysterin family is unique in that all six tandem AAA+ domains are encoded on a single polypeptide, instead of forming from six identical or similar protomers (). Dynein, extensively studied, is found in eukaryotes and associates with microtubules to transport macromolecular complexes around the cell. The linear sequence of dynein from N to C terminus consists of a tail region, an N-terminal sequence, the first four AAA+ domains, a microtubule-binding domain (MTBD), the fifth and sixth AAA+ domains, and then a C-terminal sequence(Roberts et al. Citation2009). The “head” of dynein is composed of a pseudo-hexameric arrangement of the six AAA+ domains, and the MTBD is at the end of a long stalk that points away from the pseudo-hexameric ring. Dynein functions as a dimer of these tailed, pseudo-hexameric ATPase rings, which are in turn connected by the tail domains and several light and intermediate chains. Inactivation of the first AAA+ domain renders dynein completely immobile, inactivation of the third dramatically reduces the velocity of dynein (Cho et al. Citation2008), and inactivation of the fourth domain renders dynein immotile (Liu et al. Citation2020). The first four AAA+ domains can bind nucleotide, but the fifth and sixth cannot. A comparison of structures of dynein bound to either ADP or ADP-vanadate suggests that closure and opening of the hexameric ring leads to a steric clash with the linker, thus generating movement (Schmidt et al. Citation2015).
Midasin or Rea1 mechanically removes ribosomal assembly factors to promote the maturation of the pre-60S complex, which eventually leads to its export to the cytosol (Sosnowski et al. Citation2018). The N to C arrangement of Rea1 is as follows: N-terminal domain, the six AAA+ domains, a larger tail linker region, and a tail C-terminus that includes a Metal Ion Dependent Adhesion Site (MIDAS), which interacts with the substrates that are removed from the pre-60S particles. Rea1 has several tail conformations that, like dynein, could produce force to dislodge the factors. The first and last AAA+ domains lack the Walker B motif required to hydrolyze ATP and are thus nonfunctional.
Mysterin, or RNF213 (RING finger protein 213), is a large, 500–600 kDa eukaryotic protein that consists of six linked AAA+ domains, as well as an N-arm and a multidomain E3 domain responsible for ubiquitin ligase activity (Ahel et al. Citation2020). Mutations in mysterin significantly increase the risk for Moyamoya disease, a cerebrovascular disease (Morito et al. Citation2014). It is targeted to lipid droplets, where it plays a role in lipid metabolism and other biological processes (Sugihara et al. Citation2019). Only the third and fourth AAA+ domains are catalytically competent, while the second AAA+ domain binds ATP but lacks a Walker B motif and is thus unable to hydrolyze ATP (Ahel et al. Citation2020).
The Pch2/Trip13 family is a AAA+ family that has no discernable placement in any of the seven clades (Ye et al. Citation2015). It lacks a clear β hairpin insertion before the fourth β strand after the third α helix, disqualifying it from clades 4–7. It also lacks the synapomorphic features of an “RC” finger from clade 1 members, or a long α helical insertion as in clade 2 members. It also has a functional sensor 2 motif, unlike the classical clade 3 members that have a mutated alanine at the position instead of a conserved polar residue. Based on phylogenetic and structural analysis (Ye et al. Citation2015), we postulate that it diverged from a clade 3 and clades 4–7 ancestor. This eukaryotic protein family is a key regulatory in spindle assembly. TRIP13, with its adaptor protein p31comet, loads substrate protein MAD2 in a spiral conformation (Alfieri et al. Citation2018) similar to other AAA+ proteins(Zhao et al. Citation2015; Puchades et al. Citation2017; White et al. Citation2018; Fei et al. Citation2020; Ripstein et al. Citation2020; Saunders et al. Citation2020).
N-ethylmaleimide sensitive factor (NSF) as a model system for the AAA+ superfamily
NSF (also known as Sec18 in yeast (Novick et al. Citation1980) and comatose (Siddiqi & Benzer Citation1976; Dellinger et al. Citation2000) in Drosophila) is a protein found in all eukaryotic cells. Early studies identified conditional mutants of the gene responsible for membrane transport defects at restrictive temperatures (Novick et al. Citation1980). In 1988, Rothman and colleagues discovered a protein that was essential for vesicular transport in eucaryotic cells (Block et al. Citation1988; Malhotra et al. Citation1988). This vesicle transport activity was ablated by the addition of the chemical N-ethylmaleimide (NEM), leading to the naming of the protein as N-ethylmaleimide-sensitive-factor (NSF). Rothman and colleagues demonstrated inhibition of transport by either titrating in more NEM or by using an antibody targeting NSF. Subsequently, SNARE proteins were identified as substrates for NSF in a process requiring the adapter protein SNAP (Sec17p in yeast) (Weidman et al. Citation1989).
SNARE proteins are essential factors required for vesicle fusion in a variety of cellular contexts. For example, they play a key role in fusion during intravesicular transport, such as cargo transport through the Golgi apparatus (Malsam and Söllner Citation2011), and secretion, as in the case of neurotransmitter release (Südhof Citation2013; Rothman Citation2014). Mechanistically, this is accomplished by the association of SNARE proteins on opposing membranes to form the trans SNARE complex (Sutton et al. Citation1998; Weber et al. Citation1998). Fusion is then achieved as the SNARE proteins more tightly associate to form a helical bundle. Following fusion, the SNARE proteins are found in a stable complex anchored to the same membrane, the cis SNARE complex. NSF maintains a supply of individual SNAREs by disassembly of these post-fusion cis SNARE complexes (Söllner et al. Citation1993). Moreover, NSF is involved in quality control, as it promotes the formation of fusogenic SNARE complexes by disassembly of the syntaxin-SNAP-25 binary complex as well as off-pathway complexes (Ma et al. Citation2013; Lai et al. Citation2017; Brunger et al. Citation2018).
NSF engages a variety of SNARE complexes through adaptor proteins called soluble NSF-attachment proteins (SNAPs). NSF, multiple SNAPs, and a SNARE complex form a super-complex (often referred to as 20S complex), the starting state for SNARE complex disassembly (Malhotra et al. Citation1988; Whiteheart et al. Citation1992; Söllner et al. Citation1993; Mayer et al. Citation1996; Hanson et al. Citation1997). NSF’s ability to disassemble post-fusion SNARE complexes is dependent on the hydrolysis of ATP (Nagiec et al. Citation1995). There is only one copy of NSF in most eukaryotic organisms, and null mutations known to date are largely lethal (Bayless et al. Citation2018).
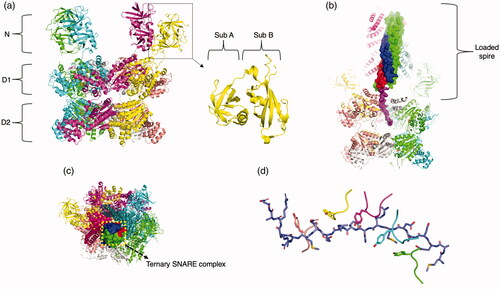
NSF is comprised of three domains, an N-domain, the first AAA+ domain (D1), and the second AAA+ domain (D2) (). These domains are discrete structural entities, as was first recognized by limited proteolysis studies (Tagaya et al. Citation1993). NSF may have branched off from the archaeo-eukaryotic CDC48 family, which also consists of an N-domain, a D1, and a D2 domain. The structure of the D2 domain was among the first two structures of the "classical" clade 3 AAA+ fold (). Moreover, the crystal structures of the NSF D2 domain provided the first insights into the oligomeric arrangements of AAA+ domains by revealing the structural basis of the formation of the NSF D2 hexameric ring (Lenzen et al. Citation1998; Yu et al. Citation1998).
Figure 10. NSF. (a) NSF hexamer with four N-domains in up position modeled with the D1 and D2 ring (PDB: 6MDN (White et al. Citation2018), trimmed to not include ternary complex and αSNAP). Each macromolecular chain is colored uniquely. Subdomain A and subdomain B of the N-terminal domain is labeled. (b) NSF/SNAP/SNARE complex of NSF with αSNAP and ternary complex SNARE (PDB: 6MDN, full structure). Synaptobrevin is colored blue, syntaxin is colored red and SNAP25 is colored green, except its interacting N-terminus, which is colored purple. (c) Top view of the NSF/SNAP/SNARE complex. (d) Depiction of the four fully engaged protomers and one partially engaged protomer to SNAP25 via tyrosine in the pore loop.
The biochemical properties of the NSF AAA+ domains point to diverging functions. The D1 domain of NSF binds ATP weakly in comparison to the D2 domain. For the D1 domain, the KD for ATP is approximately 15–20 μM, while in the case of the D2 domain, the KD is approximately 30–40 nM (Matveeva et al. Citation1997). Although both the D1 and D2 domains hydrolyze ATP, the D2 does so very slowly (Morgan et al. Citation1994). When nucleotide is entirely stripped from NSF, it reverts to a monomeric state, and upon reconstitution with ATP, it re-forms a hexamer (Zhao et al. Citation2015). Together, these observations suggest that the D2 domain is primarily responsible for oligomerization, while the D1 domain actively processes substrate over successive rounds of hydrolysis. The order of the domains also matters, as flipping the D2 domain for the D1 domain completely destroys the activity of the enzymatic complex (Whiteheart et al. Citation1994). Mutations to the Walker A or B motif of the D1 domain (K266A and E329Q) leave the complex catalytically dead (Matveeva et al. Citation1997). As mentioned above, the Walker A motif is involved in binding nucleotide, whereas the Walker B motif plays a role in hydrolysis. The D1 Walker B mutation (E329Q) is still able to form the NSF/SNAP/SNARE complex at WT levels, while the D1 Walker A mutation (K266A) only forms ∼10% of complex compared to WT. Mutation of the D2 Walker B site (D604Q) still allows for NSF/SNAP/SNARE formation and disassembly activity, which is in line with the hypothesis that the D1 domain provides the majority of ATPase activity required for function (although it is theoretically possible that ATPase activity of D2 may be stimulated by substrate). Interestingly, mutation of the D2 Walker A site (K549A) still allows for NSF/SNAP/SNARE complex formation and intermediate amounts of activity.
The N-domain interacts with the SNAP adaptor proteins to bind substrate, following another key trend in AAA+ N-domains (Hohl et al. Citation1998). The N-domain is composed of two different sub-domains, A and B, joined by a linker () (May et al. Citation1999; Yu et al. Citation1999). The A sub-domain is a double-ψ β barrel (Castillo et al. Citation1999) (DPBB), appropriately named due to the two interlocking ψ loops. The first ψ loop lies between the first and second β-strands, and the second ψ loop is between fourth and fifth β-strands. The DPBB of A domain varies from the canonical structure only by the location of the first ψ loop being between the first and second β-strands, instead of the canonical second and third. The B-domain is an α/β roll, with one primary alpha helix surrounded by four beta strands. The core of the roll between the α-helix and the four β-strands is stabilized by a row of conserved phenylalanine and tyrosine side chains, forming an aromatic ladder. The overall charge of the N-domain is positive, and it has an extremely positive “groove” which serves as a point of electrostatic interaction with the negatively-charged interface of the C-terminal region of the SNAP protein (Zhao et al. Citation2015). Deletion of the N-domain prevents formation of the NSF/SNAP/SNARE complex with the SNAPs and SNARE complex. The recombinant N-domain alone does not bind SNAP/SNARE either (Nagiec et al. Citation1995), but when fused to a single D1 or D2 domain it is able to form a complex with SNAP, suggesting that the N-domain in the context of a AAA+ domain is the minimum requirement for SNAP binding. The N-domain of NSF is similar to the CDC48/p97 family (Iyer et al. Citation2004).
The oligomeric state of NSF is primarily hexameric as supported by solution studies (Fleming et al. Citation1998), structural studies of the D2 domain (Lenzen et al. Citation1998; Yu et al. Citation1998), and, more recently, cryo-EM structures of NSF (Zhao et al. Citation2015). The number of SNAP molecules involved varies from 2 to 4 (Shah et al. Citation2015; Zhao et al. Citation2015; White et al. Citation2018).
Structure determination of full-length NSF and the NSF/SNAP/SNARE complex proved extremely challenging. Crystallization of full-length NSF as well as the NSF/SNAP/SNARE complex has been unsuccessful to date, and early cryo-EM studies resulted in low-resolution maps that did not reveal SNAREs/SNAPs or artificially symmetry averaged densities of SNAREs/SNAPs (Furst et al. Citation2003; Chang et al. Citation2012). The first near-atomic resolution cryo-EM structures of both apo NSF and the NSF/SNAP/SNARE complex were determined in 2015 (Zhao et al. Citation2015). The structure of the NSF/SNAP/SNARE complex revealed the engagement of NSF with αSNAP and a highly truncated neuronal SNARE complex composed of syntaxin-1a, synaptobrevin-2, and SNAP-25a in the presence of the non-hydrolysable nucleotide analog AMPPNP. This success was due to an extensive purification scheme; NSF was ectopically expressed in E. coli, reduced to monomeric state through repeated buffer exchange and removal of nucleotide, and then reassembled during size exclusion chromatography with re-addition of nucleotide. With this purification scheme, hexameric NSF of exceptional homogeneity and purity can be prepared in the presence of a variety of nucleotides and nucleotide analogues. Moreover, the disassembly activity of this highly purified NSF sample is much higher than that obtained by previously published methods. Advances in cryo-EM methodology and the availability of direct electron detectors also played a key role (Bammes et al. Citation2012). Subsequently, an even higher-resolution structure of the NSF/SNAP/SNARE complex was determined by cryo-EM with αSNAP and a near-full length neuronal SNARE complex, in the presence of ATP under non-hydrolyzing conditions (White et al. Citation2018) (). This success was due to slight truncation of the SNARE complex at the C-terminal end, reducing aggregation and association of particles on EM grids, and use of a more advanced electron microscope. Critically, this cryo-EM structure approached near-atomic resolution and for the first time revealed the engagement of 17 N-terminal residues of SNAP-25a by the NSF D1 ring (White et al. Citation2018). This structure was among the very first to reveal any substrate-clade 3 interactions.
Together, these cryo-EM structures (Zhao et al. Citation2015; White et al. Citation2018) provided many new insights about NSF and the NSF/SNAP/SNARE complex. The fold of the AAA+ domain is conventional for both D1 and D2, with the notable exception of a bent α2 helix in the D1 domain. This is not the case for the D2 domain and a close relative, p97, which both have the classical straight helix. In ATP-bound NSF (ATP-NSF), all six N-domains are facing upwards, opposite to direction of substrate translocation, whereas in ADP-bound NSF (ADP-NSF), four are up, and two are facing downward, hugging the side of the double-ringed hexamer (Zhao et al. Citation2015). ATP-NSF has a symmetric D2 ring and asymmetric D1 ring, with a split between the first and last protomer both in the plane of the ring and along the pore axis. ADP-NSF is more planar than ATP-NSF but maintains a larger gap between the first and last protomer. The α7 helix is also significantly translated between ATP-NSF and ADP-NSF, likely due to the difference in nucleotide state in the D1 ring.
As noted previously, NSF requires the SNAPs to mediate interactions with various SNARE complexes; the SNAPs and their yeast homolog Sec17 are composed of an extensive twisted sheet of α helical hairpins and a C-terminal helical bundle (Rice and Brunger Citation1999) that interacts with the NSF N-domain (). A hydrophobic loop in the N-terminal region of SNAP might serve as a membrane attachment site as disassembly activity is greatly enhanced in the presence of membranes mediated by this N-terminal region of SNAP (Winter et al. Citation2009). The SNAPs directly interact with the SNARE complex via primarily electrostatic interactions (Vivona et al. Citation2013; Zhao et al. Citation2015; White et al. Citation2018). The SNARE complex has a highly conserved characteristic pattern of positive and negative surface charges (conserved across all species and localizations investigated so far) with which the SNAPs interact; a minimum of two SNAPs interface with this pattern through their own complementary charged surfaces in all structures observed to date, with two additional SNAPs possibly binding as well. The presence of the SNAP-25a linker may preclude binding of an additional two αSNAPs as observed in the NSF/SNAP/SNARE structure lacking this fragment (Zhao et al. Citation2015; White et al. Citation2018). Biochemical data highlight the importance of the charged SNARE complex surface in disassembly; NSF is unable to disassemble a four-helix bundle that does not have the characteristic surface charge distribution of SNARE complexes (Cipriano et al. Citation2013). Interestingly, NSF is capable of disassembling a “double” SNARE complex created by linking two SNARE complexes in a head-to-tail fashion (Cipriano et al. Citation2013).
The position of the N-domains relative to the D1 ring is likely related to substrate recognition. N-domains in the “up” position interact with αSNAP electrostatically, typically with two N-domains associating with negative residues in the C-terminal domain of αSNAP. This series of SNAP-SNARE and SNAP-NSF interactions positions the N-terminal end of the SNARE complex proximal to the D1 pore. This pore is formed primarily by protomers A–F of the D1 ring, which are arranged to form an asymmetric staircase configuration with an average rotation of 57.5° ± 1.0° around the pore axis (). The N-terminal residues of one of the neuronal SNAREs, SNAP-25a, are gripped by a spiraling array of tyrosine residues on the NSF D1 pore loop 1’s of protomers A–E (White et al. Citation2018); this structure was determined in the presence of ATP and EDTA (i.e. under non-hydrolyzing conditions). Each tyrosine intercalates between every other amino acid of the substrate by engaging in a hydrogen bonding interaction with SNAP-25a’s backbone carbonyl, as well as apparently nonspecific packing interactions with the substrate sidechains themselves ().
The available cryo-EM structures of NSF/SNAP/SNARE complexes so far have not revealed any density inside the D2 pore. Moreover, the register of the N-terminal residues of SNAP-25 is uniquely determined by the position of the SNARE complex (White et al. Citation2018) (), precluding extension of this chain into the D2 pore. Of course, it cannot be ruled out that substrate may enter the D2 pore under hydrolyzing conditions.
The density corresponding to the D1 F protomer is poor, indicative of high mobility and thus disengagement from the rest of the D1 ring. The overall configuration of the D1 ring with respect to substrate is consistent with other classical clade 3 AAA+ proteins such as YME1, Vps4, and many others (Caillat et al. Citation2015; Monroe and Hill Citation2016; Monroe et al. Citation2017; Puchades et al. Citation2017; Sun et al. Citation2017); apical aromatic residues at the pore loop 1 intercalate between every other amino acid of the substrate. However, the NSF D2 pore loops lack a conserved aromatic residue, suggestive of a minimal role in the active processing of substrate and casting further doubt on the possibility of substrate threading through the D2 pore. Furthermore, despite the asymmetry of the D1 ring, the D2 ring remains largely symmetric in the currently available cryo-EM structures and probably serves as a rigid platform, with conformational change of the D1-D2 linker accommodating the asymmetric arrangement of the D1 domains.
The structural details of the SNARE complex disassembly process remain largely unknown. Based on the structures of the substrate-loaded complex (Zhao et al. Citation2015; White et al. Citation2018), we speculated that pulling and subsequent translocation of the SNARE protein (the N-terminal region of SNAP-25 in the available structures) engaged by the D1 ring leads to destabilization of the ternary complex, possibly through the disruption of the N-terminal portion of the SNARE bundle. This process could be further driven by the motion of the N-domains and associated ɑSNAP molecules; we note that the ɑSNAPs themselves wrap around the SNARE complex in the opposite direction of the twist in the ternary SNARE complex. Following destabilization of the SNARE complex, the SNAP-25 N-terminus could be released through a split in the D1 and/or D2 rings of NSF, as suggested by the mobility of the F-protomer, while the other SNAREs could simply disassociate.
The energetics and dynamics of SNARE complex disassembly process have been studied as well. Single molecule experiments show that NSF unravels the SNARE ternary complex in a single, fast (<10 ms) step and appears to require only the 12 ATP (Ryu et al. Citation2015), although the temporal resolution of these experiments precluded observation of possible intermediate steps during SNARE complex disassembly. This observation is in contrast to other systems, where sequential steps have been observed for AAA+ proteins involved in degradation/disaggregation which occurs on the timescale of seconds (Sen et al. Citation2013; Rodriguez-Aliaga et al. Citation2016; Olivares et al. Citation2017). Furthermore, magnetic tweezer-assisted unfolding of all four SNARE motifs is simultaneous on a millisecond timescale (Kim et al. Citation2021). ATP binding to NSF hexamers is random and negatively cooperative while in the NSF/SNAP/SNARE complex it is synchronous and cooperative (Kim et al. Citation2021). Finally, substrate binding is associated with an 8.4-fold increase in the rate of ATP hydrolysis (Kim et al. Citation2021).
The gap in knowledge in understanding how NSF (and related AAA+ complexes) performs its explosive disassembly of SNARE proteins necessitates additional studies. Single molecule experiments performed at even higher time resolutions would be worthwhile in determining intermediate conformations. These single molecule studies could then help guide structural studies to capture these intermediate conformations and help delineate principles in AAA+ action on substrate.
The p97/Cdc48 family
A close relative of the NSF protein family worth noting is the p97/Cdc48 family, also a type II, clade 3 AAA+ protein. p97 (also called VCP, valosin-containing protein) typically refers to the mammalian ortholog, and Cdc48 refers to the yeast ortholog. p97 has been associated with many cellular functions that span across many different biochemical processes (van den Boom and Meyer Citation2018), and is perhaps most well-known for its central role in the ubiquitin-proteosome system (UPS). Through the aid of adaptor proteins, p97 binds ubiquitinated proteins and assists in their degradation at the proteosome. This is highly important for alleviating proteotoxic and organelle stress where proteins become damaged or misfolded. The unfolding activity of p97 prevents the mass accumulation of these proteins in cellular sub-compartments. Studies have also linked p97 to the lysosome pathway and macroautophagy (van den Boom and Meyer Citation2018). p97 also helps to liberate proteins that are sequestered in macromolecular complexes or the membrane; these proteins are then able to become functional, so the function of p97 is not solely related to protein degradation. The loss of p97 is lethal in mice (Müller et al. Citation2007), and it is upregulated in certain cancers (Deshaies Citation2014). Its key role in several processes makes its dysregulation key for several diseases (Meyer & Weihl Citation2014).
Like NSF, p97 contains an N-domain followed by a D1 and D2 domain (DeLaBarre and Brunger Citation2003; Davies et al. Citation2008; Banerjee et al. Citation2016). Six subunits form a homohexameric oligomer that maintains interprotomer contacts through the traditional mode of arginine fingers. Corkscrew-like changes of the D1 and D2 domains along with upward motions of the N domains are observed in the presence of ATPγS (Banerjee et al. Citation2016). At variance with NSF, mutations to the D2 domain Walker A or Walker B motifs reduced activity several fold over when compared to mutations to the D1 domain Walker A or Walker B motifs. Additionally, D2 Walker A or B mutations ablated activity much more dramatically than D1 domain mutants (Song et al. Citation2003). At increased temperatures, however, mutations to D1 reduced activity, whereas D2 mutants were insensitive to temperature change. Thus, the roles of the D1 and D2 domain appear to be reversed compared to NSF, as the D1 domain is primarily responsible for oligomerization while the D2 domain appears to be the domain that carries out the bulk of the hydrolysis activity. Indeed, mutation of the Walker B motif in D1 of Cdc48 eliminated ATPase activity in the absence of substrate and reduced activity when the substrate and co-factor are present, while mutation of the Walker B motif in D2 of Cdc48 led to reduced ATPase activity without substrate and even further reduced activity upon addition of substrate and co-factor (Bodnar and Rapoport Citation2017). Substrate stimulates ATPase activity in D2 while suppressing activity in D1. Moreover, substrate is completely translocated by both D1 and D2 rings as revealed by EM structures of human p97 (Cooney et al. Citation2019; Twomey et al. Citation2019; Pan et al. Citation2021). In contrast, available structures have thus far not revealed any substrate binding to the D2 domain of NSF.
The N-terminal domain of this protein family, as briefly mentioned above, interacts with adaptor proteins that help to coordinate engagement of substrate. For example, the Ufd1-Npl4 (UN) heterodimer interacts with p97 (Bodnar et al. Citation2018). Ufd1 interacts with Npl4 through an unstructured protein segment. Two short SHP motifs in Ufd1 mediate its interaction with Cdc48. Npl4 interacts via a UBX-like domain the with N-domain of Cdc48 and two zinc fingers to orient itself above the D1 ring. Alternatively, Cdc48 can bind other mutually exclusive adaptor proteins such as Shp1 (Cooney et al. Citation2019).
The UN heterodimer binds polyubiquitinated proteins and which form a “tower” on top of the D1 ring to form a processive complex with Cdc48/p97 (Twomey et al. Citation2019; Pan et al. Citation2021) (). In all three cryo-EM structures, the substrate transverses both the D1 and the D2 pores. However, at variance with NSF, the Cdc48/p97 D1 pores lack the conserved tyrosine “ladder” that is a hallmark of the substrate-NSF interactions (White et al. Citation2018) as well as other substrate-AAA+ interactions known to date (Puchades et al. Citation2017, Citation2020). Methionine residues in D1 are involved in substrate binding (Twomey et al. Citation2019). In contrast, the Cdc48/p97 D2 pore contains conserved tyrosine residues, and the substrate is bound by the D2 pore in a fashion similar to NSF D1 (). Cooney et al. (Citation2019) determined structures both in the presence of ADP-BeFx and of the Walker B mutant with ATP, whereas both Twomey et al. (Citation2019) and Pan et al. (Citation2021) used a Walker B mutant to slow hydrolysis in the presence of ATP. Moreover, Pan et al. used a mutation in the N domain that makes the p97 preparation more homogeneous, and, importantly, let hydrolysis proceed for 5 min at room temperature before flash freezing the sample for cryo-EM.
Figure 11. p97 complex. The p97 complex processing poly-ubiquitinated substrate (PDB: 7LN5 (Pan et al. Citation2021)). Each macromolecular chain is colored uniquely. The substrate is colored purple. (b) Depiction of partially engaged D1 ring and fully engaged D2 ring in the open conformation of p97.
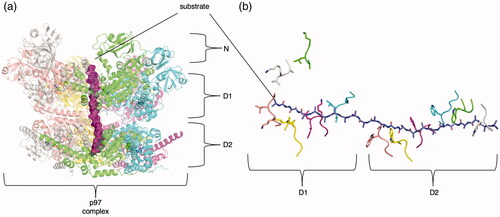
Critically, Pan et al. (Citation2021) found that the nucleotide states in D1 and D2 are not coupled. Coordination of hydrolysis between the D1 and D2 appears to be asynchronous, given that in an active state D1 and D2 domains in the same chain can bind different nucleotides. Moreover, two distinct states were identified—an open state with all six D2 pore loops engaged with substrate, and a closed state with only five engaged, (i.e. the F D2 domain does not interact with substrate). Within a single D1 or D2 ring however, it appears that hydrolysis could be sequential, as an intersubunit signaling (ISS) motif appears to insert itself into neighboring nucleotide-binding sites for several consecutive protomers; this could inhibit uncoordinated ATP hydrolysis (Pan et al. Citation2021). This ISS motif is conserved in several AAA+ proteins in different clades, suggesting that it could be widely important in different complexes (Chang et al. Citation2017). The authors, therefore, suggest that the D2 domain is likely exerting the force on the substrate by a power-stroke-like mechanism, although a stochastic Brownian ratchet mechanism cannot be entirely ruled out (see discussion below).
Mechanistic models of AAA+ engagement
A mechanistic explanation of the action of different members of the AAA+ protein superfamily has been a key goal ever since the discovery of its first members (Ogura and Wilkinson Citation2001; Harrison Citation2004). NSF and other AAA+ proteins have been used as model systems to tease apart the mechanism of oligomerized AAA+ action on substrate through a variety of methods. Despite remarkable progress, particularly with respect to understanding the biological contexts of AAA+ protein action, many questions remain. Indeed, progress in understanding the mechanistic pathway preceding, during, and following nucleotide hydrolysis—the reaction coordinate of AAA+ protein substrate processing—remains, at best, limited in most cases. Furthermore, to what degree might a given AAA+ protein reaction coordinate be conserved across members of the family? Of the proposed models so far, one can at least subdivide them into two groups based on the pattern of ATPase domain firing—that is, either sequential or stochastic hydrolysis of ATP by the protomers in any oligomeric arrangement of AAA+ proteins.
The sequential model
The sequential model (also known as the hand-over-hand model) posits that each protomer hydrolyzes ATP one at a time in a sequential fashion. One protomer hydrolyzes ATP to ADP. The conformational change that this protomer undergoes during hydrolysis and/or with the release of ADP is transmitted to its neighboring protomer, which is then primed to hydrolyze its own ATP. In this manner, hydrolysis proceeds around the ring, either continuing to cycle several times until it has completed its full action on the substrate (processive) or ending after one cycle (non-processive).
The sequential model is supported primarily by the structures and biophysical data of many different AAA+ proteins engaged to protein substrate (Cipriano et al. Citation2013; Han et al. Citation2017; Puchades et al. Citation2017; de la Peña et al. Citation2018; White et al. Citation2018; Deville et al. Citation2019; Ripstein et al. Citation2020). Some AAA+ proteins that engage with substrate form a spiral arrangement, suggesting that protomers that have already fired might transition between the top and bottom of the spiral. AAA+ proteins with two catalytically active rings show a spiral engagement of both rings(Gates et al. Citation2017), while AAA+ proteins with a single active and single inactive domain show one ring as spiral and the other as predominantly planar (Zhao et al. Citation2015; White et al. Citation2018). Most structures of AAA+ proteins engaging substrate are unfoldases, but presumably this sequential mechanism would hold true for helicases that must unwind large portions of DNA (Enemark and Joshua-Tor Citation2006).
The clamp loader clade (clade 1) is unique from the other clades as it typically arranges itself into a heteropentameric arrangement. Despite this, it follows many of the same local conformational patterns other AAA+ proteins show when bound to nucleotide(Kelch Citation2016). Upon hydrolysis, the large and small subdomains move toward each other. This conformational change could presumably promote ATP hydrolysis at the next, sequential protomer. For clamp loaders, sequential activation of the protomers would be non-processive. ATP hydrolysis would lead to clamp and DNA release. In essence, the clamp loaders would thus be an “on-off” switch.
The stochastic model
In contrast, the stochastic model proposes that protomers fire independently of each other, regardless of position, to exert force on the substrate(Fei et al. Citation2020). This model is supported by studies of synthetically linked ClpX protomers and synthetically linked HslU protomers. In both cases, the entire hexamer is present as a single fusion protein; these proteins function in vitro even with mutations to the ATP binding/hydrolysis sites of some but not all protomers (Martin et al. Citation2005; Cordova et al. Citation2014; Baytshtok et al. Citation2017). If the protomers were forced to act in sequential fashion, then an inactive protomer unable to hydrolyze ATP would presumably stall the cycle of hydrolysis, leading to a completely inactive enzyme. On the contrary, a stochastic mechanism could tolerate some inactive protomers, albeit with reduced processivity. However, mixing of unlinked, active ClpB protomers with catalytically dead ClpB protomers abolished disassembly activity after the addition of only 1–2 dead protomers, contrary to results seen with synthetically linked Clp family protomers (Martin et al. Citation2005; Cordova et al. Citation2014; Deville et al. Citation2019). Mixing of inactive ClpA protomers resulted in inactivation of complex activity with four or more dead subunits (Olivares et al. Citation2014).
Single molecule data also seems to suggest that Clp family ATPases process substrate in step sizes larger than two amino acids, which is what would be expected in the sequential model (Maillard et al. Citation2011; Sen et al. Citation2013; Avellaneda et al. Citation2020). Under a stochastic model, however, multiple protomers could fire at once and drive larger step sizes consistent with single molecule data (Qu et al. Citation2011; Sen et al. Citation2013; Rodriguez-Aliaga et al. Citation2016). Recent high-resolution structures consistently show ADP only at protomers at the top or bottom of the staircase. If protomers could fire randomly, one would expect a random distribution of ATP/ADP nucleotide. This is what was seen in a crystal structure of HslU, where three AMPPNP ligands were found bound to alternating subunits (Bochtler et al. Citation2000; Ogura and Wilkinson Citation2001). Atomic force microscopy on Abo1, without substrate, revealed random transitions from a planar to asymmetric D1 ring in the presence of ATP, suggestive of a stochastic model of activation (Cho et al. Citation2019). Presumably, clamp loaders would randomly hydrolyze ATP and release their clamp DNA once a sufficient number of protomers had fired.
Firing mechanism
We have discussed the two main models, involving either sequential or stochastic hydrolysis of ATP by the AAA+ protomers. An equally important question relates to the mode of transfer of energy to the substrate. Sequential models often assume that the protomers fire in a “power-stroke”-like manner, whereas stochastic models involve two potential firing mechanisms—one in which the protomers power stroke stochastically and one in which the AAA+ complex acts as a Brownian ratchet. However, these different firing mechanisms do not necessarily depend on the sequential or stochastic nature of the model.
The power-stroke model dictates that the AAA+ protein irreversibly performs action on substrate in one single step (Hwang and Karplus Citation2019). Biophysical studies of linked ClpX measuring the force it exerts on substrate reveal that each protomer hydrolyzes ATP with a maximum efficiency of about ∼35%, which is in the range of other power-stroke motors (Maillard et al. Citation2011; Rodriguez-Aliaga et al. Citation2016). Additionally, in optical tweezer experiments, phosphate release appears to coincide with a burst phase of disassembly (Rodriguez-Aliaga et al. Citation2016). Combination studies using single-molecule magnetic tweezers and single-molecule fluorescence suggest that NSF disassembles SNAREs in a rapid disassembly step coupled directly to ATP hydrolysis (Ryu et al. Citation2015).
Conversely, the Brownian ratchet model assumes that the AAA+ protein uses energy to rectify or bias the diffusive motion of the substrate (Hwang and Karplus Citation2019). FRET studies of ClpB pore loops show that they undergo large conformational fluctuations between discrete states on the microsecond timescale (Mazal et al. Citation2020). These microsecond transitions between the different FRET states are consistent with motions of more than 10 Å and are presumably associated with the translocation of substrate peptide by more than two residues. This observation implies translocation of substrate on a timescale faster than expected, as many residues could translocate in between pore loop transitions. The pore loops of ClpB are thus proposed to serve as “ratchets” for the substrate, which fluctuate in the pore via Brownian motion. While substrate motion through the AAA+ pore is random in this model, the stochastic hydrolysis of ATP causes the pore loops responsible for engagement with the substrate to act as a “pawl” and prevent the substrate from moving backwards through the pore. As such, each subsequent stochastic ATP hydrolysis event does not directly translocate the substrate through the pore of the AAA+ complex, but instead ensures the unidirectional motion of the substrate. We conclude that the current data do not rule out stochastic or sequential models and firing mechanisms. It is also possible that all these mechanisms exist simultaneously, or that they are specific to particular members of the AAA+ family.
Future directions
A plethora of already existing biophysical and structural data pertaining to AAA+ proteins have been reviewed here. However, many aspects of underlying molecular mechanisms remain unclear after decades of research. Capturing transition state intermediates of these AAA+ proteins during active translocation of substrate by cryo-EM would be highly informative in discerning between the sequential and stochastic model, along with their details. Cryo-EM also enables the search for new drugs targeting AAA+ proteins; new workflows will drive the discovery of drug candidates that target malfunctioning AAA+ proteins responsible for disease and enable the development of novel antibiotics that target bacterial AAA+ proteins. Additionally, employing cryo-electron tomography (cryoET) could be employed to capture unique conformational or oligomeric states of AAA+ proteins in situ, providing valuable information to the structural heterogeneity of these molecular machines.
Acknowledgements
The authors thank Minglei Zhao and Subu Subramanian for discussions and careful reading of this review.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Adams NBP, Reid JD. 2013. The allosteric role of the AAA+ domain of ChlD protein from the magnesium chelatase of synechocystis species PCC 6803. J Biol Chem. 288(40):28727–28732.
- Ahel J, Lehner A, Vogel A, Schleiffer A, Meinhart A, Haselbach D, Clausen T. 2020. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. Elife. 9:e56185.
- Alfieri C, Chang L, Barford D. 2018. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature. 559(7713):274–278.
- Aramayo RJ, Willhoft O, Ayala R, Bythell-Douglas R, Wigley DB, Zhang X. 2018. Cryo-EM structures of the human INO80 chromatin-remodeling complex. Nat Struct Mol Biol. 25(1):37–44.
- Arias-Palomo E, Puri N, O'Shea Murray VL, Yan Q, Berger JM. 2019. Physical basis for the loading of a bacterial replicative helicase onto DNA. Mol Cell. 74(1):173–184.e4.
- Arora J, Goswami K, Saha S. 2014. Characterization of the replication initiator Orc1/Cdc6 from the Archaeon Picrophilus torridus. J Bacteriol. 196(2):276–286.
- Ason B, Bertram JG, Hingorani MM, Beechem JM, O'Donnell M, Goodman MF, Bloom LB. 2000. A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends. DNA triggers a change in binding specificity of the gamma complex clamp loader. J Biol Chem. 275(4):3006–3015.
- Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. 2016. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 44(15):7007–7078.
- Ausiannikava D, Allers T. 2017. Diversity of DNA replication in the archaea. Genes (Basel). 8(2)
- Avellaneda MJ, Franke KB, Sunderlikova V, Bukau B, Mogk A, Tans SJ. 2020. Processive extrusion of polypeptide loops by a Hsp100 disaggregase. Nature. 578(7794):317–320.
- Bammes BE, Rochat RH, Jakana J, Chen D-H, Chiu W. 2012. Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. J Struct Biol. 177(3):589–601.
- Banchenko S, Arumughan A, Petrović S, Schwefel D, Wanker EE, Roske Y, Heinemann U. 2019. Common mode of remodeling AAA ATPases p97/CDC48 by their disassembling cofactors ASPL/PUX1. Structure. 27(12):1830–1841.e3.
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. 2016. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 351(6275):871–875.
- Baranov PV, Atkins JF, Yordanova MM. 2015. Augmented genetic decoding: global, local and temporal alterations of decoding processes and codon meaning. Nat Rev Genet. 16(9):517–529.
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. 2018. Structure and function of the 26S proteasome. Annu Rev Biochem. 87:697–724.
- Bayless AM, Zapotocny RW, Grunwald DJ, Amundson KK, Diers BW, Bent AF. 2018. An atypical N-ethylmaleimide sensitive factor enables the viability of nematode-resistant Rhg1 soybeans. Proc Natl Acad Sci USA. 115(19):E4512–E4521.
- Baytshtok V, Chen J, Glynn SE, Nager AR, Grant RA, Baker TA, Sauer RT. 2017. Covalently linked HslU hexamers support a probabilistic mechanism that links ATP hydrolysis to protein unfolding and translocation. J Biol Chem. 292(14):5695–5704.
- Beckwith R, Estrin E, Worden EJ, Martin A. 2013. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 20(10):1164–1172.
- Beyer A. 1997. Sequence analysis of the AAA protein family. Protein Sci. 6(10):2043–2058.
- Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, Houry WA. 2018. The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases. ACS Chem Biol. 13(6):1413–1425.
- Blinkowa AL, Walker JR. 1990. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 18(7):1725–1729.
- Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. 1988. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci U S A. 85(21):7852–7856.
- Blok NB, Tan D, Wang RY-R, Penczek PA, Baker D, DiMaio F, Rapoport TA, Walz T. 2015. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc Natl Acad Sci USA. 112(30):E4017–25.
- Blythe EE, Olson KC, Chau V, Deshaies RJ. 2017. Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP•NPLOC4•UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc Natl Acad Sci USA. 114(22):E4380–E4388.
- Bochtler M, Hartmann C, Song HK, Bourenkov GP, Bartunik HD, Huber R. 2000. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 403(6771):800–805.
- Bodnar NO, Kim KH, Ji Z, Wales TE, Svetlov V, Nudler E, Engen JR, Walz T, Rapoport TA. 2018. Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat Struct Mol Biol. 25(7):616–622.
- Bodnar NO, Rapoport TA. 2017. Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell. 169(4):722–735.e9.
- Bowman GD, O'Donnell M, Kuriyan J. 2004. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 429(6993):724–730.
- Brown RSH, Zhao C, Chase AR, Wang J, Schlieker C. 2014. The mechanism of Torsin ATPase activation. Proc Natl Acad Sci USA. 111(45):E4822–31.
- Brunger AT, Choi UB, Lai Y, Leitz J, Zhou Q. 2018. Molecular mechanisms of fast neurotransmitter release. Annu Rev Biophys. 47:469–497.
- Burroughs AM, Iyer LM, Aravind L. 2007. Comparative genomics and evolutionary trajectories of viral ATP dependent DNA-packaging systems. Genome Dyn. 3:48–65.
- Bush M, Dixon R. 2012. The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol Mol Biol Rev. 76(3):497–529.
- Caillat C, Macheboeuf P, Wu Y, McCarthy AA, Boeri-Erba E, Effantin G, Göttlinger HG, Weissenhorn W, Renesto P. 2015. Asymmetric ring structure of Vps4 required for ESCRT-III disassembly. Nat Commun. 6:8781.
- Carter JR, Franden MA, Aebersold R, McHenry CS. 1993. Identification, isolation, and characterization of the structural gene encoding the delta' subunit of Escherichia coli DNA polymerase III holoenzyme. J Bacteriol. 175(12):3812–3822.
- Castanzo DT, LaFrance B, Martin A. 2020. The AAA+ ATPase Msp1 is a processive protein translocase with robust unfoldase activity. Proc Natl Acad Sci U S A. 117(26):14970–14977.
- Castillo RM, Mizuguchi K, Dhanaraj V, Albert A, Blundell TL, Murzin AG. 1999. A six-stranded double-psi beta barrel is shared by several protein superfamilies. Structure. 7(2):227–236.
- Cattaneo M, Morozumi Y, Perazza D, Boussouar F, Jamshidikia M, Rousseaux S, Verdel A, Khochbin S. 2014. Lessons from yeast on emerging roles of the ATAD2 protein family in gene regulation and genome organization. Mol Cells. 37(12):851–856.
- Chang L-F, Chen S, Liu C-C, Pan X, Jiang J, Bai X-C, Xie X, Wang H-W, Sui S-F. 2012. Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol. 19(3):268–275.
- Chang C-W, Lee S, Tsai FTF. 2017. Structural elements regulating AAA+ protein quality control machines. Front Mol Biosci. 4:27.
- Chen B, Sysoeva TA, Chowdhury S, Guo L, De Carlo S, Hanson JA, Yang H, Nixon BT. 2010. Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase. Structure. 18(11):1420–1430.
- Cho C, Jang J, Kang Y, Watanabe H, Uchihashi T, Kim SJ, Kato K, Lee JY, Song J-J. 2019. Structural basis of nucleosome assembly by the Abo1 AAA+ ATPase histone chaperone. Nat Commun. 10(1):5764.
- Cho C, Reck-Peterson SL, Vale RD. 2008. Regulatory ATPase sites of cytoplasmic dynein affect processivity and force generation. J Biol Chem. 283(38):25839–25845.
- Chodavarapu S, Jones AD, Feig M, Kaguni JM. 2016. DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res. 44(1):210–220.
- Ciniawsky S, Grimm I, Saffian D, Girzalsky W, Erdmann R, Wendler P. 2015. Molecular snapshots of the Pex1/6 AAA+ complex in action. Nat Commun. 6:7331.
- Cipriano DJ, Jung J, Vivona S, Fenn TD, Brunger AT, Bryant Z. 2013. Processive ATP-driven substrate disassembly by the N-ethylmaleimide-sensitive factor (NSF) molecular machine. J Biol Chem. 288(32):23436–23445.
- Confalonieri F, Duguet M. 1995. A 200-amino acid ATPase module in search of a basic function. Bioessays. 17(7):639–650.
- Cooney I, Han H, Stewart MG, Carson RH, Hansen DT, Iwasa JH, Price JC, Hill CP, Shen PS. 2019. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science. 365(6452):502–505.
- Cordova JC, Olivares AO, Shin Y, Stinson BM, Calmat S, Schmitz KR, Aubin-Tam M-E, Baker TA, Lang MJ, Sauer RT. 2014. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell. 158(3):647–658.
- Costa A, Hood IV, Berger JM. 2013. Mechanisms for initiating cellular DNA replication. Annu Rev Biochem. 82:25–54.
- Cupido T, Jones NH, Grasso MJ, Pisa R, Kapoor TM. 2021. A chemical genetics approach to examine the functions of AAA proteins. Nat Struct Mol Biol. 28(4):388–397.
- Cupido T, Pisa R, Kelley ME, Kapoor TM. 2019. Designing a chemical inhibitor for the AAA protein spastin using active site mutations. Nat Chem Biol. 15(5):444–452.
- Cupo RR, Shorter J. 2020. Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. Elife. 9:e55279.
- Davies JM, Brunger AT, Weis WI. 2008. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 16(5):715–726.
- de la Peña AH, Goodall EA, Gates SN, Lander GC, Martin A. 2018. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science. 362(6418).
- DeLaBarre B, Brunger AT. 2003. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 10(10):856–863.
- Dellinger B, Felling R, Ordway RW. 2000. Genetic modifiers of the Drosophila NSF mutant, comatose, include a temperature-sensitive paralytic allele of the calcium channel alpha1-subunit gene, cacophony. Genetics. 155(1):203–211.
- Demircioglu FE, Sosa BA, Ingram J, Ploegh HL, Schwartz TU. 2016. Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia. Elife. 5:e17983.
- Demircioglu FE, Zheng W, McQuown AJ, Maier NK, Watson N, Cheeseman IM, Denic V, Egelman EH, Schwartz TU. 2019. The AAA+ ATPase TorsinA polymerizes into hollow helical tubes with 8.5 subunits per turn. Nat Commun. 10(1):3262.
- Deshaies RJ. 2014. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 12:94.
- Deville C, Franke K, Mogk A, Bukau B, Saibil HR. 2019. Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor. Cell Rep. 27(12):3433–3446.e4.
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, Finardi A, Cagnoli C, Tempia F, Frontali M, et al. 2010. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 42(4):313–321.
- Ding B, Martin DW, Rampello AJ, Glynn SE. 2018. Dissecting Substrate Specificities of the Mitochondrial AFG3L2 Protease. Biochemistry. 57(28):4225–4235.
- Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, Mao Y. 2019. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature. 565(7737):49–55.
- Dong Z, Onrust R, Skangalis M, O'Donnell M. 1993. DNA polymerase III accessory proteins. I. holA and holB encoding delta and delta. J Biol Chem. 268(16):11758–11765.
- Duderstadt KE, Berger JM. 2008. AAA+ ATPases in the initiation of DNA replication. Crit Rev Biochem Mol Biol. 43(3):163–187.
- Duderstadt KE, Chuang K, Berger JM. 2011. DNA stretching by bacterial initiators promotes replication origin opening. Nature. 478(7368):209–213.
- El Bakkouri M, Gutsche I, Kanjee U, Zhao B, Yu M, Goret G, Schoehn G, Burmeister WP, Houry WA. 2010. Structure of RavA MoxR AAA+ protein reveals the design principles of a molecular cage modulating the inducible lysine decarboxylase activity. Proc Natl Acad Sci U S A. 107(52):22499–22504.
- Enemark EJ, Joshua-Tor L. 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 442(7100):270–275.
- Erdmann R, Wiebel FF, Flessau A, Rytka J, Beyer A, Fröhlich KU, Kunau WH. 1991. PAS1, a yeast gene required for peroxisome biogenesis, encodes a member of a novel family of putative ATPases. Cell. 64(3):499–510.
- Erzberger JP, Berger JM. 2006. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 35(1):93–114.
- Erzberger JP, Mott ML, Berger JM. 2006. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 13(8):676–683.
- Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, Sauer RT. 2020. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife. 9
- Fernandez-Leiro R, Conrad J, Scheres SH, Lamers MH. 2015. cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. Elife. 4
- Fleming KG, Hohl TM, Yu RC, Müller SA, Wolpensinger B, Engel A, Engelhardt H, Brünger AT, Söllner TH, Hanson PI. 1998. A revised model for the oligomeric state of the N-ethylmaleimide-sensitive fusion protein, NSF. J Biol Chem. 273(25):15675–15681.
- Flower AM, McHenry CS. 1990. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc Natl Acad Sci USA. 87(10):3713–3717.
- Flynn JM, Levchenko I, Seidel M, Wickner SH, Sauer RT, Baker TA. 2001. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA. 98(19):10584–10589.
- Fragkos M, Ganier O, Coulombe P, Méchali M. 2015. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol. 16(6):360–374.
- Fröhlich KU, Fries HW, Rüdiger M, Erdmann R, Botstein D, Mecke D. 1991. Yeast cell cycle protein CDC48p shows full-length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation, and gene expression. J Cell Biol. 114(3):443–453.
- Fujisawa T, Filippakopoulos P. 2017. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 18(4):246–262.
- Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N. 2003. Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. Embo J. 22(17):4365–4374.
- Gardner BM, Chowdhury S, Lander GC, Martin A. 2015. The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits. J Mol Biol. 427(6 Pt B):1375–1388.
- Gates SN, Yokom AL, Lin JBei, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E, et al. 2017. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 357(6348):273–279.
- Gatsogiannis C, Balogh D, Merino F, Sieber SA, Raunser S. 2019. Cryo-EM structure of the ClpXP protein degradation machinery. Nat Struct Mol Biol. 26(10):946–954.
- Gaubitz C, Liu X, Magrino J, Stone NP, Landeck J, Hedglin M, Kelch BA. 2020. Structure of the human clamp loader reveals an autoinhibited conformation of a substrate-bound AAA+ switch. Proc Natl Acad Sci USA. 117(38):23571–23580.
- Germany EM, Zahayko N, Huebsch ML, Fox JL, Prahlad V, Khalimonchuk O. 2018. The AAA ATPase Afg1 preserves mitochondrial fidelity and cellular health by maintaining mitochondrial matrix proteostasis. J Cell Sci. 131(22):jcs219956.
- Ghosh DK, Dasgupta D, Guha A. 2012. Models, regulations, and functions of microtubule severing by katanin. ISRN Mol Biol. 2012:596289.
- Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. 2009. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 139(4):744–756.
- Goedken ER, Levitus M, Johnson A, Bustamante C, O'Donnell M, Kuriyan J. 2004. Fluorescence measurements on the E.coli DNA polymerase clamp loader: implications for conformational changes during ATP and clamp binding. J Mol Biol. 336(5):1047–1059.
- Goodchild RE, Dauer WT. 2005. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 168(6):855–862.
- Grillet M, Dominguez Gonzalez B, Sicart A, Pöttler M, Cascalho A, Billion K, Hernandez Diaz S, Swerts J, Naismith TV, Gounko NV, et al. 2016. Torsins are essential regulators of cellular lipid metabolism. Dev Cell. 38(3):235–247.
- Guenther B, Onrust R, Sali A, O'Donnell M, Kuriyan J. 1997. Crystal structure of the delta’ subunit of the clamp-loader complex of E. coli DNA polymerase III. Cell. 91(3):335–345.
- Hamon M-P, Bulteau A-L, Friguet B. 2015. Mitochondrial proteases and protein quality control in ageing and longevity. Ageing Res Rev. 23(Pt A):56–66.
- Han H, Monroe N, Sundquist WI, Shen PS, Hill CP. 2017. The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. Elife [Internet]. 6:e31324.
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE. 1997. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 90(3):523–535.
- Harrison SC. 2004. Whither structural biology? Nat Struct Mol Biol. 11(1):12–15.
- Hedglin M, Kumar R, Benkovic SJ. 2013. Replication clamps and clamp loaders. Cold Spring Harb Perspect Biol. 5(4):a010165.
- Hilbert BJ, Hayes JA, Stone NP, Duffy CM, Sankaran B, Kelch BA. 2015. Structure and mechanism of the ATPase that powers viral genome packaging. Proc Natl Acad Sci U S A. 112(29):E3792–9.
- Ho C-M, Beck JR, Lai M, Cui Y, Goldberg DE, Egea PF, Zhou ZH. 2018. Malaria parasite translocon structure and mechanism of effector export. Nature. 561(7721):70–75.
- Hohl TM, Parlati F, Wimmer C, Rothman JE, Söllner TH, Engelhardt H. 1998. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol Cell. 2(5):539–548.
- Hussain M, Zhou Y, Song Y, Hameed HMA, Jiang H, Tu Y, Zhang J. 2018. ATAD2 in cancer: a pharmacologically challenging but tractable target. Expert Opin Ther Targets. 22(1):85–96.
- Hwang W, Karplus M. 2019. Structural basis for power stroke vs. Brownian ratchet mechanisms of motor proteins. Proc Natl Acad Sci U S A. 116(40):19777–19785.
- Ishiguro H, Shimokawa T, Tsunoda T, Tanaka T, Fujii Y, Nakamura Y, Furukawa Y. 2002. Isolation of HELAD1, a novel human helicase gene up-regulated in colorectal carcinomas. Oncogene. 21(41):6387–6394.
- Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, Zhao R, St-Germain J, Heese LE, Egan G, et al. 2019. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell. 35(5):721–737.e9.
- Iyer LM, Leipe DD, Koonin EV, Aravind L. 2004. Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol. 146(1-2):11–31.
- Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 331(6153):280–283.
- Jackson AP, Laskey RA, Coleman N. 2014. Replication proteins and human disease. Cold Spring Harb Perspect Biol [Internet]. 6(1):a013060.
- Jacks T, Varmus HE. 1985. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science (80-). 230(4731):1237–1242.
- Jarvis TC, Paul LS, Hockensmith JW, von Hippel PH. 1989. Structural and enzymatic studies of the T4 DNA replication system. II. ATPase properties of the polymerase accessory protein complex. J Biol Chem. 264(21):12717–12729.
- Jeruzalmi D, O'Donnell M, Kuriyan J. 2001. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell. 106(4):429–441.
- Jessop M, Arragain B, Miras R, Fraudeau A, Huard K, Bacia-Verloop M, Catty P, Felix J, Malet H, Gutsche I. 2020. Structural insights into ATP hydrolysis by the MoxR ATPase RavA and the LdcI-RavA cage-like complex. Commun Biol. 3(1):46.
- Joly N, Zhang N, Buck M, Zhang X. 2012. Coupling AAA protein function to regulated gene expression. Biochim Biophys Acta. 1823(1):108–116.
- Kandiah E, Carriel D, Perard J, Malet H, Bacia M, Liu K, Chan SWS, Houry WA, Ollagnier de Choudens S, Elsen S, et al. 2016. Structural insights into the Escherichia coli lysine decarboxylases and molecular determinants of interaction with the AAA+ ATPase RavA. Sci Rep. 6:24601.
- Kardon JR, Moroco JA, Engen JR, Baker TA. 2020. Mitochondrial ClpX activates an essential biosynthetic enzyme through partial unfolding. Elife. 9
- Kater L, Wagener N, Berninghausen O, Becker T, Neupert W, Beckmann R. 2020. Structure of the Bcs1 AAA-ATPase suggests an airlock-like translocation mechanism for folded proteins. Nat Struct Mol Biol. 27(2):142–149.
- Kazmirski SL, Podobnik M, Weitze TF, O'Donnell M, Kuriyan J. 2004. Structural analysis of the inactive state of the Escherichia coli DNA polymerase clamp-loader complex. Proc Natl Acad Sci U S A. 101(48):16750–16755.
- Kelch BA. 2016. Review: The lord of the rings: structure and mechanism of the sliding clamp loader. Biopolymers. 105(8):532–546.
- Kelch BA, Makino DL, O'Donnell M, Kuriyan J. 2011. How a DNA polymerase clamp loader opens a sliding clamp. Science. 334(6063):1675–1680.
- Kelch BA, Makino DL, O'Donnell M, Kuriyan J. 2012. Clamp loader ATPases and the evolution of DNA replication machinery. BMC Biol. 10:34.
- Kendra JA, de la Fuente C, Brahms A, Woodson C, Bell TM, Chen B, Khan YA, Jacobs JL, Kehn-Hall K, Dinman JD. 2017. Ablation of programmed -1 ribosomal frameshifting in venezuelan equine encephalitis virus results in attenuated neuropathogenicity. J Virol. 91(3).
- Khan YA, Jungreis I, Wright JC, Mudge JM, Choudhary JS, Firth AE, Kellis M. 2020. Evidence for a novel overlapping coding sequence in POLG initiated at a CUG start codon. BMC Genet. 21(1):25.
- Khan YA, Loughran G, Atkins JF. 2019. Contesting the evidence for −1 frameshifting in immune-functioning C-C chemokine receptor 5 (CCR5) – the HIV-1 co-receptor. bioRxiv. doi:https://doi.org/10.1101/513333
- Kim G, Lee S-G, Han S, Jung J, Jeong HS, Hyun J-K, Rhee D-K, Kim HM, Lee S. 2020. ClpL is a functionally active tetradecameric AAA+ chaperone, distinct from hexameric/dodecameric ones. Faseb J. 34(11):14353–14370.
- Kim C, Shon MJ, Kim SH, Eun GS, Ryu J-K, Hyeon C, Jahn R, Yoon T-Y. 2021. Extreme parsimony in ATP consumption by 20S complexes in the global disassembly of single SNARE complexes. Nat Commun. 12(1):3206.
- Kim Y-C, Snoberger A, Schupp J, Smith DM. 2015. ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat Commun. 6:8520.
- Kiykim A, Garncarz W, Karakoc-Aydiner E, Ozen A, Kiykim E, Yesil G, Boztug K, Baris S. 2016. Novel CLPB mutation in a patient with 3-methylglutaconic aciduria causing severe neurological involvement and congenital neutropenia. Clin Immunol. 165:1–3. http://www.sciencedirect.com/science/article/pii/S1521661616300249.
- Kodaira M, Biswas SB, Kornberg A. 1983. The dnaX gene encodes the DNA polymerase III holoenzyme tau subunit, precursor of the gamma subunit, the dnaZ gene product. Mol Gen Genet. 192(1-2):80–86.
- Kress W, Maglica Z, Weber-Ban E. 2009. Clp chaperone-proteases: structure and function. Res Microbiol. 160(9):618–628.
- Kunau WH, Beyer A, Franken T, Götte K, Marzioch M, Saidowsky J, Skaletz-Rorowski A, Wiebel FF. 1993. Two complementary approaches to study peroxisome biogenesis in Saccharomyces cerevisiae: forward and reversed genetics. Biochimie. 75(3-4):209–224.
- Kyrpides N, Overbeek R, Ouzounis C. 1999. Universal protein families and the functional content of the last universal common ancestor. J Mol Evol. 49(4):413–423.
- Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, et al. 2017. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron. 95(3):591–607.e10.
- Lee S-Y, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. 2003. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 17(20):2552–2563.
- Lee YJ, Wickner RB. 1992. AFG1, a new member of the SEC18-NSF, PAS1, CDC48-VCP, TBP family of ATPases. Yeast. 8(9):787–790.
- Leipe DD, Koonin EV, Aravind L. 2003. Evolution and classification of P-loop kinases and related proteins. J Mol Biol. 333(4):781–815.
- Leipe DD, Wolf YI, Koonin EV, Aravind L. 2002. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 317(1):41–72.
- Lenzen CU, Steinmann D, Whiteheart SW, Weis WI. 1998. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell. 94(4):525–536.
- Leonard AC, Méchali M. 2013. DNA replication origins. Cold Spring Harb Perspect Biol. 5(10):a010116.
- Li N, Lam WH, Zhai Y, Cheng J, Cheng E, Zhao Y, Gao N, Tye B-K. 2018. Structure of the origin recognition complex bound to DNA replication origin. Nature. 559(7713):217–222.
- Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, Tye B-K, Gao N. 2015. Structure of the eukaryotic MCM complex at 3.8 Å. Nature. 524(7564):186–191.
- Lindeboom JJ, Nakamura M, Hibbel A, Shundyak K, Gutierrez R, Ketelaar T, Emons AMC, Mulder BM, Kirik V, Ehrhardt DW. 2013. A mechanism for reorientation of cortical microtubule arrays driven by microtubule severing. Science. 342(6163):1245533.
- Liu X, Rao L, Gennerich A. 2020. The regulatory function of the AAA4 ATPase domain of cytoplasmic dynein. Nat Commun. 11(1):5952.
- Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N, Ciechanover A. 2016. The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 26(8):869–885.
- Lo Y-H, Sobhany M, Hsu AL, Ford BL, Krahn JM, Borgnia MJ, Stanley RE. 2019. Cryo-EM structure of the essential ribosome assembly AAA-ATPase Rix7. Nat Commun. 10(1):513.
- Lopez KE, Rizo AN, Tse E, Lin J, Scull NW, Thwin AC, Lucius AL, Shorter J, Southworth DR. 2020. Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis. Nat Struct Mol Biol. 27(5):406–416.
- Lundqvist J, Elmlund H, Wulff RP, Berglund L, Elmlund D, Emanuelsson C, Hebert H, Willows RD, Hansson M, Lindahl M, et al. 2010. ATP-induced conformational dynamics in the AAA+ motor unit of magnesium chelatase. Structure. 18(3):354–365.
- Ma C, Su L, Seven AB, Xu Y, Rizo J. 2013. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science. 339(6118):421–425.
- Maillard RA, Chistol G, Sen M, Righini M, Tan J, Kaiser CM, Hodges C, Martin A, Bustamante C. 2011. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 145(3):459–469.
- Majumder P, Rudack T, Beck F, Danev R, Pfeifer G, Nagy I, Baumeister W. 2019. Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc Natl Acad Sci U S A. 116(2):534–539.
- Maki H, Kornberg A. 1985. The polymerase subunit of DNA polymerase III of Escherichia coli II. Purification of the alpha subunit, devoid of nuclease activities. J Biol Chem. 260(24):12987–12992.
- Malhotra V, Orci L, Glick BS, Block MR, Rothman JE. 1988. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell. 54(2):221–227.
- Malsam J, Söllner TH. 2011. Organization of SNAREs within the Golgi stack. Cold Spring Harb Perspect Biol. 3(10):a005249.
- Mao Y-Q, Houry WA. 2017. The role of pontin and reptin in cellular physiology and cancer etiology. Front Mol Biosci. 4:58.
- March ZM, Mack KL, Shorter J. 2019. AAA+ protein-based technologies to counter neurodegenerative disease. Biophys J. 116(8):1380–1385.
- Martin A, Baker TA, Sauer RT. 2005. Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature. 437(7062):1115–1120.
- Matveeva EA, He P, Whiteheart SW. 1997. N-Ethylmaleimide-sensitive fusion protein contains high and low affinity ATP-binding sites that are functionally distinct. J Biol Chem. 272(42):26413–26418.
- May AP, Misura KM, Whiteheart SW, Weis WI. 1999. Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nat Cell Biol. 1(3):175–182.
- May AP, Whiteheart SW, Weis WI. 2001. Unraveling the mechanism of the vesicle transport ATPase NSF, the N-ethylmaleimide-sensitive factor. J Biol Chem. 276(25):21991–21994.
- Mayer A, Wickner W, Haas A. 1996. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 85(1):83–94.
- Mazal H, Iljina M, Riven I, Haran G. 2020. Ultrafast Brownian-ratchet mechanism for protein translocation by a AAA+ machine. bioRxiv. doi:https://doi.org/10.1101/2020.11.19.384313
- McInerney P, Johnson A, Katz F, O'Donnell M. 2007. Characterization of a triple DNA polymerase replisome. Mol Cell. 27(4):527–538.
- Meagher M, Epling LB, Enemark EJ. 2019. DNA translocation mechanism of the MCM complex and implications for replication initiation. Nat Commun. 10(1):3117.
- Merrill SA, Hanson PI. 2010. Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J Biol Chem. 285(46):35428–35438.
- Meydan S, Klepacki D, Karthikeyan S, Margus T, Thomas P, Jones JE, Khan Y, Briggs J, Dinman JD, Vázquez-Laslop N, Mankin AS. 2017. Programmed ribosomal frameshifting generates a copper transporter and a copper chaperone from the same gene. Mol Cell. 65(2):207–219.
- Meyer H, Weihl CC. 2014. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. 127(Pt 18):3877–3883.
- Miller JM, Arachea BT, Epling LB, Enemark EJ. 2014. Analysis of the crystal structure of an active MCM hexamer. Elife. 3:e03433.
- Miller JM, Enemark EJ. 2016. Fundamental characteristics of AAA+ protein family structure and function. Archaea. 2016:9294307.
- Miyata T, Suzuki H, Oyama T, Mayanagi K, Ishino Y, Morikawa K. 2005. Open clamp structure in the clamp-loading complex visualized by electron microscopic image analysis. Proc Natl Acad Sci USA. 102(39):13795–13800.
- Mok M, Marians KJ. 1987. The Escherichia coli preprimosome and DNA B helicase can form replication forks that move at the same rate. J Biol Chem. 262(34):16644–16654.
- Monroe N, Han H, Shen PS, Sundquist WI, Hill CP. 2017. Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. Elife. 6
- Monroe N, Hill CP. 2016. Meiotic clade AAA ATPases: protein polymer disassembly machines. J Mol Biol. 428(9 Pt B):1897–1911.
- Moomau C, Musalgaonkar S, Khan YA, Jones JE, Dinman JD. 2016. Structural and functional characterization of programmed ribosomal frameshift signals in west nile virus strains reveals high structural plasticity among cis-acting RNA elements. J Biol Chem. 291(30):15788–15795.
- Morgan A, Dimaline R, Burgoyne RD. 1994. The ATPase activity of N-ethylmaleimide-sensitive fusion protein (NSF) is regulated by soluble NSF attachment proteins. J Biol Chem. 269(47):29347–29350.
- Morito D, Nishikawa K, Hoseki J, Kitamura A, Kotani Y, Kiso K, Kinjo M, Fujiyoshi Y, Nagata K. 2014. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci Rep. 4:4442.
- Muley PD, McNeill EM, Marzinke MA, Knobel KM, Barr MM, Clagett-Dame M. 2008. The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev Neurobiol. 68(13):1441–1453.
- Müller JMM, Deinhardt K, Rosewell I, Warren G, Shima DT. 2007. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun. 354(2):459–465.
- Mullin DA, Woldringh CL, Henson JM, Walker JR. 1983. Cloning of the Escherichia coli dnaZX region and identification of its products. Mol Gen Genet. 192(1-2):73–79.
- Nagata K, Okada A, Ohtsuka J, Ohkuri T, Akama Y, Sakiyama Y, Miyazaki E, Horita S, Katayama T, Ueda T, et al. 2020. Crystal structure of the complex of the interaction domains of Escherichia coli DnaB helicase and DnaC helicase loader: structural basis implying a distortion-accumulation mechanism for the DnaB ring opening caused by DnaC binding. J Biochem. 167(1):1–14.
- Nagiec EE, Bernstein A, Whiteheart SW. 1995. Each domain of the N-ethylmaleimide-sensitive fusion protein contributes to its transport activity. J Biol Chem. 270(49):29182–29188.
- Nano N, Ugwu F, Seraphim TV, Li T, Azer G, Isaac M, Prakesch M, Barbosa LRS, Ramos CHI, Datti A, et al. 2020. Sorafenib as an Inhibitor of RUVBL2. Biomolecules. 10(4).
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9(1):27–43.
- Nirwan N, Itoh Y, Singh P, Bandyopadhyay S, Vinothkumar KR, Amunts A, Saikrishnan K. 2019. Structure-based mechanism for activation of the AAA+ GTPase McrB by the endonuclease McrC. Nat Commun. 10(1):3058.
- Nirwan N, Singh P, Mishra GG, Johnson CM, Szczelkun MD, Inoue K, Vinothkumar KR, Saikrishnan K. 2019. Hexameric assembly of the AAA+ protein McrB is necessary for GTPase activity. Nucleic Acids Res. 47(2):868–882.
- Novick P, Field C, Schekman R. 1980. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 21(1):205–215.
- O'Donnell ME, Kornberg A. 1985. Dynamics of DNA polymerase III holoenzyme of Escherichia coli in replication of a multiprimed template. J Biol Chem. 260(23):12875–12883.
- Ogura T, Whiteheart SW, Wilkinson AJ. 2004. Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol. 146(1-2):106–112.
- Ogura T, Wilkinson AJ. 2001. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells. 6(7):575–597.
- Okreglak V, Walter P. 2014. The conserved AAA-ATPase Msp1 confers organelle specificity to tail-anchored proteins. Proc Natl Acad Sci U S A. 111(22):8019–8024.
- Olivares AO, Kotamarthi HC, Stein BJ, Sauer RT, Baker TA. 2017. Effect of directional pulling on mechanical protein degradation by ATP-dependent proteolytic machines. Proc Natl Acad Sci USA. 114(31):E6306–E6313.
- Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. 2014. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol. 21(10):871–875.
- Pan M, Yu Y, Ai H, Zheng Q, Xie Y, Liu L, Zhao M. 2021. Mechanistic insight into substrate processing and allosteric inhibition of human p97. Nat Struct Mol Biol. 28:614–625.
- Panne D, Müller SA, Wirtz S, Engel A, Bickle TA. 2001. The McrBC restriction endonuclease assembles into a ring structure in the presence of G nucleotides. Embo J. 20(12):3210–3217.
- Park S-S, Kwon H-Y, Tran TD-H, Choi M-H, Jung S-H, Lee S, Briles DE, Rhee D-K. 2015. ClpL is a chaperone without auxiliary factors. Febs J. 282(8):1352–1367.
- Patel S, Latterich M. 1998. The AAA team: related ATPases with diverse functions. Trends Cell Biol. 8(2):65–71.
- Petukhov M, Dagkessamanskaja A, Bommer M, Barrett T, Tsaneva I, Yakimov A, Quéval R, Shvetsov A, Khodorkovskiy M, Käs E, et al. 2012. Large-scale conformational flexibility determines the properties of AAA+ TIP49 ATPases. Structure. 20(8):1321–1331.
- Pietroni P, Young MC, Latham GJ, von Hippel PH. 1997. Structural analyses of gp45 sliding clamp interactions during assembly of the bacteriophage T4 DNA polymerase holoenzyme. I. Conformational changes within the gp44/62-gp45-ATP complex during clamp loading. J Biol Chem. 272(50):31666–31676.
- Prattes M, Lo Y-H, Bergler H, Stanley RE. 2019. Shaping the nascent ribosome: AAA-ATPases in eukaryotic ribosome biogenesis. Biomolecules. 9(11):715.
- Puchades C, Ding B, Song A, Wiseman RL, Lander GC, Glynn SE. 2019. Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol Cell. 75(5):1073–1085.e6.
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, Lander GC. 2017. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science. 358(6363):eaao0464.
- Puchades C, Sandate CR, Lander GC. 2020. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat Rev Mol Cell Biol. 21(1):43–58.
- Qu X, Wen JD, Lancaster L, Noller HF, Bustamante C, Tinoco I. 2011. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature. 475(7354):118–121.
- Reck-Peterson SL, Redwine WB, Vale RD, Carter AP. 2018. The cytoplasmic dynein transport machinery and its many cargoes. Nat Rev Mol Cell Biol. 19(6):382–398.
- Rice LM, Brunger AT. 1999. Crystal structure of the vesicular transport protein Sec17: implications for SNAP function in SNARE complex disassembly. Mol Cell. 4(1):85–95.
- Ripstein ZA, Vahidi S, Houry WA, Rubinstein JL, Kay LE. 2020. A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. Elife. 9:e52158.
- Roberts AJ, Numata N, Walker ML, Kato YS, Malkova B, Kon T, Ohkura R, Arisaka F, Knight PJ, Sutoh K, et al. 2009. AAA+ Ring and linker swing mechanism in the dynein motor. Cell. 136(3):485–495.
- Rodriguez-Aliaga P, Ramirez L, Kim F, Bustamante C, Martin A. 2016. Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat Struct Mol Biol. 23(11):974–981.
- Rose AE, Brown RSH, Schlieker C. 2015. Torsins: not your typical AAA+ ATPases. Crit Rev Biochem Mol Biol. 50(6):532–549.
- Rothman JE. 2014. The principle of membrane fusion in the cell (Nobel lecture). Angew Chem Int Ed Engl. 53(47):12676–12694.
- Ryu J-K, Min D, Rah S-H, Kim SJ, Park Y, Kim H, Hyeon C, Kim HM, Jahn R, Yoon T-Y. 2015. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science. 347(6229):1485–1489.
- Saffert P, Enenkel C, Wendler P. 2017. Structure and Function of p97 and Pex1/6 Type II AAA+ Complexes. Front Mol Biosci. 4:33.
- Sandate CR, Szyk A, Zehr EA, Lander GC, Roll-Mecak A. 2019. An allosteric network in spastin couples multiple activities required for microtubule severing. Nat Struct Mol Biol. 26(8):671–678.
- Santosh V, Musayev FN, Jaiswal R, Zárate-Pérez F, Vandewinkel B, Dierckx C, Endicott M, Sharifi K, Dryden K, Henckaerts E, et al. 2020. The Cryo-EM structure of AAV2 Rep68 in complex with ssDNA reveals a malleable AAA+ machine that can switch between oligomeric states. Nucleic Acids Res. 48(22):12983–12999.
- Saraste M, Sibbald PR, Wittinghofer A. 1990. The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 15(11):430–434.
- Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 80:587–612.
- Saunders RA, Stinson BM, Baker TA, Sauer RT. 2020. Multistep substrate binding and engagement by the AAA+ ClpXP protease. Proc Natl Acad Sci USA. 117(45):28005–28013.
- Schmidt H, Zalyte R, Urnavicius L, Carter AP. 2015. Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature. 518(7539):435–438.
- Schöneberg J, Pavlin MR, Yan S, Righini M, Lee I-H, Carlson L-A, Bahrami AH, Goldman DH, Ren X, Hummer G, et al. 2018. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science. 362(6421):1423–1428.
- Sen M, Maillard RA, Nyquist K, Rodriguez-Aliaga P, Pressé S, Martin A, Bustamante C. 2013. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 155(3):636–646.
- Seraphim TV, Houry WA. 2020. AAA+ proteins. Curr Biol. 30(6):R251–R257.
- Shah N, Colbert KN, Enos MD, Herschlag D, Weis WI. 2015. Three αSNAP and 10 ATP molecules are used in SNARE complex disassembly by N-ethylmaleimide-sensitive factor (NSF). J Biol Chem. 290(4):2175–2188.
- Shen J, Gai D, Patrick A, Greenleaf WB, Chen XS. 2005. The roles of the residues on the channel beta-hairpin and loop structures of simian virus 40 hexameric helicase. Proc Natl Acad Sci USA. 102(32):11248–11253.
- Shin M, Puchades C, Asmita A, Puri N, Adjei E, Wiseman RL, Karzai AW, Lander GC. 2020. Structural basis for distinct operational modes and protease activation in AAA+ protease Lon. Sci Adv. 6(21):eaba8404.
- Siddiqi O, Benzer S. 1976. Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci USA. 73(9):3253–3257.
- Singh A, Grover A. 2010. Plant Hsp100/ClpB-like proteins: poorly-analyzed cousins of yeast ClpB machine. Plant Mol Biol. 74(4-5):395–404.
- Snider J, Gutsche I, Lin M, Baby S, Cox B, Butland G, Greenblatt J, Emili A, Houry WA. 2006. Formation of a distinctive complex between the inducible bacterial lysine decarboxylase and a novel AAA+ ATPase. J Biol Chem. 281(3):1532–1546.
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. 1993. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75(3):409–418.
- Song C, Wang Q, Li C-CH. 2003. ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem. 278(6):3648–3655.
- Sosa BA, Demircioglu FE, Chen JZ, Ingram J, Ploegh HL, Schwartz TU. 2014. How lamina-associated polypeptide 1 (LAP1) activates Torsin. Elife. 3:e03239.
- Sosnowski P, Urnavicius L, Boland A, Fagiewicz R, Busselez J, Papai G, Schmidt H. 2018. The CryoEM structure of the Saccharomyces cerevisiae ribosome maturation factor Rea1. Elife. 7:e39163.
- Soules KR, LaBrie SD, May BH, Hefty PS. 2020. Sigma 54-regulated transcription is associated with membrane reorganization and type III secretion effectors during conversion to infectious forms of Chlamydia trachomatis. MBio. 11(5):e01725–20.
- Sousa MC, Trame CB, Tsuruta H, Wilbanks SM, Reddy VS, McKay DB. 2000. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 103(4):633–643.
- Sparks JL, Chistol G, Gao AO, Räschle M, Larsen NB, Mann M, Duxin JP, Walter JC. 2019. The CMG helicase bypasses DNA-protein cross-links to facilitate their repair. Cell. 176(1-2):167–181.e21.
- Story RM, Steitz TA. 1992. Structure of the recA protein-ADP complex. Nature. 355(6358):374–376.
- Subramanian S, Gorday K, Marcus K, Orellana MR, Ren P, Luo XR, O'Donnell ME, Kuriyan J. 2021. Allosteric communication in DNA polymerase clamp loaders relies on a critical hydrogen-bonded junction. Elife. 10:e66181.
- Südhof TC. 2013. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 80(3):675–690.
- Sugihara M, Morito D, Ainuki S, Hirano Y, Ogino K, Kitamura A, Hirata H, Nagata K. 2019. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J Cell Biol. 218(3):949–960.
- Sun S, Li L, Yang F, Wang X, Fan F, Yang M, Chen C, Li X, Wang H-W, Sui S-F. 2017. Cryo-EM structures of the ATP-bound Vps4E233Q hexamer and its complex with Vta1 at near-atomic resolution. Nat Commun. 8:16064.
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395(6700):347–353.
- Tagaya M, Wilson DW, Brunner M, Arango N, Rothman JE. 1993. Domain structure of an N-ethylmaleimide-sensitive fusion protein involved in vesicular transport. J Biol Chem. 268(4):2662–2666.
- Tang WK, Borgnia MJ, Hsu AL, Esser L, Fox T, de Val N, Xia D. 2020. Structures of AAA protein translocase Bcs1 suggest translocation mechanism of a folded protein. Nat Struct Mol Biol. 27(2):202–209.
- Tang WK, Xia D. 2016. Mutations in the human AAA+ chaperone p97 and related diseases. Front Mol Biosci. 3:79.
- Tsai Y-CC, Ye F, Liew L, Liu D, Bhushan S, Gao Y-G, Mueller-Cajar O. 2020. Insights into the mechanism and regulation of the CbbQO-type Rubisco activase, a MoxR AAA+ ATPase. Proc Natl Acad Sci USA. 117(1):381–387.
- Tsuchihashi Z, Kornberg A. 1990. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci USA. 87(7):2516–2520.
- Tsurimoto T, Shinozaki A, Yano M, Seki M, Enomoto T. 2005. Human Werner helicase interacting protein 1 (WRNIP1) functions as a novel modulator for DNA polymerase delta. Genes Cells. 10(1):13–22.
- Turner J, Hingorani MM, Kelman Z, O'Donnell M. 1999. The internal workings of a DNA polymerase clamp-loading machine. Embo J. 18(3):771–783.
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, Rapoport TA. 2019. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science. 365(6452). https://doi.org/10.1126/science.aax1033
- Vale RD. 2000. AAA proteins. Lords of the ring. J Cell Biol. 150(1):F13–9.
- van den Boom J, Meyer H. 2018. VCP/p97-mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell. 69(2):182–194.
- Vieux EF, Wohlever ML, Chen JZ, Sauer RT, Baker TA. 2013. Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc Natl Acad Sci USA. 110(22):E2002–8.
- Vivona S, Cipriano DJ, O'Leary S, Li YH, Fenn TD, Brunger AT. 2013. Disassembly of all SNARE complexes by N-ethylmaleimide-sensitive factor (NSF) is initiated by a conserved 1:1 interaction between α-soluble NSF attachment protein (SNAP) and SNARE complex. J Biol Chem. 288(34):24984–24991.
- Wagener N, Ackermann M, Funes S, Neupert W. 2011. A pathway of protein translocation in mitochondria mediated by the AAA-ATPase Bcs1. Mol Cell. 44(2):191–202.
- Wahle E, Lasken RS, Kornberg A. 1989. The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J Biol Chem. 264(5):2469–2475.
- Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. Embo J. 1(8):945–951.
- Wang J, Hartling JA, Flanagan JM. 1997. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 91(4):447–456.
- Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y. 2011. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 471(7338):331–335.
- Wang L, Myasnikov A, Pan X, Walter P. 2020. Structure of the AAA protein Msp1 reveals mechanism of mislocalized membrane protein extraction. Elife. 9
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92(6):759–772.
- Weidman PJ, Melançon P, Block MR, Rothman JE. 1989. Binding of an N-ethylmaleimide-sensitive fusion protein to Golgi membranes requires both a soluble protein(s) and an integral membrane receptor. J Cell Biol. 108(5):1589–1596.
- Wendler P, Ciniawsky S, Kock M, Kube S. 2012. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 1823(1):2–14.
- White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT. 2018. Structural principles of SNARE complex recognition by the AAA+ protein NSF. Elife. 7:e38888.
- Whiteheart SW, Brunner M, Wilson DW, Wiedmann M, Rothman JE. 1992. Soluble N-ethylmaleimide-sensitive fusion attachment proteins (SNAPs) bind to a multi-SNAP receptor complex in Golgi membranes. J Biol Chem. 267(17):12239–12243.
- Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. 1994. N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol. 126(4):945–954.
- Winter U, Chen X, Fasshauer D. 2009. A conserved membrane attachment site in alpha-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J Biol Chem. 284(46):31817–31826.
- Wohlever ML, Baker TA, Sauer RT. 2014. Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol Microbiol. 91(1):66–78.
- Wortmann SB, Ziętkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, Muntau AC, Rakovic A, Renkema GH, Rodenburg RJ, et al. 2015. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. 96(2):245–257.
- Yamada K, Miyata T, Tsuchiya D, Oyama T, Fujiwara Y, Ohnishi T, Iwasaki H, Shinagawa H, Ariyoshi M, Mayanagi K, et al. 2002. Crystal structure of the RuvA-RuvB complex: a structural basis for the Holliday junction migrating motor machinery. Mol Cell. 10(3):671–681.
- Yang Y, Darbari VC, Zhang N, Lu D, Glyde R, Wang Y-P, Winkelman JT, Gourse RL, Murakami KS, Buck M, et al. 2015. TRANSCRIPTION. Structures of the RNA polymerase-σ54 reveal new and conserved regulatory strategies. Science. 349(6250):882–885.
- Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, Corbett KD. 2015. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife. 4:e07367.
- Yedidi RS, Wendler P, Enenkel C. 2017. AAA-ATPases in protein degradation. Front Mol Biosci. 4:42.
- Yoshimura A, Seki M, Enomoto T. 2017. The role of WRNIP1 in genome maintenance. Cell Cycle. 16(6):515–521.
- Yu RC, Hanson PI, Jahn R, Brünger AT. 1998. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol. 5(9):803–811.
- Yu RC, Jahn R, Brunger AT. 1999. NSF N-terminal domain crystal structure: models of NSF function. Mol Cell. 4(1):97–107.
- Zehr E, Szyk A, Piszczek G, Szczesna E, Zuo X, Roll-Mecak A. 2017. Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat Struct Mol Biol. 24(9):717–725.
- Zhang K, Li S, Hsieh K-Y, Su S-C, Pintilie GD, Chiu W, Chang C-I. 2020. Molecular basis for the ATPase-powered substrate translocation by the Lon AAA+ protease. bioRxiv.
- Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT. 2015. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 518(7537):61–67.
- Zimmermann F, Serna M, Ezquerra A, Fernandez-Leiro R, Llorca O, Luders J. 2020. Assembly of the asymmetric human γ-tubulin ring complex by RUVBL1-RUVBL2 AAA ATPase. Sci Adv. 6(51):eabe0894.